New Chiral Phosphoramidite Complexes of Iron as Catalytic Precursors in the Oxidation of Activated Methylene Groups

Abstract
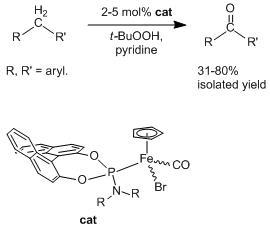
:
1. Introduction
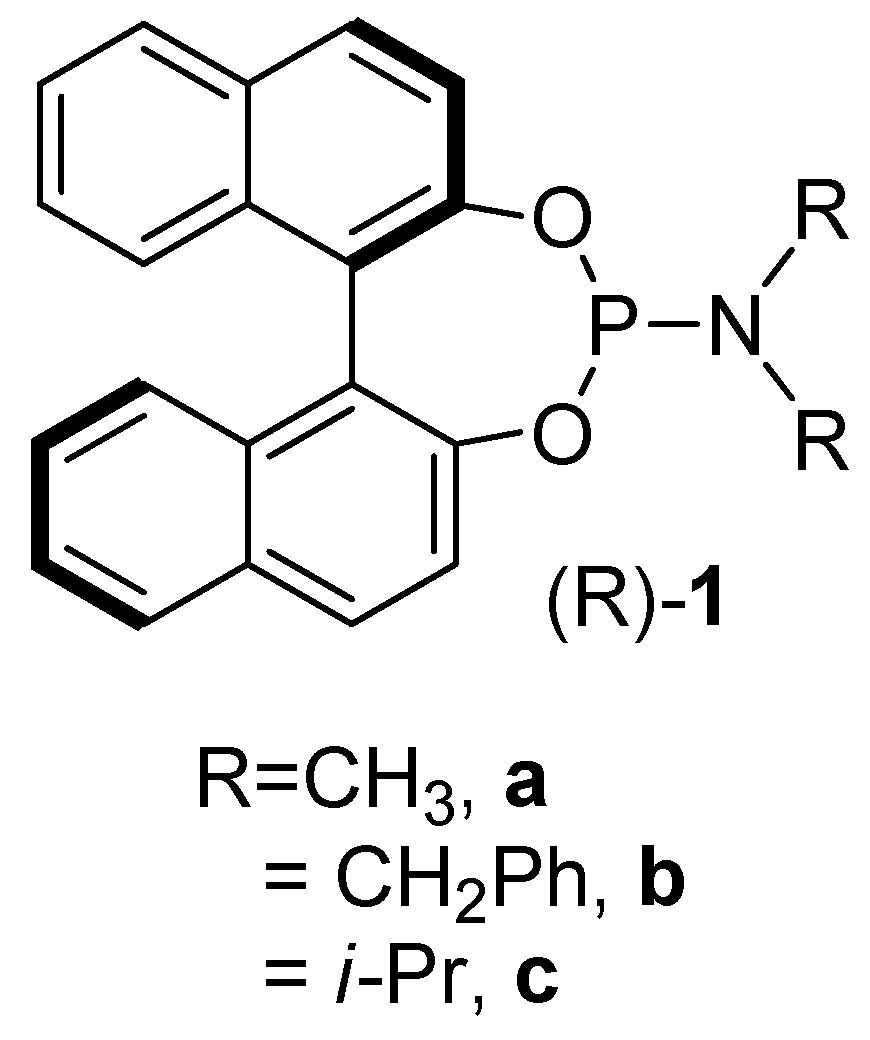
2. Results and Discussion
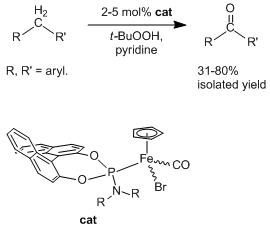
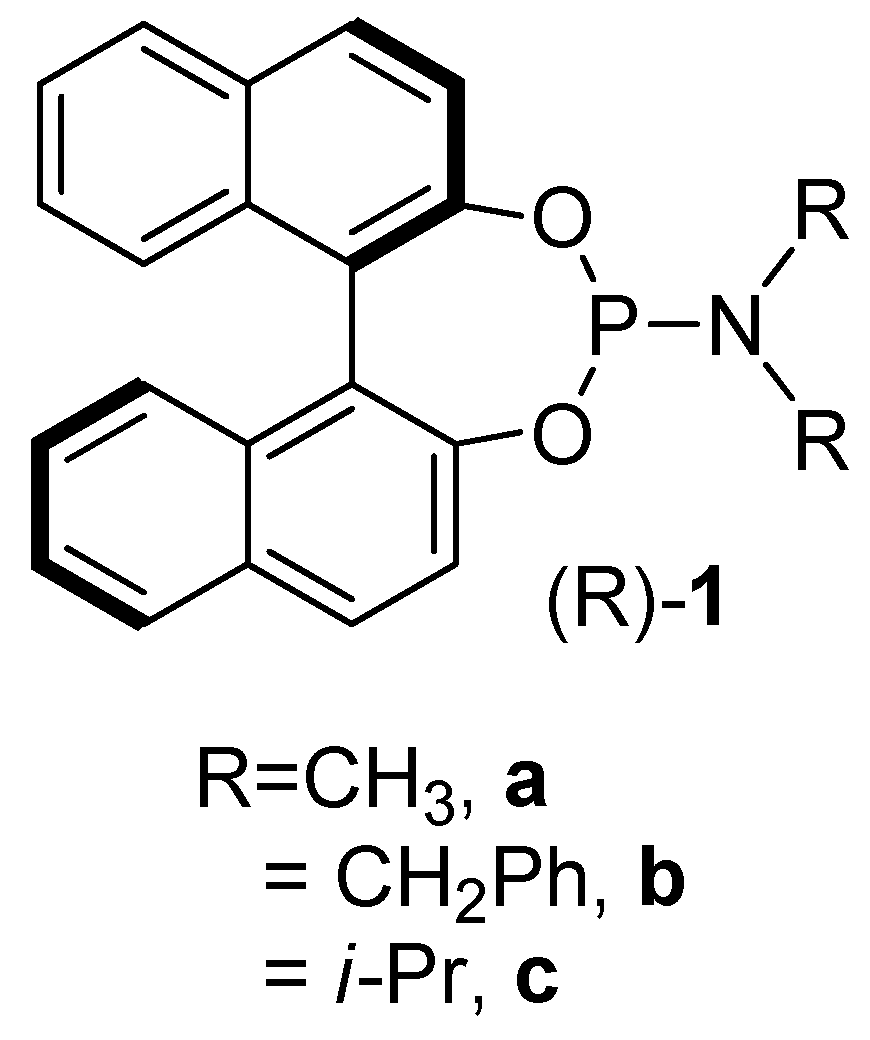
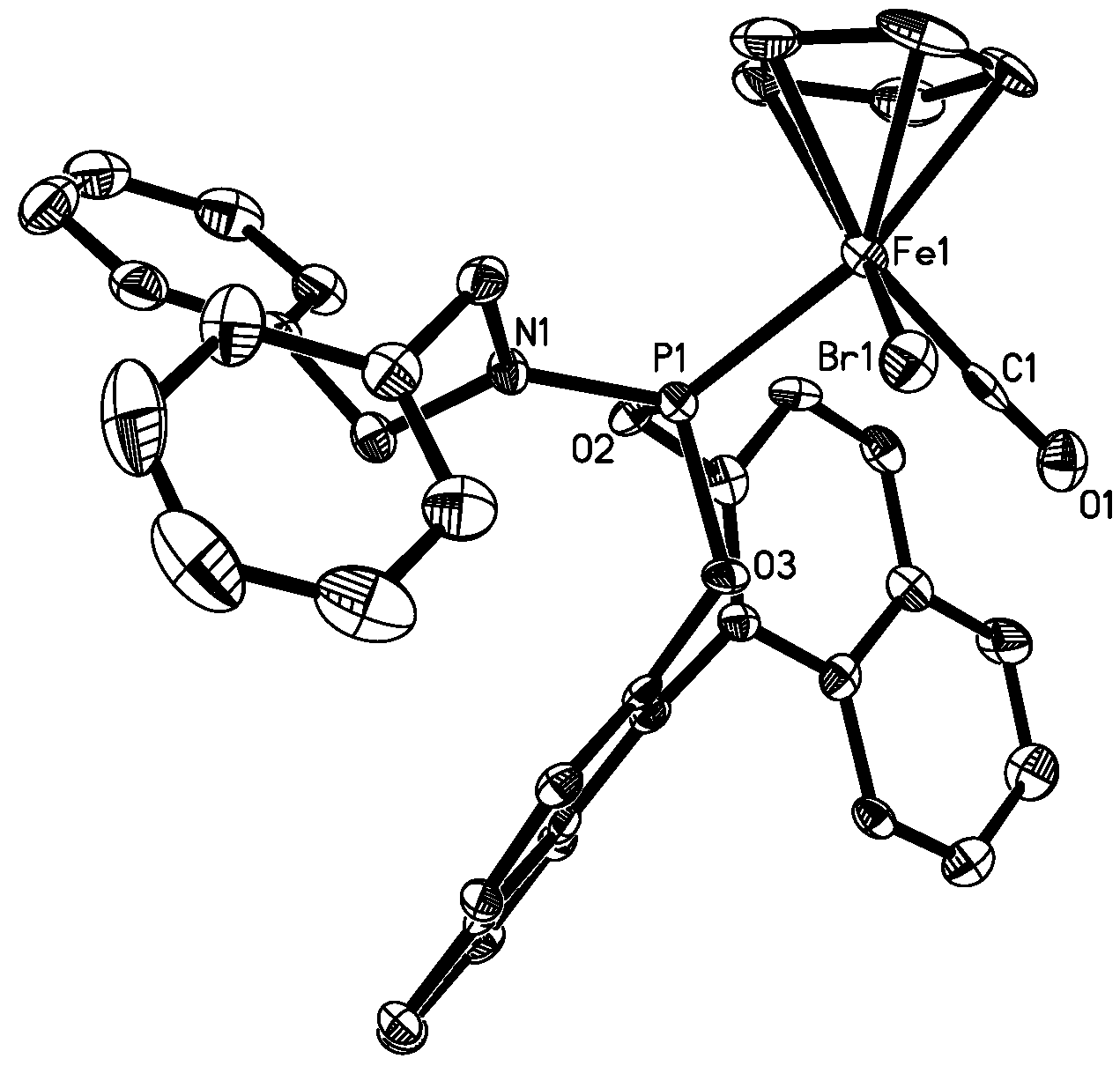
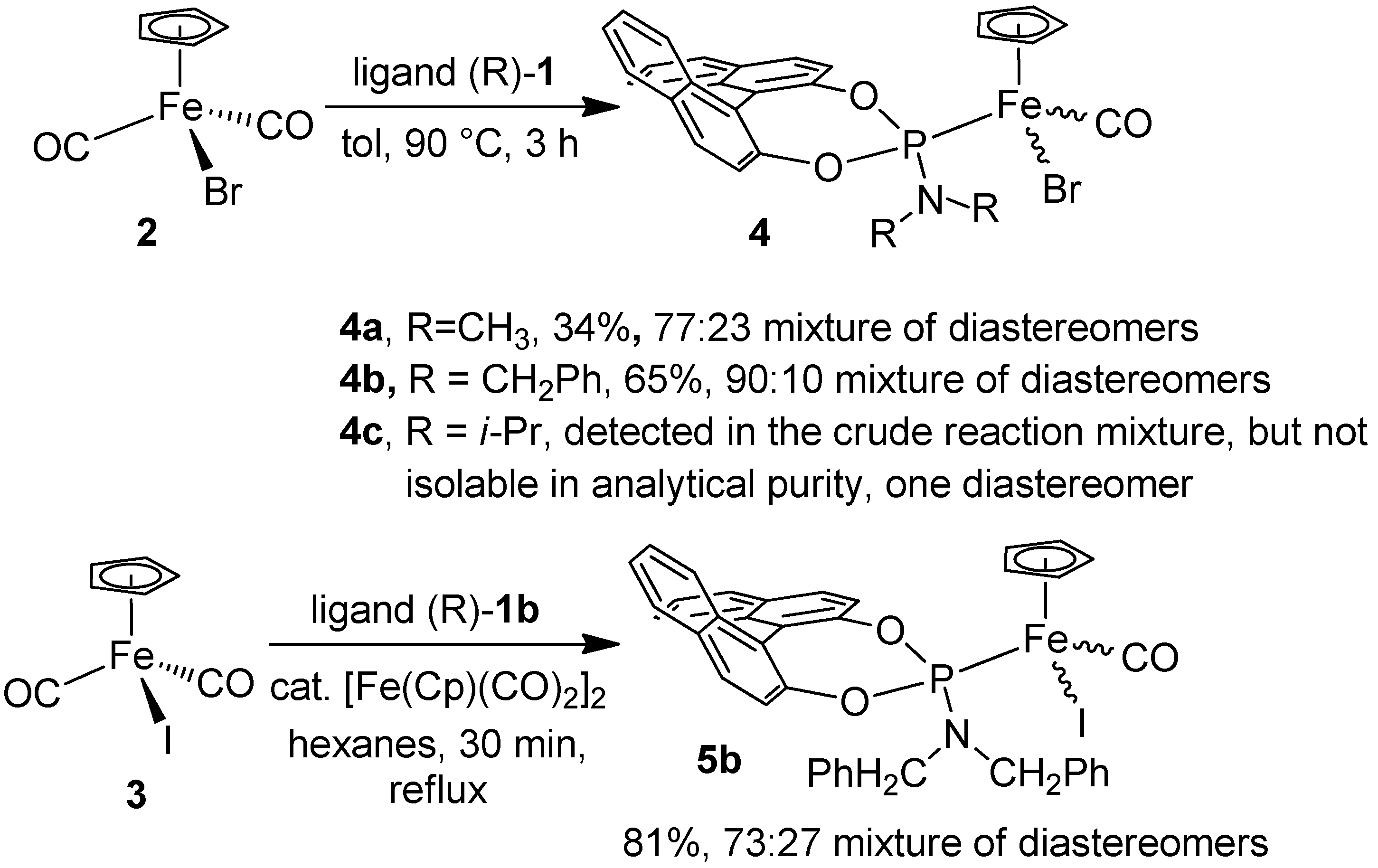
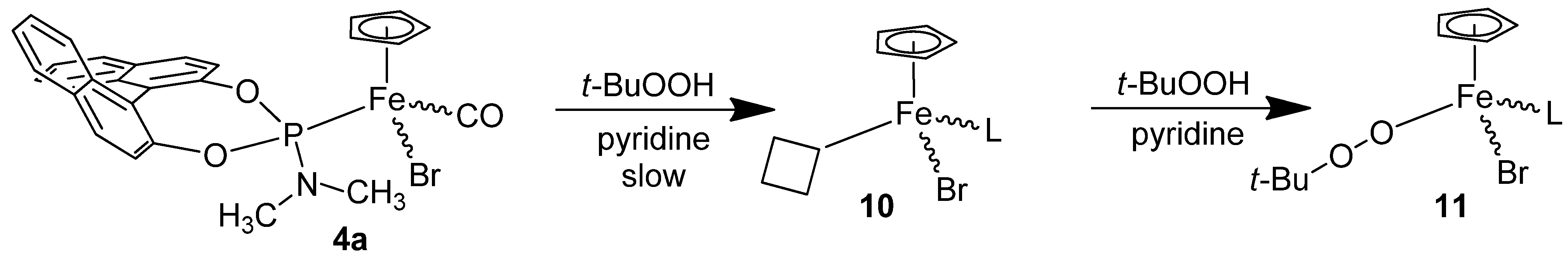
2.1. Synthesis of the Iron Phosphoramidite Complexes


| Empirical formula | C40H31BrFeNO3P (CH2Cl2)2 | C40H31FeINO3P (C6H14) |
|---|---|---|
| Formula weight | 910.24 | 873.55 |
| Temperature, Wavelength | 100(2) K, 0.71073 Å | 100(2) K, 0.71073 Å |
| Crystal system, Space group | Orthorhombic, P212121 | Orthorhombic, P212121 |
| Unit cell dimensions | a = 10.3609(5) Å | a = 10.2311(8) Å |
| b = 17.6708(8) Å | b = 14.9124(11) Å | |
| c = 21.2889(10) Å | c = 26.1055(19) Å | |
| α = β = γ = 90 ° | α = β = γ = 90 ° | |
| Volume, Z | 3897.7(3) Å3, 4 | 3982.9(5) Å3, 4 |
| Density (calculated) | 1.551 Mg/m3 | 1.457 Mg/m3 |
| Absorption coefficient | 1.769 mm−1 | 1.236 mm−1 |
| Crystal size | 0.57 x 0.13 x 0.08 mm3 | 0.31 x 0.07 x 0.06 mm3 |
| Theta range for data collection | 1.50 to 26.78° | 2.07 to 24.99° |
| Reflections collected | 40286 | 56634 |
| Independent reflections | 8268 [R(int) = 0.0520] | 7016 [R(int) = 0.1303] |
| Absorption correction | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max. and min. transmission | 0.8714 and 0.4316 | 0.9340 and 0.7044 |
| Data / restraints / parameters | 8268 / 0 / 478 | 7016 / 72 / 472 |
| Goodness-of-fit on F2 | 1.02 | 1.06 |
| Final R indices [I>2sigma(I)] | R1 = 0.0304, wR2 = 0.0555 | R1 = 0.0566, wR2 = 0.1052 |
| R indices (all data) | R1 = 0.0444, wR2 = 0.0593 | R1 = 0.0922, wR2 = 0.1176 |
| Absolute structure parameter | 0.003(5) | 0.04(3) |
| Largest diff. peak and hole | 0.553 and -0.353 e.Å−3 | 0.993 and -0.932 e.Å−3 |
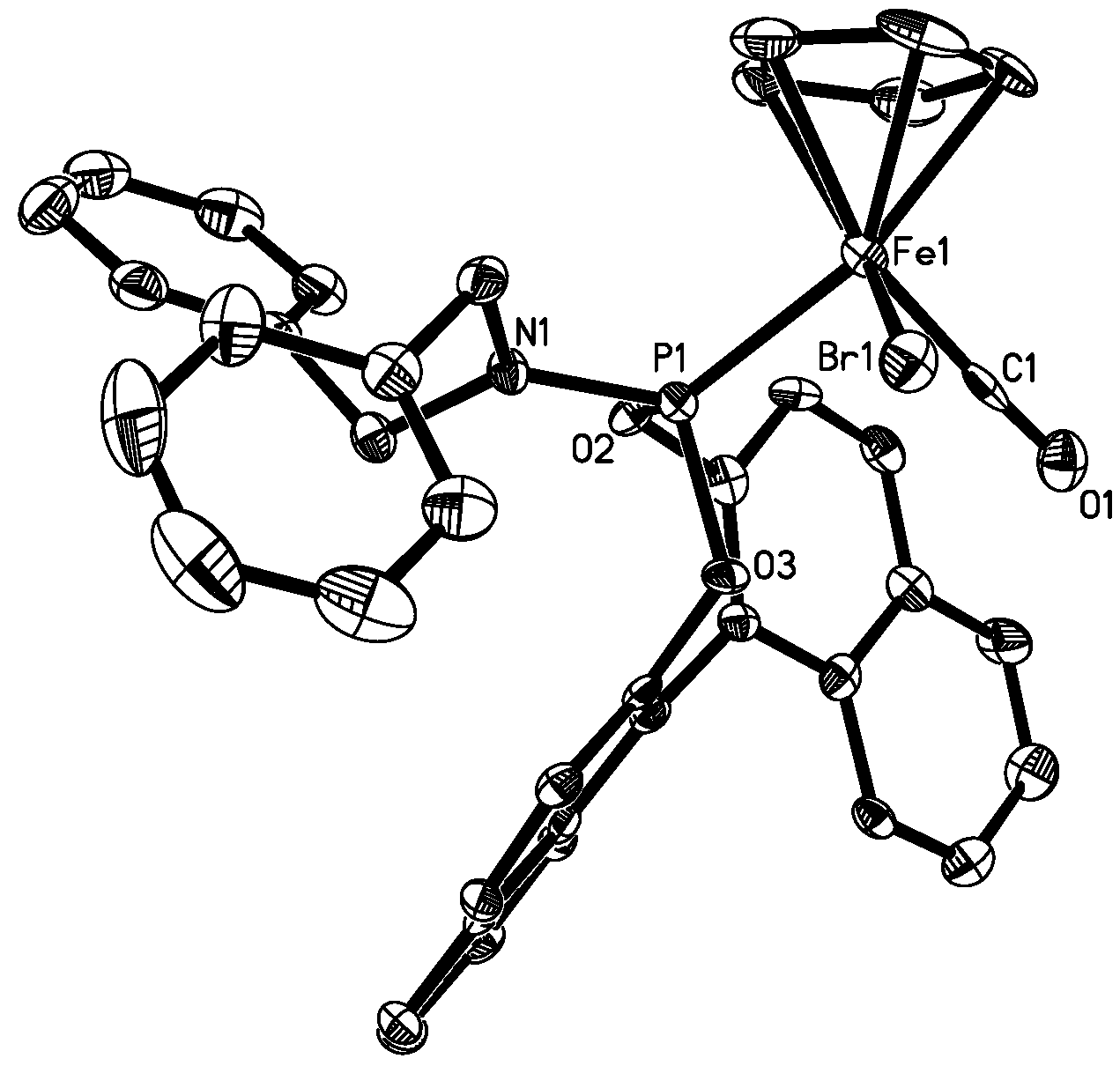
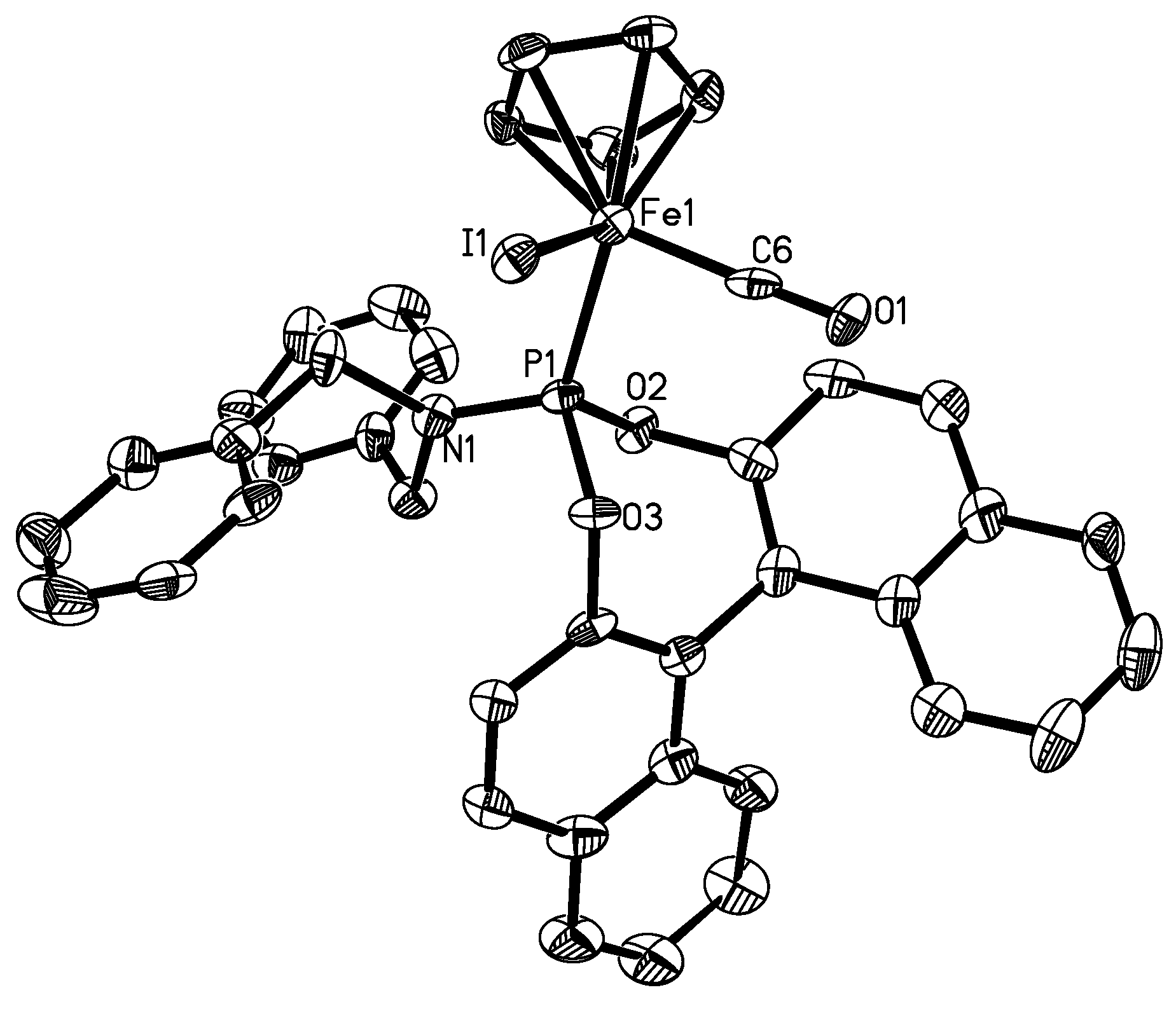
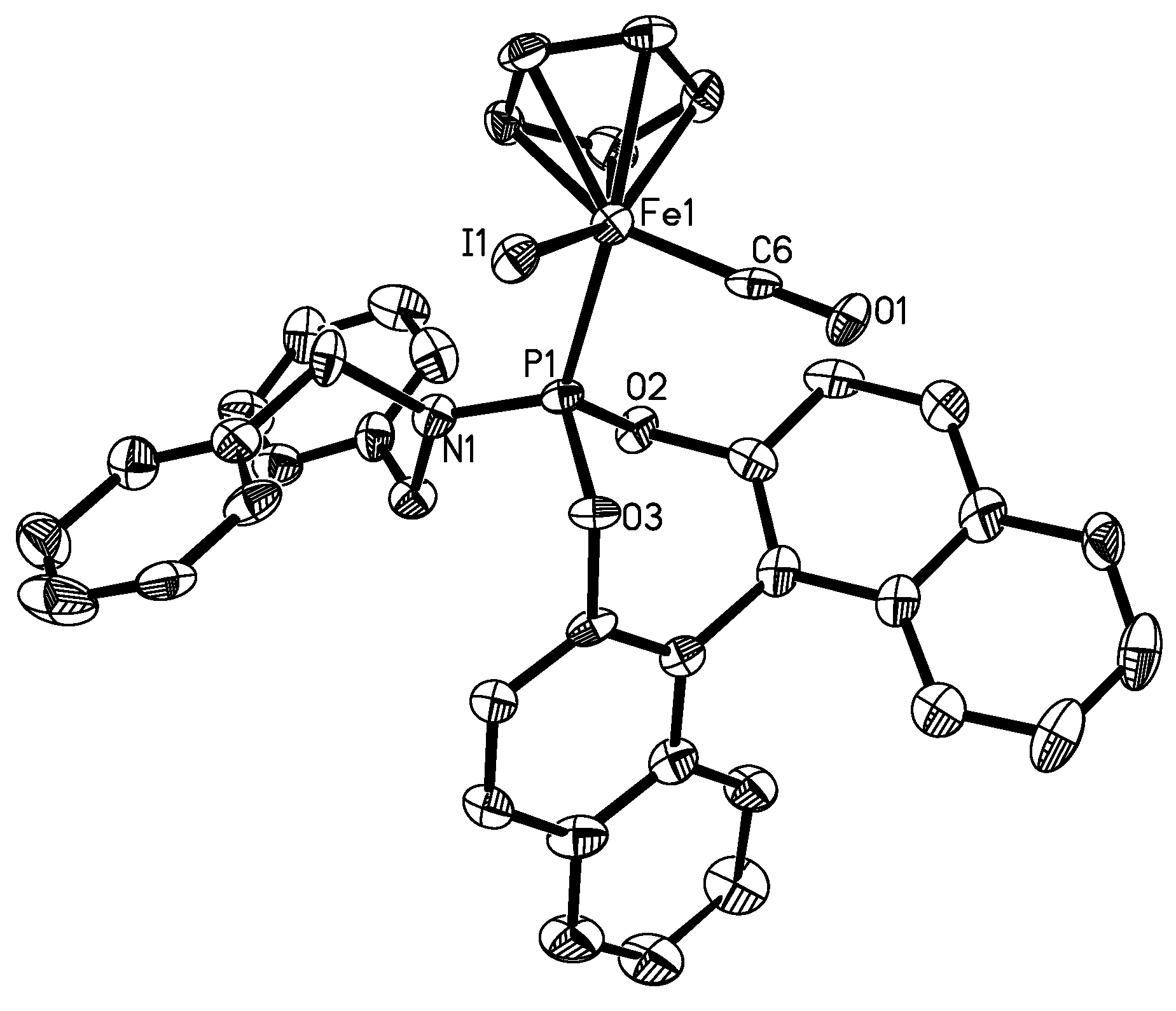
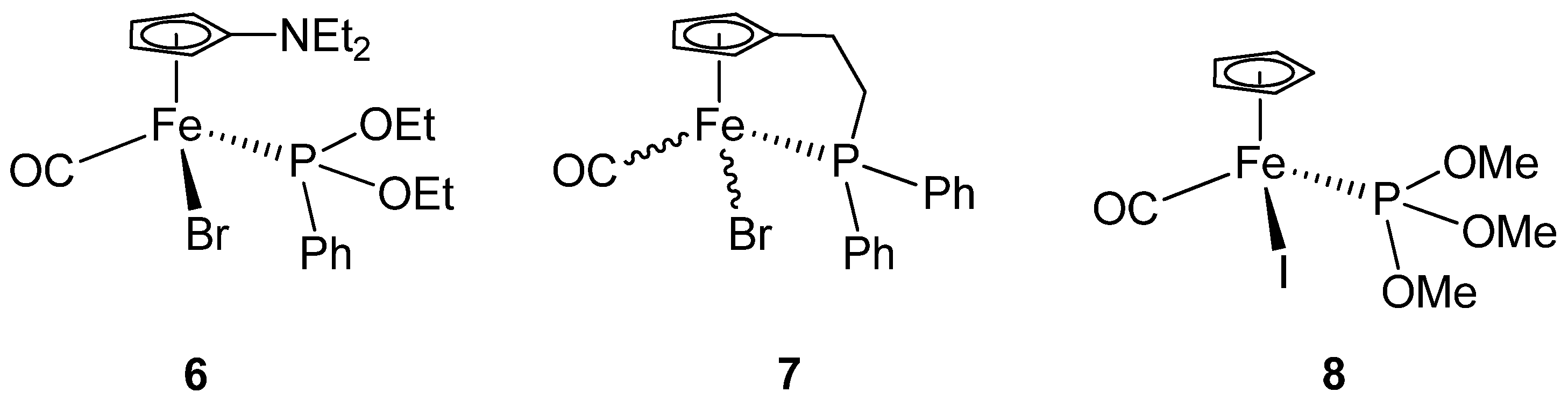
| Complex 4b (X=Br, Y=1) | 5b (X=I, Y=6) | 6 (X=Br) | 7 (X=Br) | 8 (X=I) | |
|---|---|---|---|---|---|
| X-Fe | 2.4399(5) | 2.5992(13) | 2.437(0) | 2.433 | 2.605(2) |
| Fe-C(Y) | 1.778(3) | 1.770(10) | 1.744(3) | 1.740 | 1.764(6) |
| C(Y)-O(1) | 1.118(3) | 1.149(10) | 1.136(4) | 1.195 | 1.077(7) |
| Fe-P | 2.1501(7) | 2.150(3) | 2.163(1) | 2.201 | 2.149(2) |
| P-N | 1.642(2) | 1.632(7) | - | - | |
| C(Y)-Fe-P | 90.76(8) | 91.1(3) | 95.0(1) | 95.20 | 92.9(2) |
| C(Y)-Fe-X | 93.17(9) | 89.5(3) | 96.02 | 91.47 | 92.8(2) |
| P-Fe-X | 92.03(2) | 95.58(8) | 91.73(3) | 96.20 | 93.3(1) |
