Norcoclaurine Synthase: Mechanism of an Enantioselective Pictet-Spengler Catalyzing Enzyme

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Enzymatic Bifunctional Catalysis
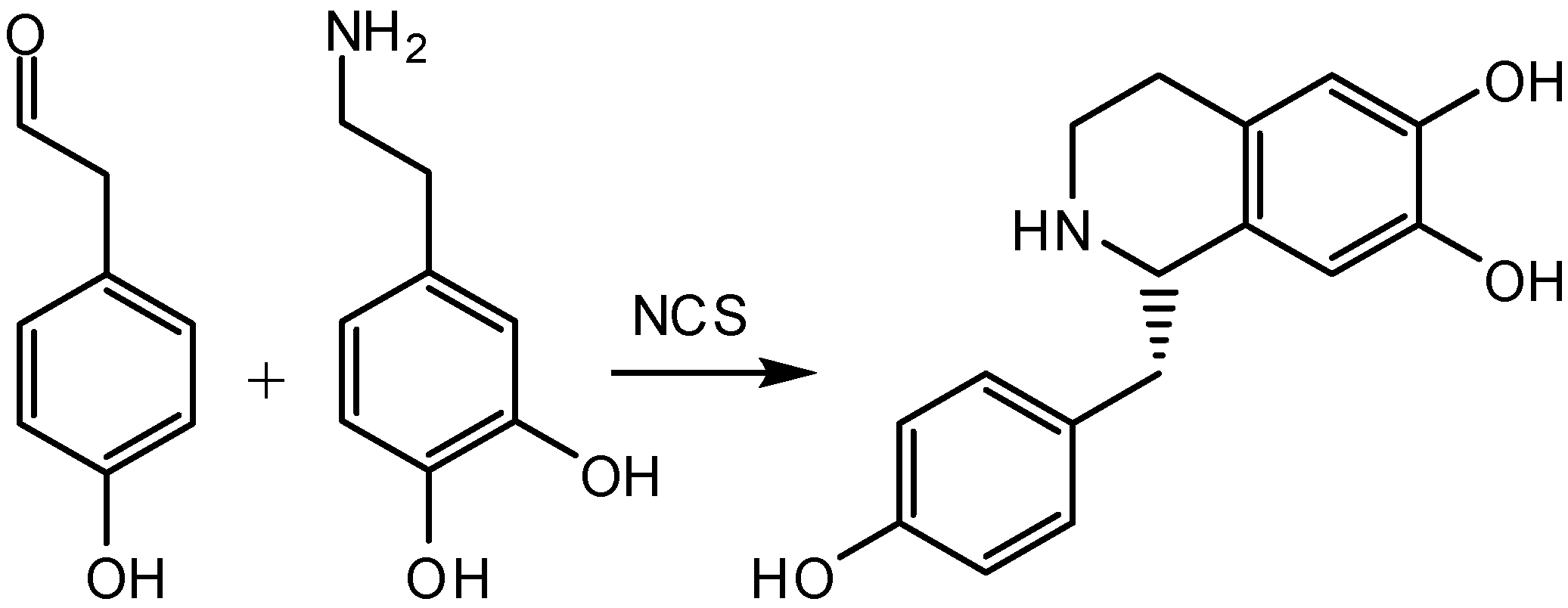
2. Benzylisoquinoline Alkaloids Production
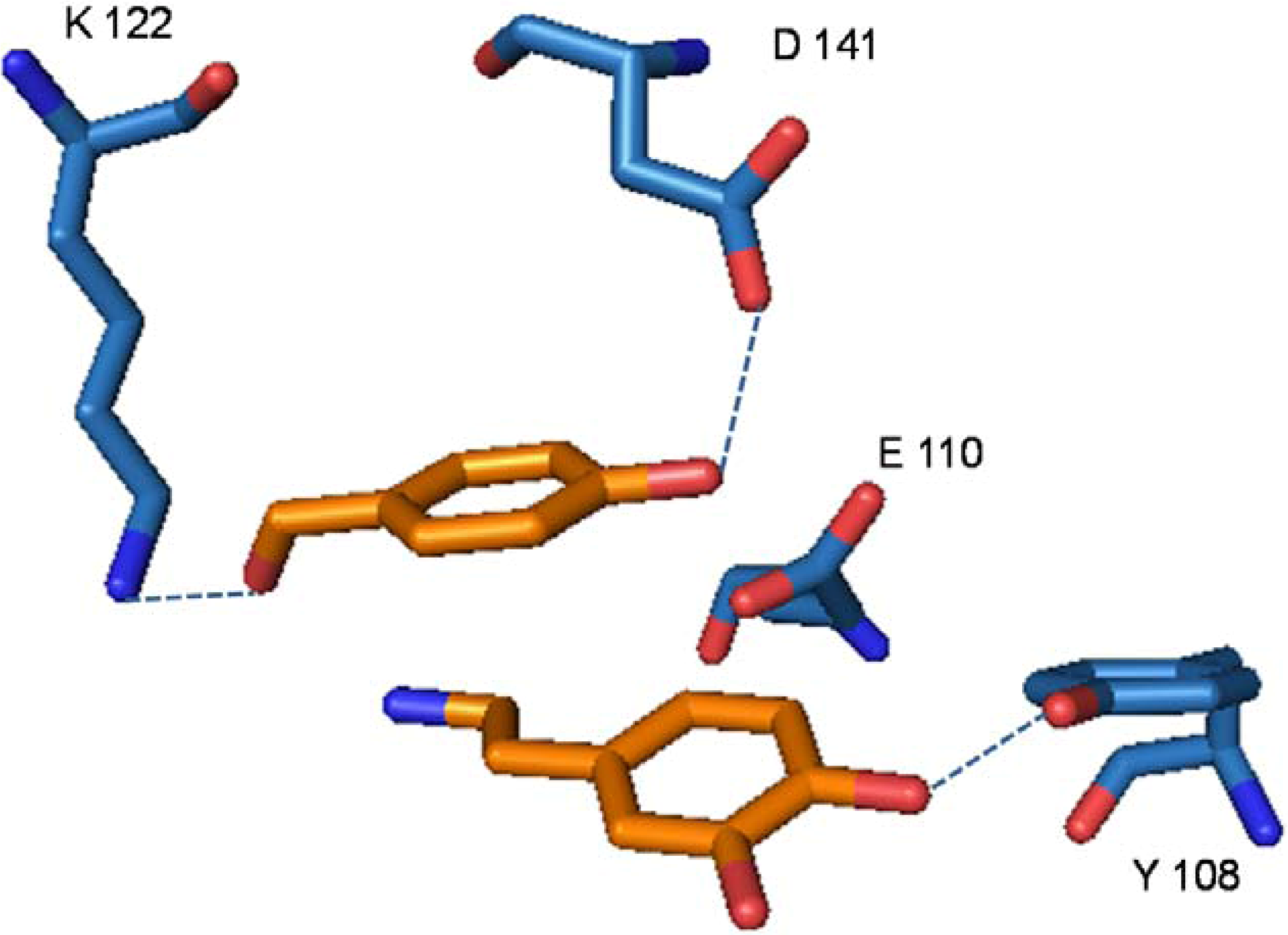
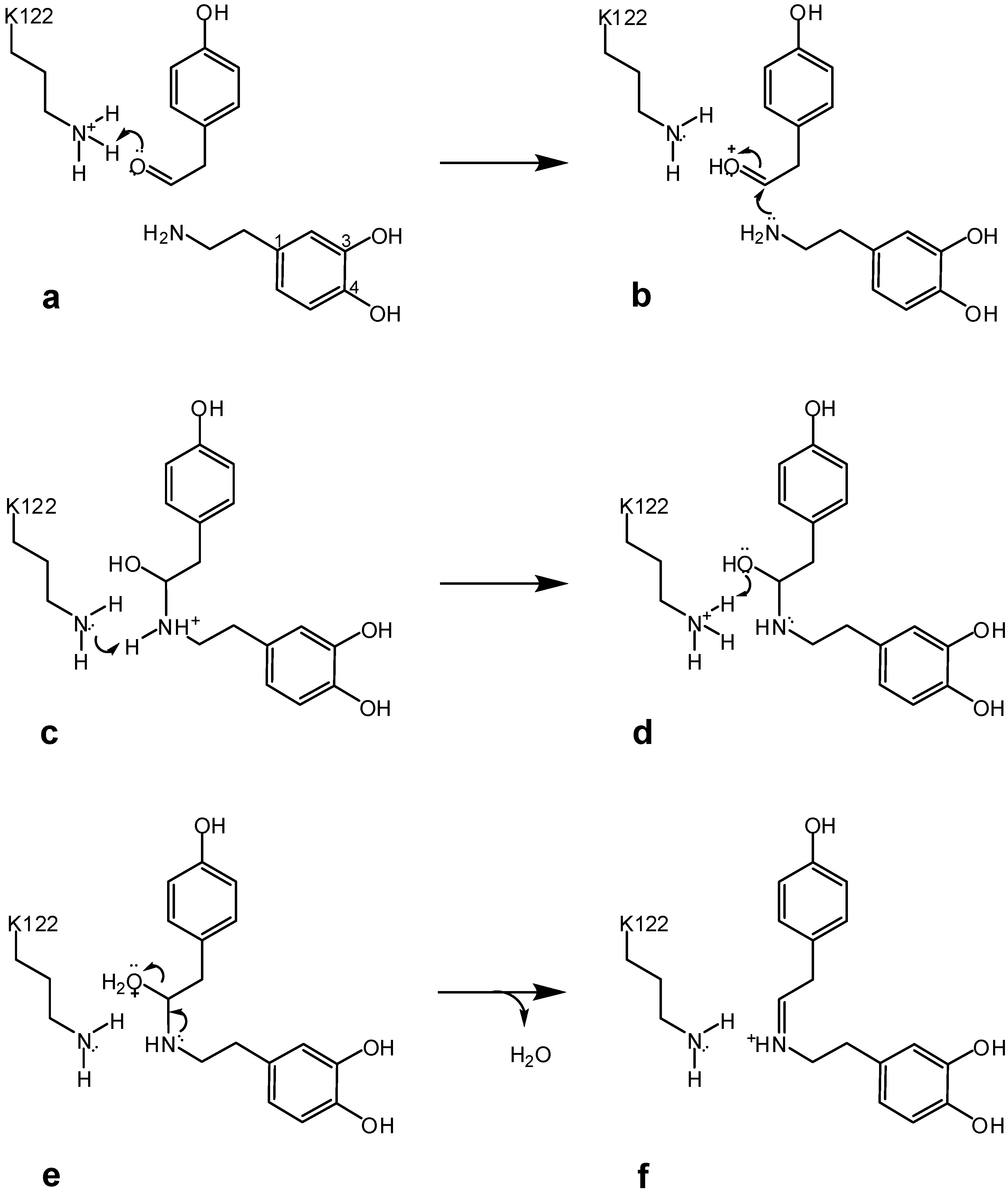
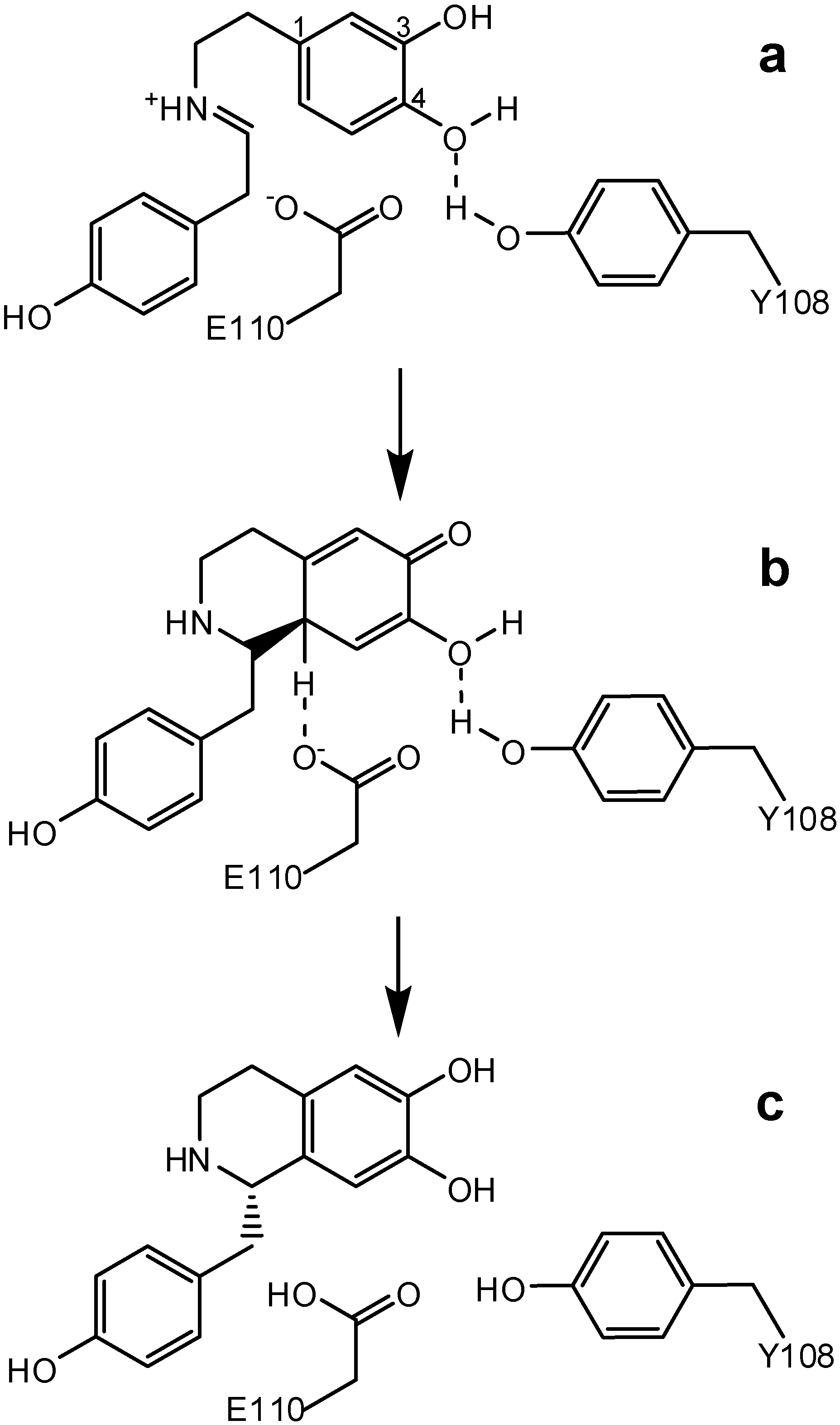
3. Bifunctional Catalysis in the Enzymatic Synthesis of (S)-Norcoclaurine

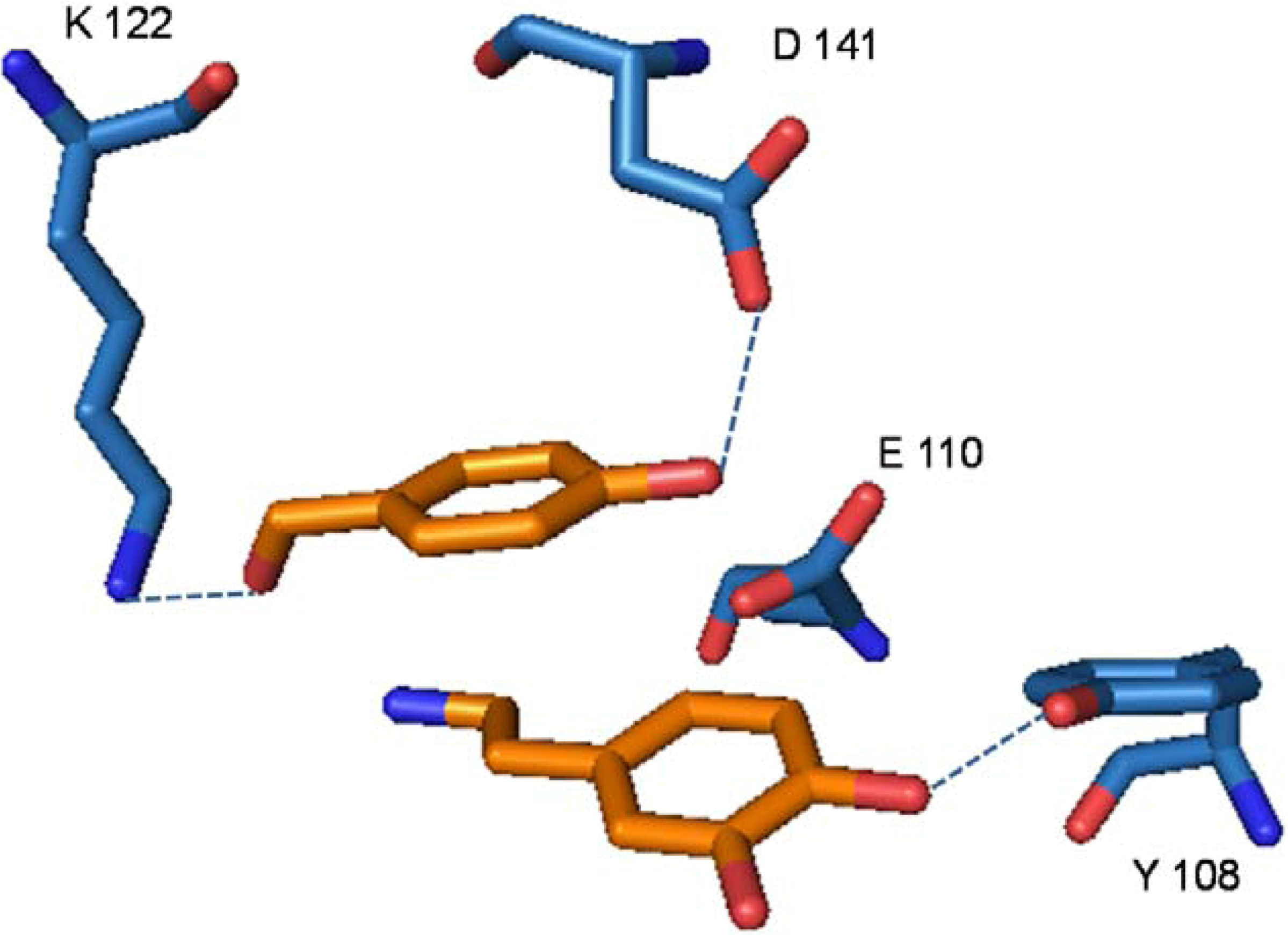


4. Conclusions
References and Notes
- Cucinotta, C.S.; Kosa, M.; Melchiorre, P.; Cavalli, A.; Gervasio, F. Bifunctional Catalysis by Natural Cinchona Alkaloids: a Mechanism Explained. Chem. Eur. J. 2009, 15, 7913–7921. [Google Scholar]
- Setoyama, T. Acid-Base Bifunctional Catalysis: an Industrial Point of View. Catal. Today 2006, 116, 250–262. [Google Scholar] [CrossRef]
