Alcohol Withdrawal and Brain Injuries: Beyond Classical Mechanisms

Abstract
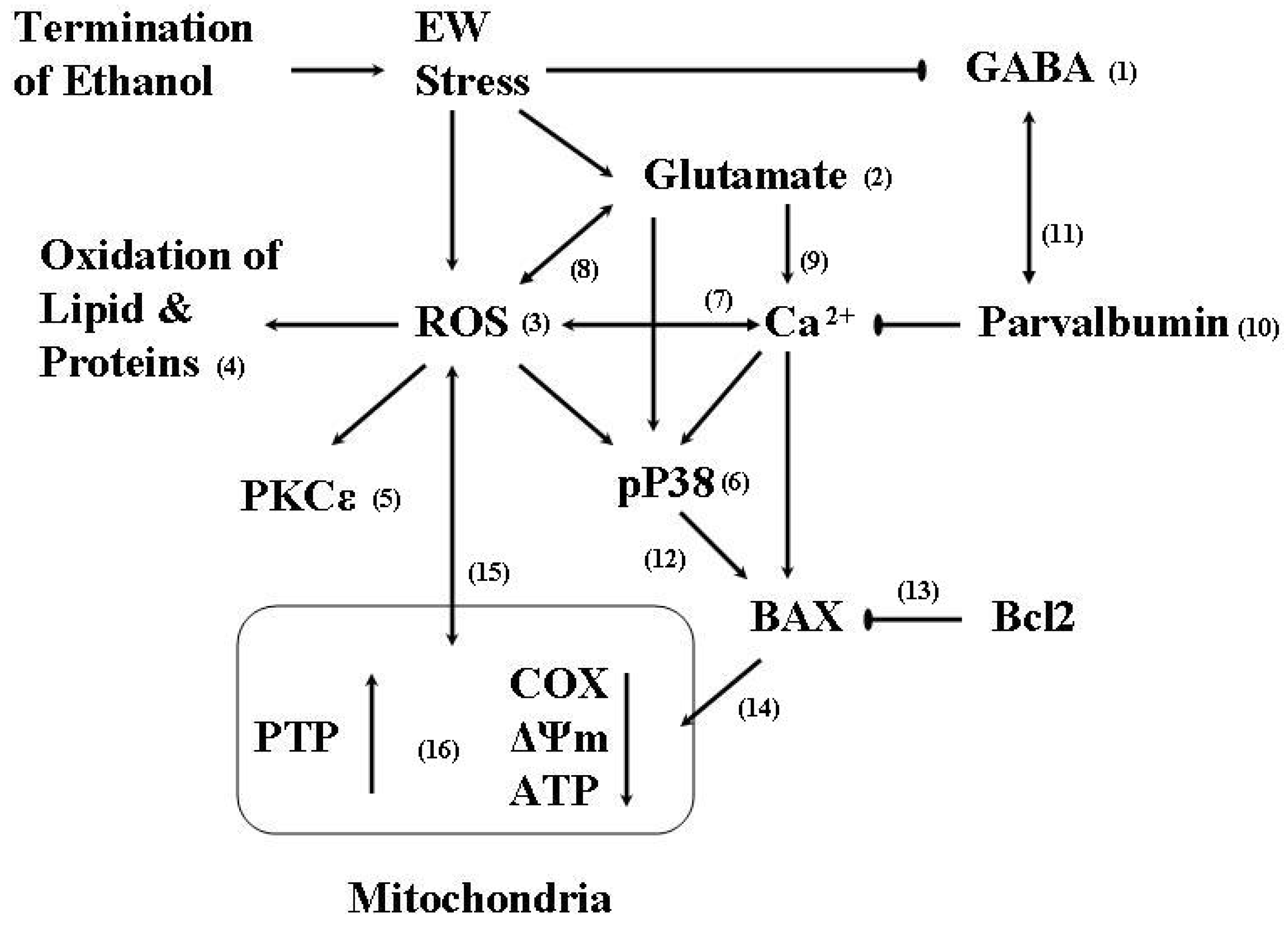
:1. Ethanol Withdrawal (EW)
2. EW & Oxidative Stress
2.1. Oxidative stress
2.2. Protein oxidation
2.3. Antioxidant mechanisms of E2



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % of control | EW only | EW + Test Compound (1 µM) | |||
|---|---|---|---|---|---|
| E2 | BHT | OP | 3-OBu-E2 | ||
| Cell survival | 30 ± 1 | 49 ± 2* | 50 ± 1* | 48 ± 3* | 28 ± 3 |
| MDA content | 219 ± 2 | 157 ± 3* | 152 ± 1* | 161 ± 3* | 220 ± 2 |
| Protein carbonyl | 229 ± 3 | 171 ± 3* | 174 ± 2* | 175 ± 2* | 230 ± 3 |
3. Protein Kinase
3.1. PKC
3.2. P38
4. EW & Mitochondria

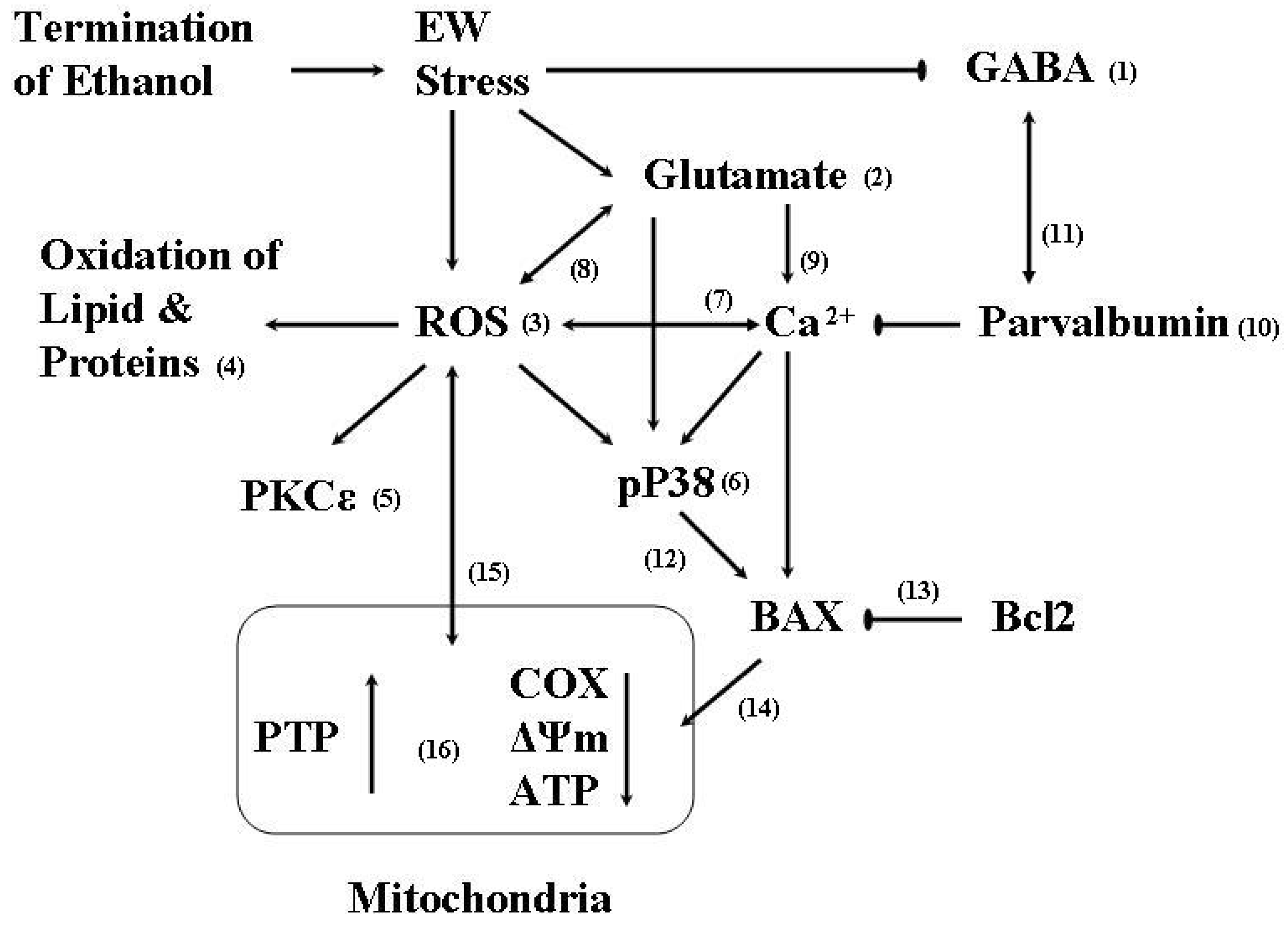
5. EW & Brain Aging
6. Conclusions
- Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- Barrett, R. Behavioral Approaches to Individual Differences in Substance Abuse. In Determinants of Substance Abuse: Biological, Pyschological, and Environmental Factors; Schaffer, H., Galizio, M., Maisto, S.A., Eds.; Plenum Press: New York, NY, USA, 1985; pp. 125–174. [Google Scholar]
- De Witte, P.; Pinto, E.; Ansseau, M.; Verbanck, P. Alcohol and withdrawal: from animal research to clinical issues. Neurosci. Biobehav. Rev. 2003, 27, 189–197. [Google Scholar] [CrossRef]
- Mandrekar, P.; Catalano, D.; Jeliazkova, V.; Kodys, K. Alcohol exposure regulates heat shock transcription factor binding and heat shock proteins 70 and 90 in monocytes and macrophages: Implication for TNF-alpha regulation. J. Leukoc. Biol. 2008, 84, 1335–1345. [Google Scholar] [CrossRef]
- Li, S.Y.; Ren, J. Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: Role of insulin signaling and ER stress. J. Mol. Cell Cardiol. 2008, 44, 992–1001. [Google Scholar] [CrossRef]
- Fadda, F.; Rossetti, Z.L. Chronic ethanol consumption: From neuroadaptation to neurodegeneration. Prog. Neurobiol. 1998, 56, 385–431. [Google Scholar] [CrossRef]
- Jaatinen, P.; Rintala, J. Mechanisms of ethanol-induced degeneration in the developing, mature, and aging cerebellum. Cerebellum 2008, 7, 332–347. [Google Scholar] [CrossRef]
- Alele, P.E.; Devaud, L.L. Sex differences in steroid modulation of ethanol withdrawal in male and female rats. J. Pharmacol. Exp. Ther. 2007, 320, 427–436. [Google Scholar]
- Phillips, S.C.; Cragg, B.G. Chronic consumption of alcohol by adult mice: Effect on hippocampal cells and synapses. Exp. Neurol. 1983, 80, 218–226. [Google Scholar] [CrossRef]
- Jung, M.E.; Yang, S.H.; Brun-Zinkernagel, A.M.; Simpkins, J.W. Estradiol protects against cerebellar damage and motor deficit in ethanol-withdrawn rats. Alcohol 2002, 26, 83–93. [Google Scholar] [CrossRef]
- Nagy, J.; Muller, F.; Laszlo, L. Cytotoxic effect of alcohol-withdrawal on primary cultures of cortical neurones. Drug Alcohol Depend 2001, 61, 155–162. [Google Scholar] [CrossRef]
- Pandey, S.C.; Roy, A.; Mittal, N. Effects of chronic ethanol intake and its withdrawal on the expression and phosphorylation of the creb gene transcription factor in rat cortex. J. Pharmacol. Exp. Ther. 2001, 296, 857–868. [Google Scholar]
