HTS-Driven Discovery of New Chemotypes with West Nile Virus Inhibitory Activity

Abstract
:1. Introduction
2. Results and Discussion
2.1. Assay optimization and implementation of live WNV for HTS

2.2. Single dose HTS results

2.3. Dose response assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Supplier ID | EC50 | CC50 | SI50 |
|---|---|---|---|
| SRI-2176 | 1.9 | 16.21 | 8.53 |
| SRI-10806 | 6.15 | 36.75 | 5.98 |
| SRI-22003 | 12.18 | >100 | >8.2 |
| SRI-11928 | 12.83 | 23.39 | 1.82 |
| SRI-795809 | 13.88 | >100 | >7.2 |
| SRI-1001309 | 16.94 | >100 | >5.9 |
| SRI-16635 | 18.4 | >100 | >5.4 |
| SRI-16537 | 18.64 | >100 | >5.4 |
| SMR000370276 | 18.9 | >100 | >5.3 |
| SRI-1665808 | 19.58 | >100 | >5.1 |
| SMR000394098 * | 19.6 | >100 | >5.1 |
| SMR000059052 * | 20.01 | >100 | >5.0 |
| SRI-12784 | 22.91 | >100 | >4.4 |
| SRI-12896 | 23.43 | >100 | >4.3 |
| SMR000027739 * | 29.58 | >100 | >3.4 |
| SMR000171908 | 30.35 | >100 | >3.3 |
| SRI-4578 * | 30.39 | >100 | >3.3 |
| SRI-2967 | 34.11 | >100 | >2.9 |
| SRI-18457 | 34.22 | >100 | >2.9 |
| SRI-6050 | 37.34 | 60.02 | 1.61 |
| SRI-19093 | 40.96 | >100 | >2.4 |
| SRI-6488 | 41.99 | >100 | >2.4 |
| SRI-1055 | 51.58 | >100 | >1.9 |
| SRI-13037 | 58.68 | >100 | >1.7 |