| O(1)-C(Y)-Fe | 176.5(2) | 177.8(7) | 176.06 | 170.20 | 177.3(7) |
| N-P-Fe | 121.54(8) | 121.0(3) | - | - | - |
| O(3)-P-O(2) | 100.36(9) | 100.0(3) | 104.32 | - | - |

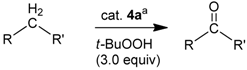
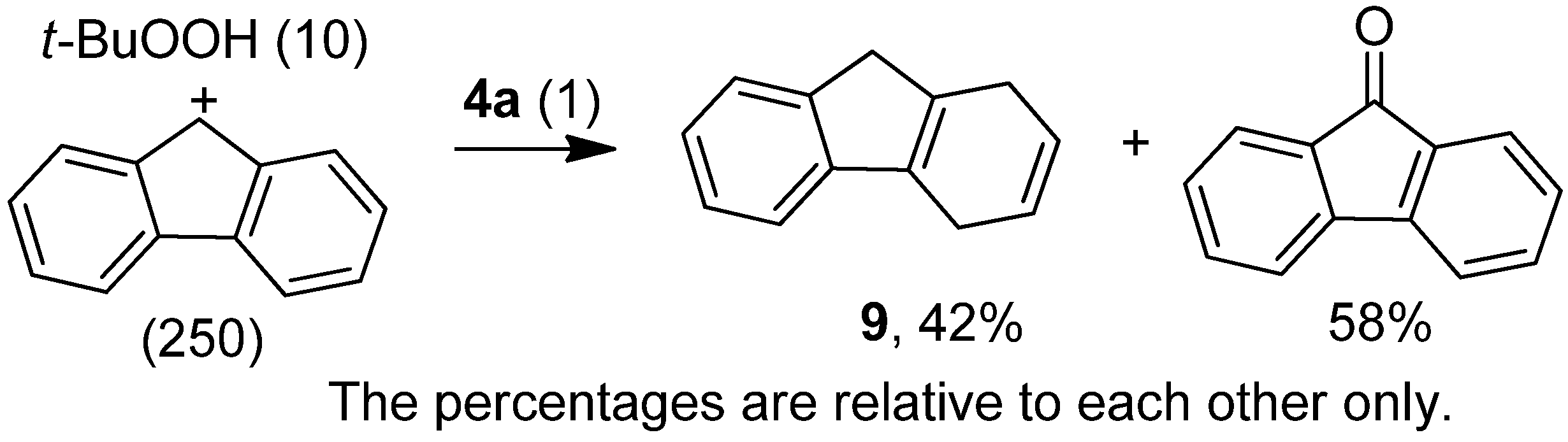
2.2. Application of the New Iron Phosphoramidite Complex 4a in Catalysis

| Entry | Substrate | Oxidant a | Time / Temperature | Catalyst Loading | Solvent | Product | Yield (%) b |
|---|---|---|---|---|---|---|---|
| 1 | toluene | t-BuOOH | 48 h / 90 °C | 2 mol% 4a | pyridine | NR e | |
| 2 | cinnamyl alcohol | t-BuOOH | 24 h / 80 °C | – | acetonitrile | cinnamaldehyde | 30 |
| 3 | cinnamyl alcohol | H2O2 | 24 h / rt | 10 mol% 4a | CH2Cl2 | benzaldehyde | 100 |
| 4 | cinnamyl alcohol | t-BuOOH | 24 h / rt | 10 mol% 4a | pyridine | cinnamaldehyde benzaldehyde | 80 ~20 |
| 5 | tetrahydro-naphthalene | t-BuOOH | 18 h / 90 °C | 2 mol% 4a | pyridine | tetrahydronaph-thalene-1-one | 100 |
| 6 | tetrahydronaph-thalene-1-ol | t-BuOOH | 16 h / rt | – | pyridine | tetrahydronaph-thalene-1-one | 100 |
| 7 | diphenyl-methane | t-BuOOH | 36 h / 82 °C | 2 mol% 4a | pyridine | benzophenone | 100 |
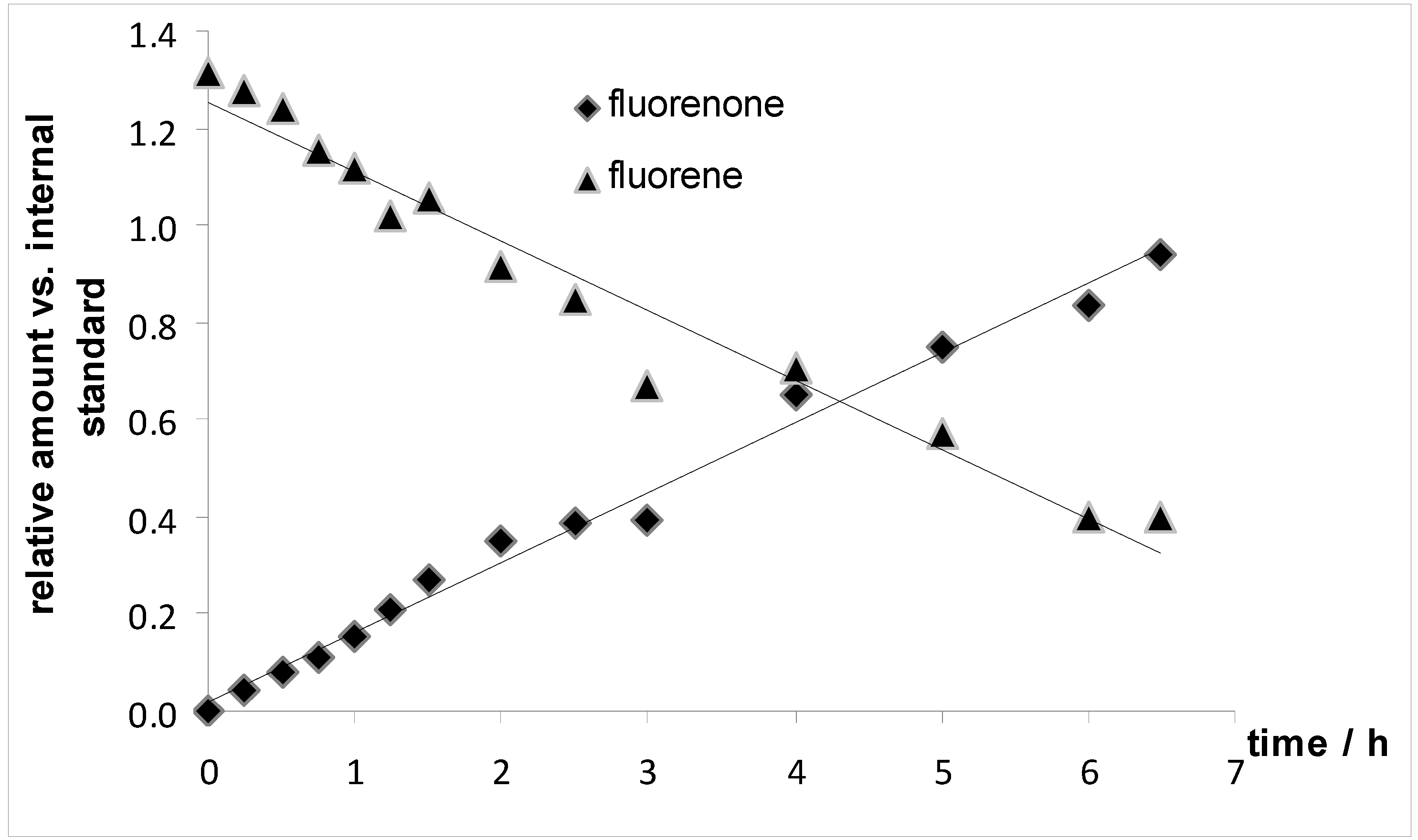
| 8 | fluorene | t-BuOOH | 36 h / rt | 2 mol% 4a | pyridine | fluorenone | 100 |
| 9 | fluorene | mCPBA c | 36 h / rt | 2 mol% 4a | pyridine | fluorenone | traces |
| 10 | fluorene | CH3COOOH d | 36 h / rt | 2 mol% 4a | pyridine | fluorenone | traces |
| 11 | fluorene | t-BuOOH | 36 h / rt | 2 mol% 4a | pyridine | fluorenone | 100 |
| 12 | dihydro-anthracene | t-BuOOH | 36 h / rt | 2 mol% 4a | pyridine | anthraquinone | 100 |
| 13 | adamantane | t-BuOOH | 36 h / rt | 2 mol% 4a | pyridine | NR e | |
| 14 | adamantane | t-BuOOH | 36 h / 90 °C | 2 mol% 4a | pyridine | NR e | |
| 15 | diphenyl-methane | H2O2 | 36 h / rt | 10 mol% 4a | CH2Cl2 | NR e | |
| 16 | cyclooctene | t-BuOOH | 42 h / rt | 2 mol% 4a | pyridine | NR e | |
| 17 | fluorene | t-BuOOH | 36 h / rt | 2 mol% 2 | pyridine | fluorenone | 30 |
| 18 | dihydro-anthracene | t-BuOOH | 36 h / rt | 2 mol% 2 | pyridine | anthraquinone | 9 f |
| 19 | diphenyl-methane | t-BuOOH | 36 h / rt | 2 mol% 2 | pyridine | benzophenone | 27 |
| 20 | fluorene | t-BuOOH | 36 h / rt | 2 mol% 3 | pyridine | fluorenone | 21 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Starting Material | Product | Yield b | TOF / h−1 c |
|---|---|---|---|---|
| 1 | diphenylmethane | benzo-phenone | 56% | 0.78 |
| 2 | fluorene | fluorenone | 80% | 1.11 |
| 3 | dihydroanthracene | anthra-quinone | 54% d | 1.39 |
| 4 | cinnamylalcohol | cinnamyl aldehyde | 31% e | 0.37 e |
| 5 | phenylmethanol | benzaldehyde | 47% e | 0.56 e |
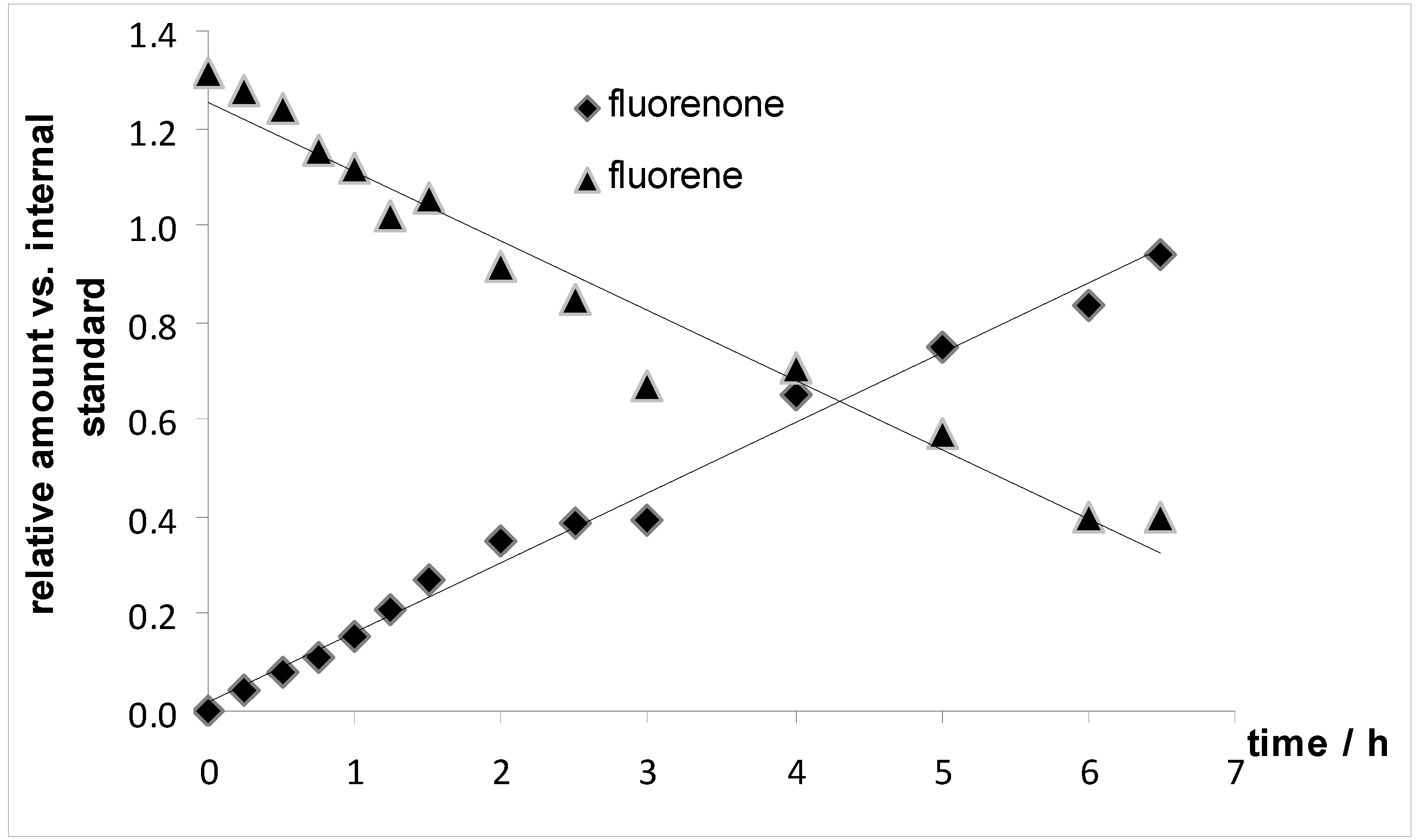