- Koeller, K.M.; Wong, C.H. Enzymes for Chemical Synthesis. Nature 2001, 409, 232–240. [Google Scholar] [CrossRef]
- Toney, M.D.; Kirsch, J.F. Brønsted Analysis of Aspartate Aminotransferase via Exogenous Catalysis of Reactions of an Inactive Mutant. Prot. Sci. 1992, 1, 107–119. [Google Scholar]
- Li, Y.K.; Chir, J.; Tanaka, S.; Chen, F.Y. Identification of the General Acid/Base Catalyst of a Family 3 β-glucosidase from Flavobacterium Meningosepticum. Biochemistry 2002, 41, 2751–2759. [Google Scholar] [CrossRef]
- Schafer, S.L.; Barrett, W.C.; Kallarakal, A.T.; Mitra, B.; Kozarich, J.W.; Gerlt, J.A. Mechanism of the Reaction Catalyzed by Mandelate Racemase: Structure and Mechanistic Properties of the D270N Mutant. Biochemistry 1996, 35, 5662–5669. [Google Scholar]
- Rodríguez, S.M.; Las Heras-Vázquez, F.J.; Mingorance-Cazorla, L.; Clemente-Jiménez, J.M.; Rodríguez-Vico, F. Molecular Cloning, Purification, and