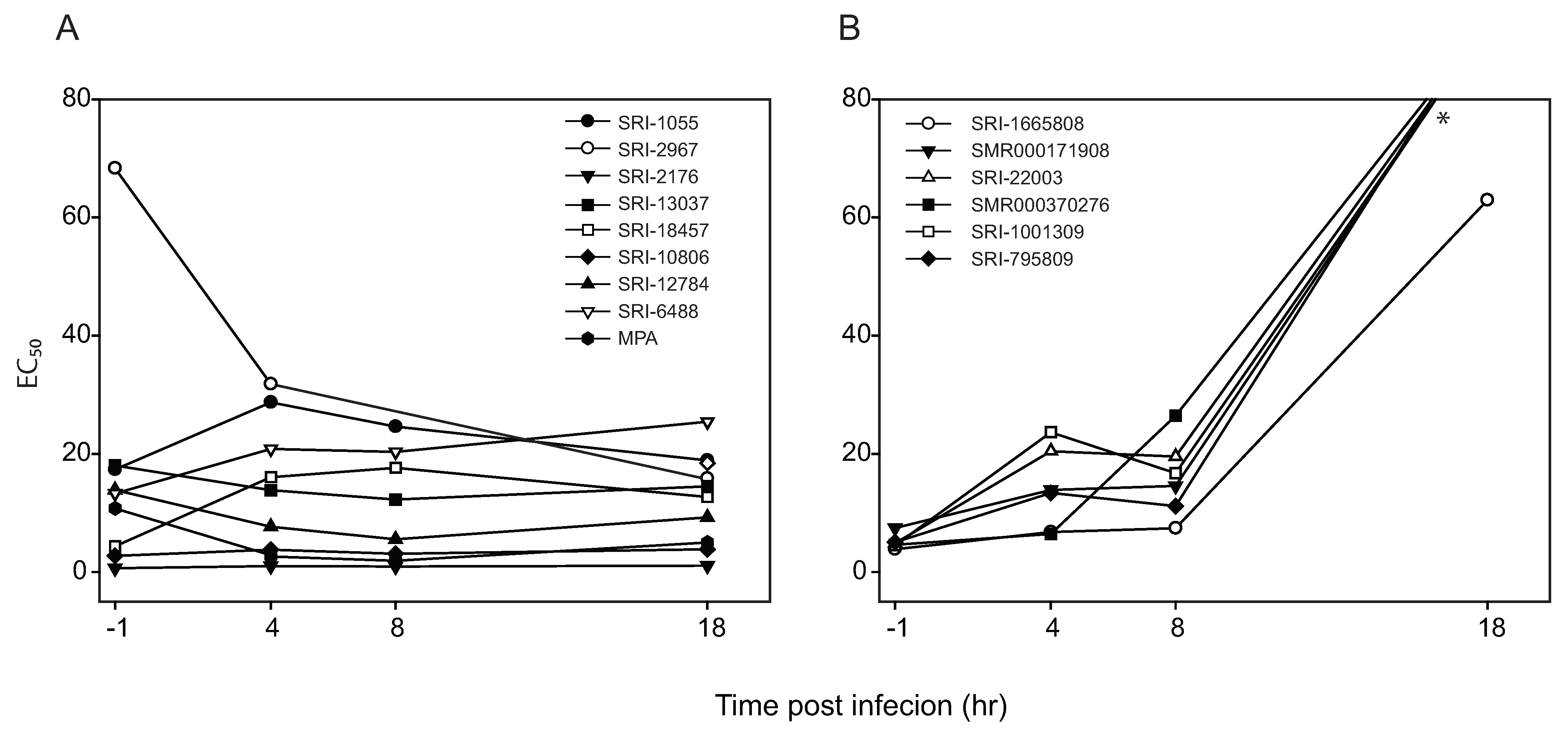
2.4. Time of addition assay

| EC50 change pattern | Example |
|---|---|
| No Change : EC501hr ≈ EC504hr ≈ EC508hr ≈ EC5018hr | MPA, SRI-12784 |
| Increase: EC501hr ≈ EC504hr ≤ EC508hr < EC5018hr | SRI-1665808, SRI-795809, SRI-1001309, SRI-22003, SMR00171908, SMR000370276 |
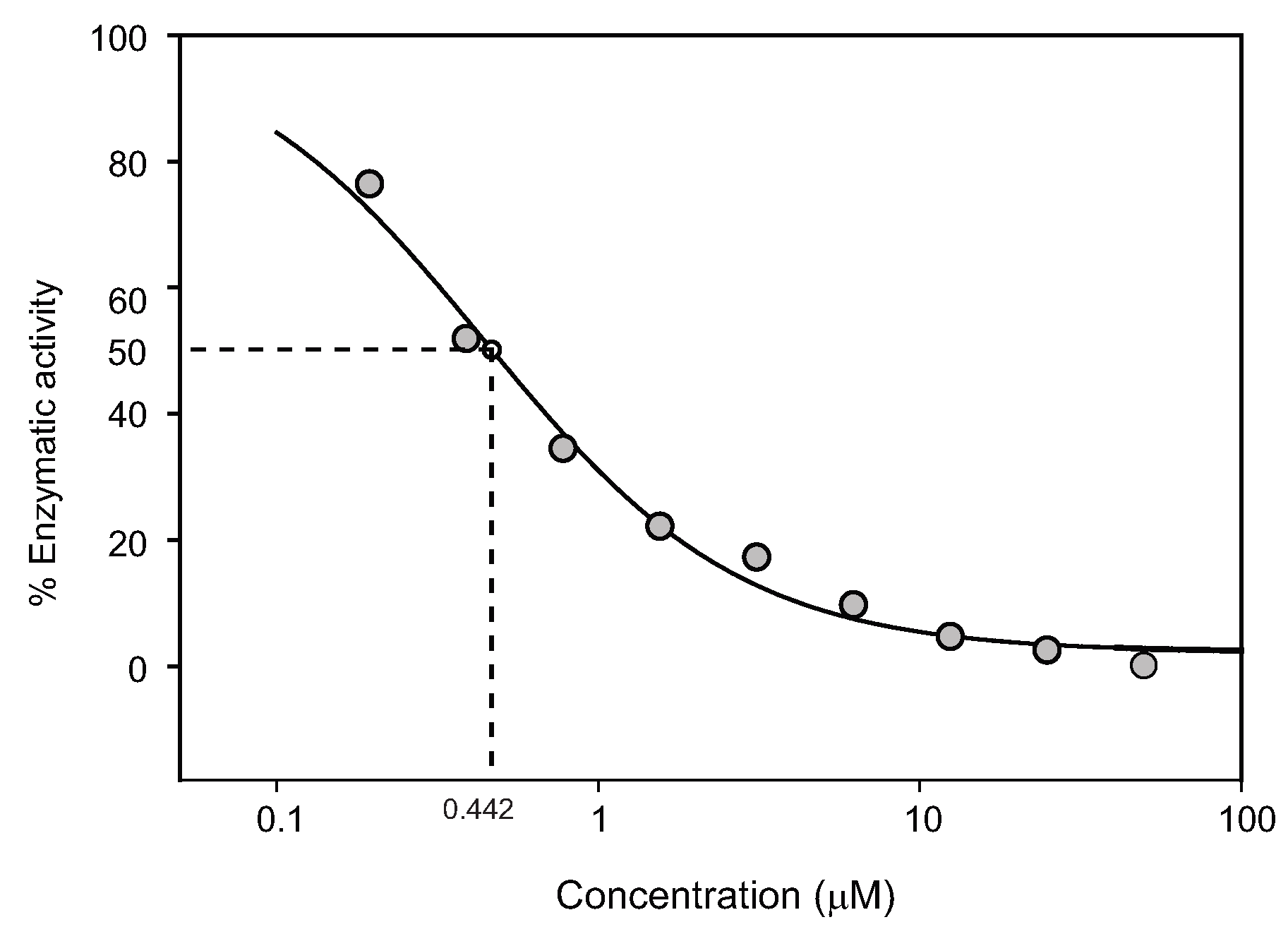
2.5. WNV NS2b-NS3pro assay

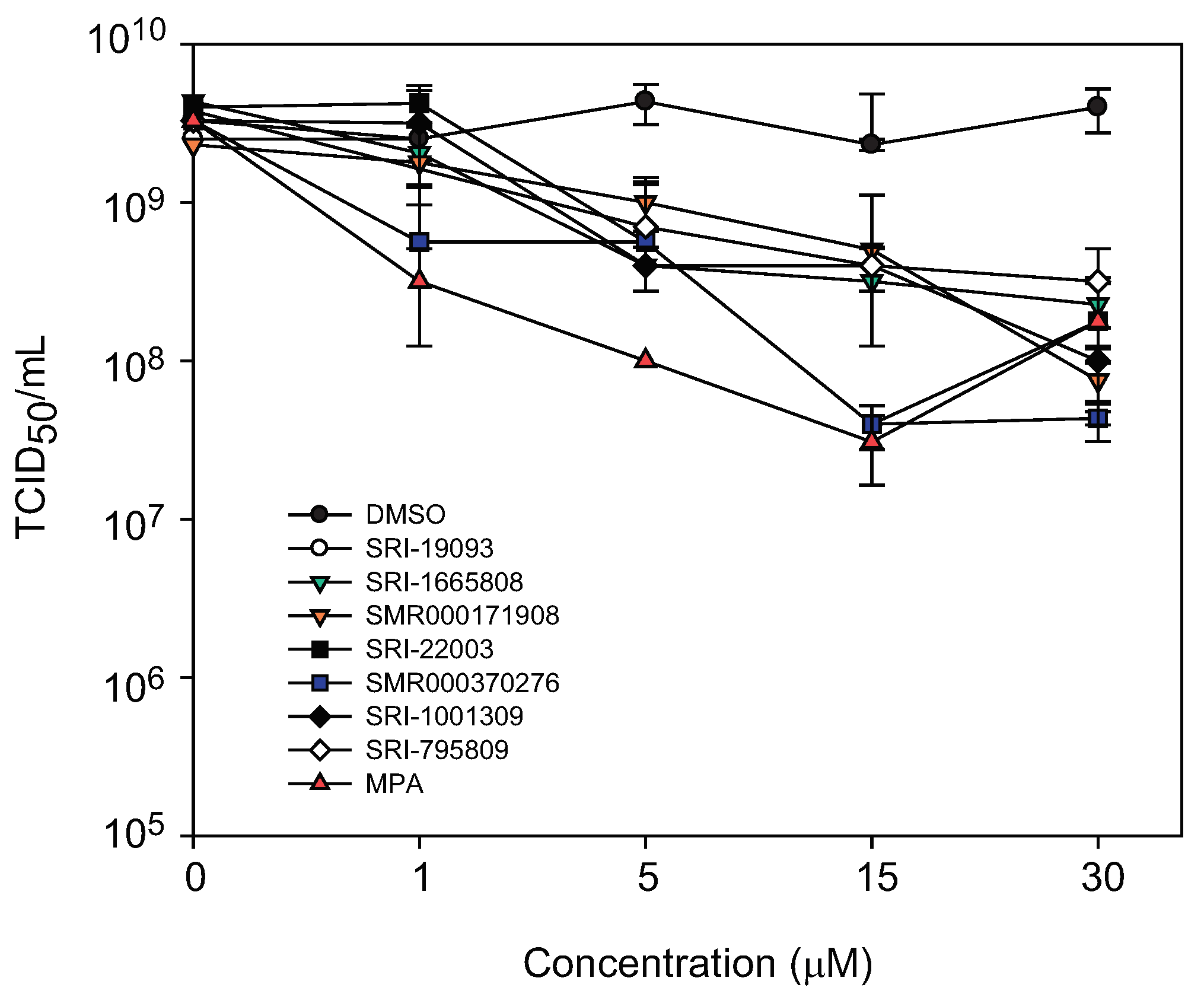
2.6. Titer reduction assay


- * All of the compounds with weak correlation in the correlation analysis have turned out to be “replication dependent” in the time of addition assay.