2.3. Further Experiment to Better Understand the Oxidation Reactions


3. Experimental
3.1. General
3.2. Synthesis of “[FeBrCp(CO)(1a)]”, 4a
3.3. Synthesis of “[FeBrCp(CO)(1b)]”, 4b
3.4. Attempted synthesis of “[FeBrCp(CO)(1c)]”, 4c
3.5. Synthesis of “[Fe(Cp)I(CO)(1b)]”, 5b
3.6. Typical Procedure for the Catalytic Experiments
3.7. Monitoring of the Oxidation of Fluorene to Fluorenone over Time (Figure 5)
3.8. Reaction of the Catalyst 4a with the Oxidant t-BuOOH without Substrate.
3.9. X-ray Structure Determination for 4b and 5b
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Czaplik, W.M.; Mayer, M.; Cvengroš, J.; von Wangelin, A.J. Coming of Age: Sustainable Iron-Catalyzed Cross-Coupling Reactions. ChemSusChem 2009, 2, 396–417. [Google Scholar] [CrossRef]
- Bauer, E.B. Recent Advances in Iron Catalysis in Organic Synthesis. Curr. Org. Chem. 2008, 12, 1341–1369. [Google Scholar] [CrossRef]
- Enthaler, S.; Junge, K.; Beller, M. Sustainable Metal Catalysis with Iron: From Rust to a Rising Star? Angew. Chem. Int. Ed. 2008, 47, 3317–3321. [Google Scholar] [CrossRef]
- Correra, A.; Garcia Mancheño, O.; Bolm, C. Iron-catalysed carbon–heteroatom and heteroatom–heteroatom bond forming processes. Chem. Soc. Rev. 2008, 37, 1108–1117. [Google Scholar] [CrossRef]
- Sherry, B.D.; Fürstner, A. The Promise and Challenge of Iron-Catalyzed Cross Coupling. Acc. Chem. Res. 2008, 41, 1500–1511. [Google Scholar] [CrossRef]
- Bolm, C.; Legros, J.; Le Paih, J.; Zani, L. Iron-catalyzed reactions in organic synthesis. Chem. Rev. 2004, 104, 6217–6254. [Google Scholar] [CrossRef]
- Costas, M.; Mehn, M.P.; Jensen, M.P.; Que, L., Jr. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. [Google Scholar]
- Tshuva, E.Y.; Lippard, S.J. Synthetic Models for Non-Heme Carboxylate-Bridged Diiron Metalloproteins: Strategies and Tactics. Chem. Rev. 2004, 104, 987–1012. [Google Scholar]
- Czaplik, W.M.; Mayer, M.; Jacobi von Wangelin, A. Domino Iron Catalysis: Direct Aryl-Alkyl Cross-Coupling. Angew. Chem. Int. Ed. 2009, 48, 607–610. [Google Scholar] [CrossRef]
- Cahiez, G.; Foulgoc, L.; Moyeux, A. Iron-Catalyzed Oxidative Heterocoupling Between Aliphatic and Aromatic Organozinc Reagents: A Novel Pathway for Functionalized Aryl-Alkyl Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2009, 48, 2969–2972. [Google Scholar] [CrossRef]
- Loska, R.; Rao Volla, C.M.; Vogel, P. Iron-Catalyzed Mizoroki-Heck Cross-Coupling Reaction with Styrenes. Adv. Synth. Catal. 2008, 350, 2859–2864. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Kondo, Y.; Fujiwara, Y.; Takaya, H.; Ito, S.; Nakamura, E.; Nakamura, M. Iron-catalysed fluoroaromatic coupling reactions under catalytic modulation with 1,2-bis(diphenylphosphino)benzene. Chem. Commun. 2009, 1216. [Google Scholar]