Biochemical Characterization of Hydantoin Racemase from the Legume Symbiont Sinorhizobium meliloti CECT 4114. Appl. Environ. Microbiol. 2004, 70, 625–630. [Google Scholar] [CrossRef]
- Strauss, U.T.; Faber, K. Deracemization of (±)-Mandelic Acid Using a Lipase–Mandelate Racemase Two-Enzyme System. Tetrahedron Asymmetry 1999, 10, 4079–4081. [Google Scholar] [CrossRef]
- Miyazawa, T.; Yukawa, T.; Koshiba, T.; Sakamoto, H.; Ueji, S.; Yanagihara, R.; Yamada, T. Resolution of 2-Aryloxy-1-Propanols via Lipase-Catalyzed Enantioselective Acylation in Organic Media. Tetrahedron Asymmetry 2001, 12, 1595–1602. [Google Scholar] [CrossRef]
- Facchini, P.J. Alkaloids Biosinthesys in Plants: Biochemistry, Cell Biology, Molecular Regulation, and Metabolic Engineering Applications. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 29–66. [Google Scholar] [CrossRef]
- Facchini, P.J.; St-Pierre, B. Synthesis and Trafficking of Alkaloid Biosynthetic Enzymes. Curr. Opin. Plant. Biol. 2005, 8, 657–666. [Google Scholar] [CrossRef]
- Stadler, R.; Zenk, M.H. A Revision of the Generally Accepted Pathway for the Biosynthesis of the Benzyltetrahydroisoquinoline Alkaloid Reticuline. Liebigs Ann. Chem. 1990, 6, 555–562. [Google Scholar] [CrossRef]
- Pasquo, A.; Bonamore, A.; Franceschini, S.; Macone, A.; Boffi, A.; Ilari, A. Cloning, Expression, Crystallization and Preliminary X-ray Data Analysis of Norcoclaurine Synthase from Thalictrum Flavum. Acta Crystallogr., Struct. Biol. Cryst. Commun. 2008, 64, 281–283. [Google Scholar]
- Ilari, A.; Franceschini, S.; Bonamore, A.; Arenghi, F.; Botta, B.; Macone, A.; Pasquo, A.; Bellucci, L.; Boffi, A. Structural Basis of Enzymatic (S)-Norcoclaurine Biosynthesis. J. Biol. Chem. 2009, 284, 897–904. [Google Scholar]