- ** SRI-16635 was inactive in the time of addition assay (data not shown) but showed anti-NS2b-NS3pro activity in the protease assay.
| Compound I.D. | Structure | Dose response in CPE based | Dose response in Titer reduction | SI50 * | NS2B-NS3pro | EC50 change pattern over the course of infection | ||
|---|---|---|---|---|---|---|---|---|
| EC50 (μM) | CC50 (μM) | EC50 (μM) | EC90 (μM) | EC50 (μM) | ||||
| SRI-22003 |  | 12.18 | >30 | 4.70 | 5.13 | >6 | Not active | Increase |
| SRI-795809 | Structure not disclosed | 13.88 | >30 | 1.14 | 26.16 | >30 | Not active | Increase |
| SRI-1001309 | Structure not disclosed | 16.94 | >30 | 5.46 | 16.69 | >6 | Not active | Increase |
| SMR000370276 |  | 18.9 | >30 | No solution | 6.07 | >5 | Not active | Increase |
| SRI-1665808 | Structure not disclosed | 19.58 | >30 | 1.29 | 7.22 | >30 | Not active | Increase |
| SMR000171908 |  | 30.35 | >30 | 1.16 | 6.73 | >25 | Not active | Increase |
| SRI-19093 |  | 40.96 | >30 | 0.13 | 2.14 | >200 | 0.44 | Increase |
| MPA |  | >30 | 0.03 | 0.94 | >1000 | Not active | no change | |
| N-Phenylanthranilic acid |  | NT | NT | NT | NT | NT | >50 | not active |
2.7. Discussion
3. Experimental
3.1. Cells and Virus
3.2. Viral Assay
3.3. Compound Library Composition and Plating
3.4. High-Throughput Screen
3.5. Antiviral Efficacy and Cytotoxicity in Dose-Response and Time of Addition Assays
3.6. Statistics and Data Analysis
3.7. WNV NS2b-NS3pro Assay
3.8. Titer Reduction Assay
4. Conclusion
Acknowledgement
References
- Guharoy, R.; Gilroy, S.A.; Noviasky, J.A.; Ference, J. West Nile virus infection. Am. J. Health Syst. Pharm. 2004, 61, 1235–1241. [Google Scholar] [PubMed]
- Briese, T.; Xi.-Yu, J.; Huang, C.; Grady, L.J.; Lipkin, W.I. Identification of a Kunjin/West Nile-like flavivirus in brains of patients with New York encephalitis. Lancet 1999, 354, 1261–1262. [Google Scholar] [CrossRef]
- Outbreak of West Nile-like viral encephalitis. MMWR: Morbidity & Mortality Weekly Report 1999, 48, 845–849.
- West Nile Virus Activity – United States, 2007. MMWR: Morbidity & Mortality Weekly Report 2008, 57, 720–723.