- Casey, C.P.; Guan, H. Cyclopentadienone Iron Alcohol Complexes: Synthesis, Reactivity, and Implications for the Mechanism of Iron Catalyzed Hydrogenation of Aldehydes. J. Am. Chem. Soc. 2009, 131, 2499–2507. [Google Scholar] [CrossRef]
- Moreau, B.; Wu, J.Y.; Ritter, T. Iron-Catalyzed 1,4-Addition of α-Olefins to Dienes. Org. Lett. 2009, 11, 337–339. [Google Scholar] [CrossRef]
- Miranda, P.O.; Carballo, R.M.; Martín, V.S.; Padrón, J.I. A New Catalytic Prins Cyclization Leading to Oxa- and Azacycles. Org. Lett. 2009, 11, 357–360. [Google Scholar] [CrossRef]
- Feng, Y.; Ke, C.; Xue, G.; Que, L., Jr. Bio-inspired arene cis-dihydroxylation by a non-haem iron catalyst modeling the action of naphthalene dioxygenase. Chem. Commun. 2009, 50–52. [Google Scholar]
- Carril, M.; Correa, A.; Bolm, C. Iron-Catalyzed Sonogashira Reactions. Angew. Chem. Int. Ed. 2008, 47, 4862–4865. [Google Scholar]
- Bedford, R.B.; Hall, M.A.; Hodges, G.R.; Huwe, M.; Wilkinson, M.C. Simple mixed Fe–Zn catalysts for the Suzuki couplings of tetraarylborates with benzyl halides and 2-halopyridines. Chem. Commun. 2009, 6430–6432. [Google Scholar]
- Xiang, S.-K.; Zhang, L.-H.; Jiao, N. Sp–sp3 C–C bond formation via Fe(OTf)3/TfOH cocatalyzed coupling reaction of terminal alkynes with benzylic alcohols. Chem. Commun. 2009, 6487–6489. [Google Scholar]
- Wu, J.-R.; Lin, C.-H.; Lee, C.-F. Iron-catalyzed thioetherification of thiols with aryl iodides. Chem. Commun. 2009, 4450–4452. [Google Scholar]
- Han, W.; Ofial, A.R. Iron-catalyzed dehydrogenative phosphonation of N,N-dimethylanilines. Chem. Commun. 2009, 6023–6025. [Google Scholar]
- Gelalcha, F.G.; Anilkumar, G.; Tse, M.K.; Brückner, A.; Beller, M. Biomimetic Iron-Catalyzed Asymmetric Epoxidation of Aromatic Alkenes by Using Hydrogen Peroxide. Chem. Eur. J. 2008, 14, 7687–7698. [Google Scholar] [CrossRef]
- Nakanishia, M.; Bolm, C. Iron-Catalyzed Benzylic Oxidation with Aqueous tert-Butyl Hydroperoxide. Adv. Synth. Catal. 2007, 349, 861–864. [Google Scholar] [CrossRef]
- Lenze, M.; Bauer, E.B. Oxidation of activated methylene groups to ketones catalyzed by new iron phosphinooxazoline complexes and by iron(II) triflate. J. Mol. Catal. A: Chem. 2009, 309, 117–123. [Google Scholar] [CrossRef]
- Chen, M.S.; White, M.C. Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, W.; Zhang, Z.; Zhang, X. Synthesis of New Monodentate Spiro Phosphoramidite Ligand and Its Application in Rh-Catalyzed Asymmetric Hydrogenation Reactions. Org. Lett. 2004, 6, 3565–3567. [Google Scholar] [CrossRef]
- Feringa, B.L. Phosphoramidites: Marvellous Ligands in Catalytic Asymmetric Conjugate Addition. Acc. Chem. Res. 2000, 33, 346–353. [Google Scholar] [CrossRef]