- Rewal, M.; Wen, Y.; Wilson, A.; Simpkins, J.W.; Jung, M.E. Role of parvalbumin in estrogen protection from ethanol withdrawal syndrome. Alcohol Clin. Exp. Res. 2005, 29, 1837–1844. [Google Scholar] [CrossRef]
- Ripley, T.L.; O'Shea, M.; Stephens, D.N. Repeated withdrawal from ethanol impairs acquisition but not expression of conditioned fear. Eur. J. Neurosci. 2003, 18, 441–448. [Google Scholar]
- Stephens, D.N.; Brown, G.; Duka, T.; Ripley, T.L. Impaired fear conditioning but enhanced seizure sensitivity in rats given repeated experience of withdrawal from alcohol. Eur. J. Neurosci. 2001, 14, 2023–2031. [Google Scholar] [CrossRef]
- Jung, M.E.; Wilson, A.M.; Simpkins, J.W. A nonfeminizing estrogen analog protects against ethanol withdrawal toxicity in immortalized hippocampal cells. J. Pharmacol. Exp. Ther. 2006, 319, 543–550. [Google Scholar] [CrossRef]
- Jung, M.E.; Yan, L.J.; Forster, M.J.; Simpkins, J.W. Ethanol withdrawal provokes mitochondrial injury in an estrogen preventable manner. J. Bioenerg. Biomembr. 2008, 40, 35–44. [Google Scholar] [CrossRef]
- Kim, J.; Jang, H.S.; Park, K.M. Reactive oxygen species generated by renal ischemia and reperfusion trigger protection against subsequent renal ischemia and reperfusion injury in mice. Am. J. Physiol. Renal. Physiol. 2009, 289, F158–F166. [Google Scholar]
- Mallet, R.T.; Ryou, M.G.; Manukhina, E.B.; Downey, H.F. Intermittent Hypoxia Conditioning of Canine Myocardium: Robust Protection against Ischemia-reperfusion Injury. In Adaptation Biology and Medicine, 1st; Lukyanova, L., Takeda, N., Singal, P.K., Eds.; Narosa Pub. House: New Delhi, India, 2008; Volume 5, pp. 59–78. [Google Scholar]
- Fujikawa, D.G. Prolonged seizures and cellular injury: Understanding the connection. Epilepsy Behav. 2005, 7, S3–S11. [Google Scholar] [CrossRef]
- Huang, M.C.; Chen, C.H.; Peng, F.C.; Tang, S.H.; Chen, C.C. Alterations in oxidative stress status during early alcohol withdrawal in alcoholic patients. J. Formos. Med. Assoc. 2009, 108, 560–569. [Google Scholar] [CrossRef]
- Grattagliano, I.; Vendemiale, G.; Errico, F.; Bolognino, A.E.; Lillo, F.; Salerno, M.T.; Altomare, E. Chronic ethanol intake induces oxidative alterations in rat testis. J. Appl. Toxicol. 1997, 17, 307–311. [Google Scholar] [CrossRef]
- Tsai, G.E.; Ragan, P.; Chang, R.; Chen, S.; Linnoila, V.M.; Coyle, J.T. Increased glutamatergic neurotransmission and oxidative stress after alcohol withdrawal. Am. J. Psychiat. 1998, 155, 726–732. [Google Scholar]
- Vallett, M.; Tabatabaie, T.; Briscoe, R.J.; Baird, T.J.; Beatty, W.W.; Floyd, R.A.; Gauvin, D.V. Free radical production during ethanol intoxication, dependence, and withdrawal. Alcohol Clin. Exp. Res. 1997, 21, 275–285. [Google Scholar] [CrossRef]
- Adachi, J. Membrane disorder and free radical. Nihon Hoigaku Zasshi 2000, 54, 356–360. [Google Scholar]
- Lehotsky, J.; Kaplan, P.; Matejovicova, M.; Murin, R.; Racay, P.; Raeymaekers, L. Ion transport systems as targets of free radicals during ischemia reperfusion injury. Gen. Physiol. Biophys. 2002, 21, 31–37. [Google Scholar]