- Samuel, M.A.; Diamond, M.S. Pathogenesis of West Nile Virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J. Virol. 2006, 80, 9349–9360. [Google Scholar] [CrossRef] [PubMed]
- Hayes, E.B.; Gubler, D.J. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu. Rev. Med. 2006, 57, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Gubler, D.J. The continuing spread of West Nile virus in the western hemisphere. Clin. Infect. Dis. 2007, 45, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Brinton, M.A. The molecular biology of west nile virus: a new invader of the western hemisphere. Annu. Rev. Microbiol. 2002, 56, 371–402. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J.; Weir, R.C.; Grakoui, A.; McCourt, D.W.; Bazan, J.F.; Fletterick, R.J.; Rice, C.M. Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein. Proc. Natl. Acad. Sci. USA 1990, 87, 8898–8902. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.K.; Kuhn, R.J.; Smith, J.L. Functional characterization of cis and trans activity of the Flavivirus NS2B-NS3 protease. J. Biol. Chem. 2007, 282, 12883–12892. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.J.; Ng, M.L. The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J. Gen. Virol. 2003, 84, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt, M.C.; Michaelis, M.; Ogbomo, H.; Doerr, H.W.; Cinatl, J., Jr. Inhibition of apoptosis prevents West Nile virus induced cell death. BMC Microbiol. 2007, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Parquet, M.C.; Kumatori, A.; Hasebe, F.; Morita, K.; Igarashi, A. West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 2001, 500, 17–24. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S.; Ahmed, A.A.; Valentine, L.E.; Davis, C.W.; Samuel, M.A.; Hanna, S.L.; Puffer, B.A.; Doms, R.W. An infectious West Nile virus that expresses a GFP reporter gene. Virology 2005, 334, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.K.; Tilgner, M.; Shi, P.Y. Potential high-throughput assay for screening inhibitors of West Nile virus replication. J. Virol. 2003, 77, 12901–12906. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.L.; Zhao, Q.; O’Donnell, V.K.; Mason, P.W. Adaptation of West Nile virus replicons to cells in culture and use of replicon-bearing cells to probe antiviral action. Virology 2005, 331, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Puig-Basagoiti, F.; Deas, T.S.; Ren, P.; Tilgner, M.; Ferguson, D.M.; Shi, P.Y. High-throughput assays using a luciferase-expressing replicon, virus-like particles, and full-length virus for West Nile virus drug discovery. Antimicrob. Agents Chemother. 2005, 49, 4980–4988. [Google Scholar] [CrossRef] [PubMed]
- Goodell, J.R.; Puig-Basagoiti, F.; Forshey, B.M.; Shi, P.Y.; Ferguson, D.M. Identification of compounds with anti-West Nile Virus activity. J. Med. Chem. 2006, 49, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Ouzunov, S.; Wang, L.; Mason, P.; Bourne, N.; Cuconati, A.; Block, T.M. Discovery of small molecule inhibitors of West Nile virus using a high-throughput sub-genomic replicon screen. Antiviral Res. 2006, 70, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Noah, J.W.; Severson, W.; Noah, D.L.; Rasmussen, L.; White, E.L.; Jonsson, C.B. A cell-based luminescence assay is effective for high-throughput screening of potential influenza antivirals. Antiviral Res. 2007, 73, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Severson, W.E.; McDowell, M.; Ananthan, S.; Chung, D.H.; Rasmussen, L.; Sosa, M.I.; White, E.L.; Noah, J.; Jonsson, C.B. High-throughput screening of a 100,000-compound library for inhibitors of influenza A virus (H3N2). J. Biomol. Screen 2008, 13, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Morrey, J.D.; Smee, D.F.; Sidwell, R.W.; Tseng, C. Identification of active antiviral compounds against a New York isolate of West Nile virus. Antiviral Res. 2002, 55, 107–116. [Google Scholar] [CrossRef]
- TAN, Y.H.; Driscoll, J.S.; Sim, M.M. Compounds for the Treatment of viral-mediated disease. WO 01/24785 A2; 9/29/2000,
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Severson, W.E.; Shindo, N.; Sosa, M.; Fletcher, T., 3rd; White, E.L.; Ananthan, S.; Jonsson, C.B. Development and validation of a high-throughput screen for inhibitors of SARS CoV and its application in screening of a 100,000–compound library. J. Biomol. Screen 2007, 12, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 1938, 27, 493–497. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chung, D.H.; Jonsson, C.B.; Maddox, C.; McKellip, S.N.; Moore, B.P.; Heil, M.; White, E.L.; Ananthan, S.; Li, Q.; Feng, S.; et al. HTS-Driven Discovery of New Chemotypes with West Nile Virus Inhibitory Activity. Molecules 2010, 15, 1690-1704. https://doi.org/10.3390/molecules15031690
Chung DH, Jonsson CB, Maddox C, McKellip SN, Moore BP, Heil M, White EL, Ananthan S, Li Q, Feng S, et al. HTS-Driven Discovery of New Chemotypes with West Nile Virus Inhibitory Activity. Molecules. 2010; 15(3):1690-1704. https://doi.org/10.3390/molecules15031690
Chicago/Turabian StyleChung, Dong Hoon, Colleen B. Jonsson, Clinton Maddox, Sara N. McKellip, Blake. P. Moore, Marintha Heil, E. Lucile White, Subramaniam Ananthan, Qianjun Li, Shuang Feng, and et al. 2010. "HTS-Driven Discovery of New Chemotypes with West Nile Virus Inhibitory Activity" Molecules 15, no. 3: 1690-1704. https://doi.org/10.3390/molecules15031690