- Hulst, R.; de Vries, N.K.; Feringa, B.L. α-henylethylamine based chiral phospholidines; new agents for the determination of the enantiomeric excess of chiral alcohols, amines and thiols by means of 31P NMR. Tetrahedron Asymmetry 1994, 5, 699–708. [Google Scholar] [CrossRef]
- Maciá, B.; Fernández-Ibáñez, A.; Mršič, N.; Minnaard, A.J.; Feringa, B.L. Copper-catalyzed enantioselective conjugate addition of Grignard reagents to acyclic enones using monodentate phosphoramidite ligands. Tetrahedron Lett. 2008, 49, 1877–1880. [Google Scholar]
- Kostas, I.D.; Vallianattou, K.A.; Holz, J.; Börner, A. A new easily accessible chiral phosphite–phosphoramidite ligand based on 2-anilinoethanol and R-BINOL moieties for Rh-catalyzed asymmetric olefin hydrogenation. Tetrahedron Lett. 2008, 49, 331–334. [Google Scholar]
- Spiess, S.; Welter, C.; Franck, G.; Taquet, J.-P.; Helmchen, G. Iridium-Catalyzed Asymmetric Allylic Substitutions - Very High Regioselectivity and Air Stability with a Catalyst Derived from Dibenzo[a,e]cyclooctatetraene and a Phosphoramidite. Angew. Chem. Int. Ed. 2008, 47, 7652–7655. [Google Scholar] [CrossRef]
- Lee, E.E.; Rovis, T. Enantioselective Synthesis of Indolizidines Bearing Quaternary Substituted Stereocenters via Rhodium-Catalyzed [2 + 2 + 2] Cycloaddition of Alkenyl Isocyanates and Terminal Alkynes. Org. Lett. 2008, 10, 1231–1234. [Google Scholar] [CrossRef]
- Saha, B.; Smith, C.R.; RajanBabu, T.V. Ligand Tuning in Asymmetric Hydrovinylation of 1,3-Dienes: A Stereoselective Route to Either Steroid-C20 (S) or -C20 (R) Derivatives. J. Am. Chem. Soc. 2008, 130, 9000–9005. [Google Scholar] [CrossRef]
- Costin, S.; Rath, N.P.; Bauer, E.B. Conversion of Propargylic Alcohols to β-Oxo Esters Catalyzed by Novel Ruthenium-Phosphoramidite Complexes. Adv. Synth. Catal. 2008, 350, 2414–2424. [Google Scholar] [CrossRef]
- Costin, S.; Rath, N.P.; Bauer, E.B. Synthesis and structural characterization of new chiral mixed phosphine phosphoramidite complexes of ruthenium. Inorg. Chim. Acta 2009, 1935–1942. [Google Scholar]
- Vierling, P.; Riess, J.G. New molybdenum and iron phosphoranides. Mechanism and stereochemistry of the rearrangement and migration of a phosphorus-bound allylic group into an iron-bound vinylic group. Organometallics 1986, 5, 2543–2550. [Google Scholar] [CrossRef]
- Vierling, P.; Riess, J.G.; Grand, A. Coordination chemistry of the bicycloaminophosphorane Ph(H)P[(OCH2CH2)2N], a potential P/N ligand. Access to iron and ruthenium phosphoranides. Reversible migration of a phenyl group between iron and phosphorus: crystal and molecular structure of η5-CpFe(CO)[P(OCH2CH2)2N](η5-C6H5). Inorg Chem. 1986, 25, 4144–4152. [Google Scholar] [CrossRef]