- Shulman, R.G.; Rothman, D.L.; Behar, K.L.; Hyder, F. Energetic basis of brain activity: Implications for neuroimaging. Trends Neurosci. 2004, 27, 489–495. [Google Scholar] [CrossRef]
- Jung, M.E.; Rewal, M.; Perez, E.; Wen, Y.; Simpkins, J.W. Estrogen protects against brain lipid peroxidation in ethanol-withdrawn rats. Pharmacol. Biochem. Behav. 2004, 79, 573–586. [Google Scholar] [CrossRef]
- Thompson, P. Platelet and erythrocyte membrane fluidity changes in alcohol-dependent patients undergoing acute withdrawal. Alcohol Alcohol. 1999, 34, 349–354. [Google Scholar]
- Marotta, F.; Reizakovic, I.; Tajiri, H.; Safran, P.; Ideo, G. Abstinence-induced oxidative stress in moderate drinkers is improved by bionormalizer. Hepatogastroenterology 1997, 44, 1360–1366. [Google Scholar]
- Bleich, S.; Spilker, K.; Kurth, C.; Degner, D.; Quintela-Schneider, M.; Javaheripour, K.; Ruther, E.; Kornhuber, J.; Wiltfang, J. Oxidative stress and an altered methionine metabolism in alcoholism. Neurosci. Lett. 2000, 293, 171–174. [Google Scholar] [CrossRef]
- Ulrichsen, J.; Woldbye, D.P.; Olsen, C.H.; Haugbol, S.; Bolwig, T.G.; Hemmingsen, R. No loss of somatostatin-immunoreactive neurons in the hippocampal dentate hilus of alcohol-withdrawal-kindled rats. Alcohol Alcohol. 1996, 31, 411–419. [Google Scholar]
- Nagatomo, K.; Ueda, Y.; Doi, T.; Nakajima, A. An acute dysfunction of the glutamate transport activity has been shown to generate free radicals and suppress the anti-oxidant ability in the hippocampus of rats. Neurosci. Res. 2007, 57, 477–480. [Google Scholar] [CrossRef]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorder. Science 1993, 262, 689–695. [Google Scholar]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef]
- Levine, R.L. Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radical Biol. Med. 2002, 32, 790–796. [Google Scholar] [CrossRef]
- Rauniyar, N.; Stevens, S.M., Jr.; Prokai-Tatrai, K.; Prokai, L. Characterization of 4-hydroxy-2-nonenal-modified peptides by liquid chromatography−tandem mass spectrometry using data-dependent acquisition: Neutral loss-driven MS3 versus neutral loss-driven electron capture dissociation. Anal. Chem. 2009, 81, 782–789. [Google Scholar] [CrossRef]
- Prokai, L.; Yan, L-Y.; Vera-Serrano, J,V.; Stevens, S.M., Jr.; Forster, M.J. Mass spectrometry-based survey of age-associated protein carbonylation in rat brain mitochondria. J. Mass Spectrom. 2007, 42, 1583–1589. [Google Scholar] [CrossRef]
- Mutlu-Turkoglu, U.; Dogru-Abbasoglu, S.; Aykac-Toker, G.; Mirsal, H.; Beyazyurek, M.; Uysal, M. Increased lipid and protein oxidation and DNA damage in patients with chronic alcoholism. J. Lab. Clin. Med. 2000, 136, 287–291. [Google Scholar] [CrossRef]
- Bailey, S.M.; Patel, V.B.; Young, T.A.; Asayama, K.; Cunningham, C.C. Chronic ethanol consumption alters the glutathione/glutathione peroxidase-1 system and protein oxidation status in rat liver. Alcohol Clin. Exp. Res. 2001, 25, 726–733. [Google Scholar] [CrossRef]