- Luk, L.Y.; Bunn, S.; Liscombe, D.K.; Facchini, P.J.; Tanner, M.E. Mechanistic Studies on Norcoclaurine Synthase of Benzylisoquinoline Alkaloid Biosynthesis: an Enzymatic Pictet-Spengler Reaction. Biochemistry 2007, 46, 10153–10161. [Google Scholar] [CrossRef]
- Pyo, M.K.; Lee, D.H.; Kim, D.H.; Lee, J.H.; Moon, C.H.; Chang, K.C.; Yun-Choi, H.S. Enantioselctive Synthesis of (R)-(+) and (S)-(-) Higenamine and their Analogues with Effects on Platelet Aggregation and Experimental Animal Model of Disseminated Intravascular Coagulation. Bioorg. Med. Chem. Lett. 2008, 18, 4110–4114. [Google Scholar] [CrossRef]
- Park, C.W.; Chang, K.C.; Lim, J.K. Effects of Higenamine on Isolated Heart Adrenoceptor of Rabbit. Arch. Int. Pharmacodyn. 1984, 167, 279–288. [Google Scholar]
- Chang, K.C.; Chong, W.S.; Lee, I.J. Different Pharmacological Characteristics of Structurally Similar Benzylisoquinoline Analogs, Papaverine, Higenamine, and GS 839, on Isolated Rat Aorta and Heart. Can. J. Physiol. Pharmacol. 1994, 72, 327–334. [Google Scholar] [CrossRef]