- Nakazawa, H.; Kadoi, Y.; Miyoshi, K. Formation of amino-substituted phosphinyliron complexes [(η5-C5H5)(CO)2Fe{P(O)(NR2)n(OR')2-n}] (n = 1, 2) by the Arbuzov-like dealkylation reaction. Organometallics 1989, 8, 2851–2865. [Google Scholar] [CrossRef]
- Kubo, K.; Baba, Y.; Mizuta, T.; Miyoshi, K. Trapping of Unsaturated Small Molecules through Cyclization with Iron-Iminophosphorane Complexes. Organometallics 2006, 25, 3238–3244. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Sobkowiak, A.; Matsushita, T. Metal [MLx; M = Fe, Cu, Co, Mn]/Hydroperoxide-Induced Activation of Dioxygen for the Oxygenation of Hydrocarbons: Oxygenated Fenton Chemistry. Acc. Chem. Res. 1996, 29, 409–416. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Doller, D. The selective functionalisation of saturated hydrocarbons: Gif and all that. Pure Appl. Chem. 1991, 63, 1567–1576. [Google Scholar] [CrossRef]
- Hallam, B.F.; Pauson, P.L. Ferrocene Derivatives. Part III.* cycloPentadienlyiron Carbonyls. J. Chem. Soc. 1956, 3030–3037. [Google Scholar]
- Treichel, P.M.; Shubkin, R.L.; Barnett, K.W.; Reichard, D. Chemistry of the Cyclopentadienylmetal Carbonyls. II. Cyclopentadienyliron Carbonyl Derivatives. Inorg. Chem. 1966, 5, 1177. [Google Scholar] [CrossRef]
- Brun, P.; Vierling, P.; Rims, J.G.; Le Borgne, G. Study of the amination reaction of the coordinated cyclopentadienyl ring in (η5-Cp)FeL1L2X derivatives. Crystal and molecular structure of (η5-C5H4NEt2)Fe(CO)[PhP(OEt)2]Br. Organometallics 1987, 6, 1032–1040. [Google Scholar] [CrossRef]
- Lang, H.; Blau, S.; Rheinwald, G. Thermolyseverhalten alkinsubstituierter Cyclopentadienyl-Dicarbonyl-Eisen-Verbindungen. J. Organomet. Chem. 1995, 492, 81–85. [Google Scholar]
- Gamasa, M.P.; Gimeno, J.; Lastra, E.; Lanfranchi, M.; Tiripicchio, A. Synthesis and characterization of novel σ-alkynyl cyclopentadienyl iron(II) complexes [Fe(C≡CR)L2(η-C5H5)] [L=CO; L2=bis(diphenylphosphino)methane (dppm)]. Crystal structure of [Fe(C≡CC6H5)(dppm)(η-C5H5)]. J. Organomet. Chem. 1991, 405, 333–345. [Google Scholar]
- King, R.B.; Stone, F.G.A.; Jolly, W.L.; Austin, G.; Covey, W.; Rabinovich, D.; Steinberg, H.; Tsugawa, R. Cyclopentadienlyiron Dicarbonyl Dimer And Cyclopentadienyliron dicarbonyl Iodide. Inorg. Synth. 1963, 7, 110–112. [Google Scholar]
- Garringer, S.M.; Hesse, A.J.; Magers, J.R.; Pugh, K.R.; O’Reilly, S.A.; Wilson, A.M. Microwave Synthesis of Benchmark Organo-Iron Complexes. Organometallics 2009, 28, 6841–6844. [Google Scholar] [CrossRef]
- Munyaneza, A.; Bala, M.D.; Coville, N.J. Substitution reactions of Fe and Ru complexes with liquid phosphite ligands in the absence of solvents. Inorg. Chem. Commun. 2008, 11, 1082–1084. [Google Scholar] [CrossRef]