- Cano, M.J.; Ayala, A.; Murillo, M.L.; Carreras, O. Protective effect of folic acid against oxidative stress produced in 21-day postpartum rats by maternal-ethanol chronic consumption during pregnancy and lactation period. Free Radical Res. 2001, 34, 1–8. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, L.; Song, Z.; Saari, J.T.; McClain, C.J.; Kang, Y.J. Zinc supplementation prevents alcoholic liver injury in mice through attenuation of oxidative stress. Am. J. Pathol. 2005, 166, 1681–1690. [Google Scholar] [CrossRef]
- Khomenko, I.P.; Bakhtina, L.Y.; Zelenina, O.M.; Kruglov, S.V.; Manukhina, E.B.; Bayda, L.A.; Malyshev, I.Y. Role of heat shock proteins HSP70 and HSP32 in the protective effect of adaptation of cultured HT22 hippocampal cells to oxidative stress. Bull. Exp. Biol. Med. 2007, 144, 174–177. [Google Scholar] [CrossRef]
- Rackova, L.; Snirc, V.; Jung, T.; Stefek, M.; Karasu, C.; Grune, T. Metabolism-induced oxidative stress is a mediator of glucose toxicity in HT22 neuronal cells. Free Radical Res. 2009, 43, 876–886. [Google Scholar] [CrossRef]
- Jung, M.E.; Wilson, A.M.; Ju, X.; Wen, Y.; Metzger, D.B.; Simpkins, J.W. Ethanol withdrawal provokes opening of the mitochondrial membrane permeability transition pore in an estrogen-preventable manner. J. Pharmacol. Exp. Ther. 2009, 328, 692–698. [Google Scholar] [CrossRef]
- Merchanthaler, I.; Dellovade, T.L.; Shughrue, P.J. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann. NY Acad. Sci. 2006, 10007, 89–100. [Google Scholar]
- Garcia-Segura, L.M.; Azcoitia, I.; DonCarlos, L.L. Neuroprotection by estradiol. Prog. Neurobiol. 2001, 63, 29–60. [Google Scholar] [CrossRef] [Green Version]
- Riu, R.; Yang, S.H.; Perez, E.; Yi, K.D.; Wu, S.S.; Eberst, K.; Prokai, L.; Prokai-Tatrai, K.; Cai, Z.Y.; Covey, D.F.; Day, A.L.; Simpkins, J.W. Neuroprotective effects of a novel non-receptor binding estrogen analogue: in vitro and in vivo analysis. Stroke 2002, 33, 2485–2491. [Google Scholar] [CrossRef]
- Behl, C.; Skutella, T.; Lezoualc'h, F.; Post, A.; Widmann, M.; Newton, C.J.; Holsboer, F. Neuroprotection against oxidative stress by estrogens: Structure-activity relationship. Mol. Pharmacol. 1997, 51, 535–554. [Google Scholar]
- Topcuoglu, A.; Uzun, H.; Balci, H.; Karakus, M.; Coban, I.; Altug, T.; Aydin, S.; Topcuoglu, D.; Cakatay, U. Effects of estrogens on oxidative protein damage in plasma and tissues in ovariectomised rats. Clin. Invest. Med. 2009, 32, E133–E143. [Google Scholar]
- Moosmann, B.; Behl, C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc. Natl. Acad. Sci. USA 1999, 96, 8867–8872. [Google Scholar] [CrossRef]
- Prokai, L.; Simpkins, J.W. Structure-nongenomic neuroprotection relationship of estrogens and estrogen-derived compounds. Pharmacol. Therapeut. 2007, 114, 1–12. [Google Scholar] [CrossRef]
- Branna, D.W.; Dhandapania, K.; Wakadea, C.; Mahesha, V.B.; Khan, M.M. Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids 2007, 72, 382–405. [Google Scholar]