- Chong, W.S.; Lee, Y.S.; Kang, Y.J.; Lee, D.H.; Ryu, J.C.; Yun-Choi, H.S.; Chang, K.C. Comparison of Inodilator Effect of Higenamine, YS-49, YS-51, Tetrahydroisoquinoline Analogs, and Dobutamine in the Rat. Korean J. Physiol. Pharmacol. 1998, 2, 323–340. [Google Scholar]
- Yun-Choi, H.S.; Pyo, M.K.; Park, K.M.; Chang, K.C.; Lee, D.H. Antithrombotic Effects of Higenamine. Planta Med. 2001, 67, 619–622. [Google Scholar] [CrossRef]
- Bao, Y.X.; Yu, G.R.; Xu, J.M.; Xu, Y.Q.; Bian, Y.T.; Zheng, D.S. Effect of Acute Higenamine Administration on Bradyarrhythmias and His Bundle: a Clinical Study of 14 Cases and Animal Experiment on Dogs. Chin. Med. J. 1982, 95, 781–784. [Google Scholar]
- Zhang, Z.; Liu, X.; Tao, Z.; Shi, R.; Zhang, X.; Yao, Z.; Liu, Y.; Zhu, K.; Chen, B. Effects of Higenamine on Hemodynamics and its Tolerability and Safety, an Experimental Study. Zhonghua Yi Xue Za Zhi 2002, 82, 352–355. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bonamore, A.; Barba, M.; Botta, B.; Boffi, A.; Macone, A. Norcoclaurine Synthase: Mechanism of an Enantioselective Pictet-Spengler Catalyzing Enzyme. Molecules 2010, 15, 2070-2078. https://doi.org/10.3390/molecules15042070
Bonamore A, Barba M, Botta B, Boffi A, Macone A. Norcoclaurine Synthase: Mechanism of an Enantioselective Pictet-Spengler Catalyzing Enzyme. Molecules. 2010; 15(4):2070-2078. https://doi.org/10.3390/molecules15042070
Chicago/Turabian StyleBonamore, Alessandra, Marco Barba, Bruno Botta, Alberto Boffi, and Alberto Macone. 2010. "Norcoclaurine Synthase: Mechanism of an Enantioselective Pictet-Spengler Catalyzing Enzyme" Molecules 15, no. 4: 2070-2078. https://doi.org/10.3390/molecules15042070