- Becker, E.; Kirchner, K.; Mereiter, K. CCDC number 658098. 2007; The compound was found in the CCDC database, but has not yet been published in the literature. It shows a position disorder between the C≡O and the bromo ligand, and averaged values for the structural parameters are given in Table 2. [Google Scholar]
- King, J.E.; Simpson, S.J. Synthesis and crystal structures of the isoelectronic monocyclopentadienyliron complexes [η5-C5H5)Fe(P{OMe}3)(CO)(I)] and [(η5-C5H5)Fe(P{OMe}3)(NO)(I)]BF4. J. Organomet. Chem. 1992, 424, 57–63. [Google Scholar]
- Company, A.; Gomez, L.; Fontrodona, X.; Ribas, X.; Costas, M. A Novel Platform for Modeling Oxidative Catalysis in Non-Heme Iron Oxygenases with Unprecedented Efficiency. Chem. Eur. J. 2008, 14, 5727–5731. [Google Scholar] [CrossRef]
- Gozzo, F. Radical and non-radical chemistry of the Fenton-like systems in the presence of organic substrates. J. Mol. Catal. A: Chem. 2001, 171, 1–22. [Google Scholar] [CrossRef]
- Barton, D.H.R. Gif Chemistry: The Present Situation. Tetrahedron 1998, 54, 5805–5817. [Google Scholar]
- Minisci, F.; Fontana, F.; Araneo, S.; Recupero, F.; Banfi, S.; Quici, S. Kharasch and Metalloporphyrin Catalysis in the Functionalization of Alkanes, Alkenes, and Alkylbenzenes by t-BuOOH. Free Radical Mechanisms, Solvent Effect, and Relationship with the Gif Reaction. J. Am. Chem. Soc. 1995, 117, 226–232. [Google Scholar] [CrossRef]
- Stavropoulos, P.; Çelenligil-Çetin, R.; Tapper, A.E. The Gif Paradox. Acc. Chem. Res. 2001, 34, 745–752. [Google Scholar] [CrossRef]
- Schuchardt, U.; Jannini, M.J.D.M.; Richens, D.T.; Guerreiro, M.C.; Spinacé, E.V. Gif chemistry: new evidence for a non-radical process. Tetrahedron 2001, 57, 2685–2688. [Google Scholar]
- Jensen, M.P.; Costas, M.; Ho, R.Y.N.; Kaizer, J.; Mairata i Payeras, A.; Münck, E.; Que, L., Jr.; Rohde, J.U.; Stubna, A. High-Valent Nonheme Iron. Two Distinct Iron(IV) Species Derived from a Common Iron(II) Precursor. J. Am. Chem. Soc. 2005, 127, 10512–10525. [Google Scholar]
- Jensen, M.P.; Lange, S.J.; Mehn, M.P.; Que, E.L.; Que, L., Jr. Biomimetic Aryl Hydroxylation Derived from Alkyl Hydroperoxide at a Nonheme Iron Center. Evidence for an FeIV=O Oxidant. J. Am. Chem. Soc. 2003, 125, 2113–2128. [Google Scholar]
- Jensen, M.P.; Mairata i Payeras, A.; Fiedler, A.T.; Costas, M.; Kaizer, J.; Stubna, A.; Münck, E.; Que, L., Jr. Kinetic Analysis of the Conversion of Nonheme (Alkylperoxo)iron(III) Species to Iron(IV) Complexes. Inorg. Chem. 2007, 46, 2398–2408. [Google Scholar]
- Namuswe, F.; Kasper, G.D.; Narducci Sarjeant, A.A.; Hayashi, T.; Krest, C.M.; Green, M.T.; Moënne-Loccoz, P.; Goldberg, D.P. Rational Tuning of the Thiolate Donor in Model Complexes of Superoxide Reductase: Direct Evidence for a trans Influence in FeIII-OOR Complexes. J. Am. Chem. Soc. 2008, 130, 14189–14200. [Google Scholar]