- Prokai, L.; Prokai-Tatrai, K.; Perjesi, P.; Simpkins, J.W. Mechanistic insights into the direct antioxidant effects of estrogens. Drug Dev. Res. 2005, 66, 118–125. [Google Scholar] [CrossRef]
- Prokai, L.; Prokai-Tatrai, K.; Perjesi, P.; Zharikova, A.D.; Perez, J.; Liu, R. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc. Natl. Acad. Sci. USA 2003, 100, 11741–11746. [Google Scholar]
- Prokai-Tatrai, K.; Perjesi, P.; Rivera-Portalatin, N.M.; Simpkins, J.; Prokai, L. Mechanistic investigations on the antioxidant action of a neuroprotective estrogen derivative. Steroids 2008, 73, 280–288. [Google Scholar] [CrossRef]
- Sagara, Y. Induction of reactive oxygen species in neurons by haloperidol. J. Neurochem. 1998, 71, 1002–1012. [Google Scholar] [CrossRef]
- Rewal, M.; Jung, M.E.; Wen, Y.; Brun-Zinkernagel, A.M.; Simpkins, J.W. Role of the GABAA system in behavioral, motoric, and cerebellar protection by estrogen during ethanol withdrawal. Alcohol 2003, 31, 49–61. [Google Scholar] [CrossRef]
- Singer, C.A.; Figueroa-Masot, X.A.; Batchelor, R.H.; Dorsa, D.M. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J. Neurosci. 1999, 19, 2455–2463. [Google Scholar]
- Sribnick, E.A.; Ray, S.K.; Nowak, M.W.; Li, L. 17β-estradiol attenuates glutamate-induced apoptosis and preserves electrophysiologic function in primary cortical neurons. J. Neurosci. Res. 2004, 76, 688–696. [Google Scholar] [CrossRef]
- Strehlow, K.; Rotter, S.; Wassmann, S.; Adam, O.; Grohe, C.; Laufs, K.; Bohm, M.; Nickenig, G. Modulation of antioxidant enzyme expression and function by estrogen. Circ. Res. 2003, 93, 170–177. [Google Scholar] [CrossRef]
- Green, P.S.; Gridley, K.E.; Simpkins, J.W. Nuclear estrogen receptor-independent neuroprotection by estratrienes: A novel interaction with glutathione. Neuroscience 1998, 84, 7–10. [Google Scholar] [CrossRef]
- Gridley, K.E.; Green, P.S.; Simpkins, J.W. A novel, synergistic interaction between 17 beta-estradiol and glutathione in the protection of neurons against beta-amyloid 25-35-induced toxicity in vitro. Mol. Pharmacol. 1998, 54, 874–880. [Google Scholar]
- Prokai, L.; Prokai-Tatrai, K.; Perjesi, P.; Zharikova, A.D.; Simpkins, J.W. Quinol-based metabolic cycle for estrogens in rat liver microsomes. Drug Metab. Dispos. 2003, 31, 701–704. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Prokai, L.; Simpkins, J.W.; Jung, M.E. Phenolic compounds protect cultured hippocampal neurons against ethanol-withdrawal induced oxidative stress. Int. J. Mol. Sci. 2009, 10, 1773–1787. [Google Scholar] [CrossRef]
- Prokai, L.; Oon, S.M.; Prokai-Tatrai, K.; Abboud, K.; Simpkins, J.W. Synthesis and biological evaluation of 17β-alkoxyestra-1,3,5(10)-trienes as potential neuroprotectants against oxidative stress. J. Med. Chem. 2001, 44, 110–114. [Google Scholar] [CrossRef]
- Lee, T.Y.; Lee, K.C.; Chen, S.Y.; Chang, H.H. 6-Gingerol inhibits ROS and iNOS through the suppression of PKC-alpha and NF-kappaB pathways in lipopolysaccharide-stimulated mouse macrophages. Biochem. Biophys. Res. Commun. 2009, 382, 134–139. [Google Scholar] [CrossRef]
- Inagaki, K.; Hahn, H.S.; Dorn, G.W., 2nd; Mochly-Rosen, D. Additive protection of the ischemic heart ex vivo by combined treatment with delta-protein kinase C inhibitor and epsilon-protein kinase C activator. Circulation 2003, 108, 869–875. [Google Scholar] [CrossRef]
- Maher, P. How protein kinase C activation protects nerve cells from oxidative stress-induced cell death. J. Neurosci. 2001, 21, 2929–2938. [Google Scholar]
- Novotny, J.L.; Simpson, A.M.; Tomicek, N.J.; Lancaster, T.S.; Korzick, D.H. Rapid estrogen receptor-alpha activation improves ischemic tolerance in aged female rats through a novel protein kinase C epsilon-dependent mechanism. Endocrinology 2009, 150, 889–896. [Google Scholar]
- Gorin, M.A.; Pan, Q. Protein kinase C epsilon: An oncogene and emerging tumor biomarker. Mol. Cancer 2009, 8, 9. [Google Scholar] [CrossRef]
- Das, J.; Pany, S.; Rahman, G.M.; Slater, S.J. PKC epsilon has an alcohol-binding site in its second cysteine-rich regulatory domain. Biochem. J. 2009, 421, 405–413. [Google Scholar] [CrossRef]
- Olive, M.F.; Mehmert, K.K.; Nannini, M.A.; Camarini, R.; Messing, R.O.; Hodge, C.W. Reduced ethanol withdrawal severity and altered withdrawal-induced c-fos expression in various brain regions of mice lacking protein kinase C-epsilon. Neuroscience 2001, 103, 171–179. [Google Scholar] [CrossRef]
- Zhou, H.Z.; Karliner, J.S.; Gray, M.O. Moderate alcohol consumption induces sustained cardiac protection by activating PKC-epsilon and Akt. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H165–H174. [Google Scholar]
- Chen, C.; Mochly-Rosen, D. Opposing effects of delta and xi PKC in ethanol-induced cardioprotection. J. Mol. Cell Cardiol. 2001, 33, 581–585. [Google Scholar] [CrossRef]
- Pascale, A.; Battaini, F.; Govoni, S.; Persichella, M.; De Salvia, M.A.; Cuomo, V. Chronic low doses of ethanol affect brain protein kinase C and ultrasonic calls in rats. Alcohol 1997, 14, 557–561. [Google Scholar] [CrossRef]
- Jung, M.E.; Watson, D.G.; Wen, Y.; Simpkins, J.W. Role of protein kinase C in estrogen protection against apoptotic cerebellar cell death in ethanol-withdrawn rats. Alcohol 2003, 31, 39–48. [Google Scholar] [CrossRef]
- Csukai, M.; Mochly-Rosen, D. Pharmacologic modulation of protein kinase C isozymes: The role of RACKs and subcellular localisation. Pharmacol. Res. 1999, 39, 253–259. [Google Scholar] [CrossRef]
- Mochly-Rosen, D.; Kauvar, L.M. Modulating protein kinase C signal transduction. Adv. Pharmacol. 1998, 44, 91–145. [Google Scholar] [CrossRef]
- Solem, M.; Almas, J.; Rubin, E.; Thomas, A. Changes in activity and regulation of the cardiac Ca2+ channel (L-type) by protein kinase C in chronic alcohol-exposed rats. Alcohol Clin. Exp. Res. 2000, 24, 1145–1152. [Google Scholar] [CrossRef]
- Ron, D.; Mochly-Rosen, D. An autoregulatory region in protein kinase C: The pseudoanchoring site. Proc. Natl. Acad. Sci. USA 1995, 92, 492–496. [Google Scholar] [CrossRef]