- Vermeulen, N.A.; Chen, M.S.; White, M.C. The Fe(PDP)-catalyzed aliphatic C–H oxidation: a slow addition protocol. Tetrahedron 2009, 65, 3078–3084. [Google Scholar] [CrossRef]
- Hulst, R.; de Vries, N.K.; Feringa, B.L. α-henylethylamine based chiral phospholidines; new agents for the determination of the enantiomeric excess of chiral alcohols, amines and thiols by means of 31P NMR. Tetrahedron Asymmetry 1994, 5, 699–708. [Google Scholar] [CrossRef]
- Duursma, A.; Boiteau, J.-G.; Lefort, L.; Boogers, J.A.F.; de Vries, A.H.M.; de Vries, J.G.; Minnaard, A.J.; Feringa, B.L. Highly Enantioselective Conjugate Additions of Potassium Organotrifluoroborates to Enones by Use of Monodentate Phosphoramidite Ligands. J. Org. Chem. 2004, 69, 8045–8052. [Google Scholar]
- All efforts to obtain correct elemental analyses for 4a and 4b have failed thus far. The NMR spectra of both compounds show baseline purity, and 1H and 13C NMR spectra are given in the Supplementary material. After intensive drying, the product 2a contained still varying amounts of CHCl3 as seen in the NMR. The calculated values for C28H23BrPFe(CHCl3)0.5 are C, 52.83; H, 3.66 and thus match the found values much closer.
- Only the sodium adduct of the compound was observed in the MS as molecular ion.
- The peaks correspond to the most intense signal of the isotope envelope.
- Due to overlaps, assignment of the aromatic 1H-NMR signals to individual diastereomers was not possible. In the 13C-NMR for 4a, not all 13C-NMR signals for the minor diastereomer were observed. In the 13C-NMR for 4b, only the 13C-NMR signal for the Cp ligand was observed for the minor diastereomer.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar]
- Sample Availability: Samples of the compounds 4a, 4b and 5b are available from the authors.
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shejwalkar, P.; Rath, N.P.; Bauer, E.B. New Chiral Phosphoramidite Complexes of Iron as Catalytic Precursors in the Oxidation of Activated Methylene Groups. Molecules 2010, 15, 2631-2650. https://doi.org/10.3390/molecules15042631
Shejwalkar P, Rath NP, Bauer EB. New Chiral Phosphoramidite Complexes of Iron as Catalytic Precursors in the Oxidation of Activated Methylene Groups. Molecules. 2010; 15(4):2631-2650. https://doi.org/10.3390/molecules15042631
Chicago/Turabian StyleShejwalkar, Pushkar, Nigam P. Rath, and Eike B. Bauer. 2010. "New Chiral Phosphoramidite Complexes of Iron as Catalytic Precursors in the Oxidation of Activated Methylene Groups" Molecules 15, no. 4: 2631-2650. https://doi.org/10.3390/molecules15042631