- Sweitzer, S.M.; Wong, S.M.; Peters, M.C.; Mochly-Rosen, D.; Yeomans, D.C.; Kendig, J.J. Protein kinase C epsilon and gamma: Involvement in formalin-induced nociception in neonatal rats. J. Pharmacol. Exp. Ther. 2004, 309, 616–625. [Google Scholar] [CrossRef]
- Dina, O.A.; Barletta, J.; Chen, X.; Mutero, A.; Martin, A.; Messing, R.O.; Levine, J.D. Key role for the epsilon isoform of protein kinase C in painful alcoholic neuropathy in the rat. J. Neurosci. 2000, 20, 8614–8619. [Google Scholar]
- Dina, O.A.; Aley, K.O.; Isenberg, W.; Messing, R.O.; Levine, J.D. Sex hormones regulate the contribution of PKCepsilon and PKA signalling in inflammatory pain in the rat. Eur. J. Neurosci. 2001, 13, 2227–2233. [Google Scholar] [CrossRef]
- Hucho, T.B.; Dina, O.A.; Kuhn, J.; Levine, J.D. Estrogen controls PKCepsilon-dependent mechanical hyperalgesia through direct action on nociceptive neurons. Eur. J. Neurosci. 2006, 24, 527–534. [Google Scholar] [CrossRef]
- Watson, D.G.; Watterson, J.M.; Lenox, R.H. Sodium valproate down-regulates the myristoylated alanine-rich C kinase substrate (MARCKS) in immortalized hippocampal cells: A property of protein kinase C-mediated mood stabilizers. J. Pharmacol. Exp. Ther. 1998, 285, 307–316. [Google Scholar]
- Behl, C.; Widmann, M.; Trapp, T.; Holsboer, F. 17-beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem. Biophys. Res. Commun. 1995, 216, 473–482. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free Radical Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
- Arimoto, K.; Fukuda, H.; Imajoh-Ohmi, S.; Saito, H.; Takekawa, M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell. Biol. 2008, 10, 1324–1332. [Google Scholar] [CrossRef]
- Barca, O.; Costoya, J.A.; Senaris, R.M.; Arce, V.M. Interferon-beta protects astrocytes against tumour necrosis factor-induced apoptosis via activation of p38 mitogen-activated protein kinase. Exp. Cell Res. 2008, 314, 2231–2237. [Google Scholar] [CrossRef]
- Du, J.; Yang, S.; Wang, Z.; Zhai, C.; Yuan, W.; Lei, R.; Zhang, J.; Zhu, T. Bone morphogenetic protein 6 inhibit stress-induced breast cancer cells apoptosis via both Smad and p38 pathways. J. Cell Biochem. 2008, 103, 1584–1597. [Google Scholar] [CrossRef]
- Giordano, G.; Klintworth, H.M.; Kavanagh, T.J.; Costa, L.G. Apoptosis induced by domoic acid in mouse cerebellar granule neurons involves activation of p38 and JNK MAP kinases. Neurochem. Int. 2008, 52, 1100–1105. [Google Scholar] [CrossRef]
- Moriguchi, T.; Toyoshima, F.; Gotoh, Y.; Iwamatsu, A.; Irie, K.; Mori, E.; Kuroyanagi, N.; Hagiwara, M.; Matsumoto, K.; Nishida, E. Purification and identification of a major activator for p38 from osmotically shocked cells. Activation of mitogen-activated protein kinase kinase 6 by osmotic shock, tumor necrosis factor-alpha, and H2O2. J. Biol. Chem. 1996, 271, 26981–26988. [Google Scholar]
- Lee, Y.J.; Soh, J.W.; Jeoung, D.I.; Cho, C.K.; Jhon, G.J.; Lee, S.J.; Lee, Y.S. PKC epsilon -mediated ERK1/2 activation involved in radiation-induced cell death in NIH3T3 cells. Biochim. Biophys. Acta 2003, 1593, 219–229. [Google Scholar] [CrossRef]
- Chen, N.Y.; Ma, W.Y.; Huang, C.; Ding, M.; Dong, Z. Activation of PKC is required for arsenite-induced signal transduction. J. Environ. Pathol. Toxicol. Oncol. 2000, 19, 297–305. [Google Scholar]
- Aydin, M.V.; Sen, O.; Kayaselcuk, F.; Bolat, F.; Tufan, K.; Caner, H.; Altinors, N. Analysis and prevalence of inflammatory cells in subtypes of lumbar disc herniations under cyclooxygenase-2 inhibitor therapy. Neurol. Res. 2005, 27, 609–612. [Google Scholar] [CrossRef]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [Google Scholar]
- Jiang, Y.; Chen, C.; Li, Z.; Guo, W.; Gegner, J.A.; Lin, S.; Han, J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). J. Biol. Chem. 1996, 271, 17920–17926. [Google Scholar]
- Stein, B.; Yang, M.X.; Young, D.B.; Janknecht, R.; Hunter, T.; Murray, B.W.; Barbosa, M.S. p38-2, a novel mitogen-activated protein kinase with distinct properties. J. Biol. Chem. 1997, 272, 19509–19517. [Google Scholar]
- Lechner, C.; Zahalka, M.A.; Giot, J.F.; Moller, N.P.; Ullrich, A. ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 4355–4359. [Google Scholar]
- Li, H.L.; Wu, S.; Rottenberg, H. Alcohol inhibits the depolarization-induced stimulation of oxidative phosphorylation in synaptosomes. J. Neurochem. 1996, 66, 1691–1697. [Google Scholar]
- Jiang, Y.; Gram, H.; Zhao, M.; New, L.; Gu, J.; Feng, L.; Di Padova, F.; Ulevitch, R.J.; Han, J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J. Biol. Chem. 1997, 272, 30122–30128. [Google Scholar]
- Lee, S.H.; Park, J.; Che, Y.; Han, P.L.; Lee, J.K. Constitutive activity and differential localization of p38alpha and p38beta MAPKs in adult mouse brain. J. Neurosci. Res. 2000, 60, 623–631. [Google Scholar] [CrossRef]
- Nonaka, Y.; Miyajima, M.; Ogino, I.; Nakajima, M.; Arai, H. Analysis of neuronal cell death in the cerebral cortex of H-Tx rats with compensated hydrocephalus. J. Neurosurg. Pediatr. 2008, 1, 68–74. [Google Scholar] [CrossRef]
- Xiong, W.; Kojic, L.Z.; Zhang, L.; Prasad, S.S.; Douglas, R.; Wang, Y.; Cynader, M.S. Anisomycin activates p38 MAP kinase to induce LTD in mouse primary visual cortex. Brain Res. 2006, 1085, 68–76. [Google Scholar] [CrossRef]
- Ku, B.M.; Lee, Y.K.; Jeong, J.Y.; Mun, J.; Han, J.Y.; Roh, G.S.; Kim, H.J.; Cho, G.J.; Choi, W.S.; Yi, G.S.; Kang, S.S. Ethanol-induced oxidative stress is mediated by p38 MAPK pathway in mouse hippocampal cells. Neurosci. Lett. 2007, 419, 64–67. [Google Scholar] [CrossRef]
- Norkina, O.; Dolganiuc, A.; Shapiro, T.; Kodys, K.; Mandrekar, P.; Szabo, G. Acute alcohol activates STAT3, AP-1, and Sp-1 transcription factors via the family of Src kinases to promote IL-10 production in human monocytes. J. Leukoc. Biol. 2007, 82, 752–762. [Google Scholar] [CrossRef]
- Drechsler, Y.; Dolganiuc, A.; Norkina, O.; Romics, L.; Li, W.; Kodys, K.; Bach, F.H.; Mandrekar, P.; Szabo, G. Heme oxygenase-1 mediates the anti-inflammatory effects of acute alcohol on IL-10 induction involving p38 MAPK activation in monocytes. J. Immunol. 2006, 177, 2592–2600. [Google Scholar]
- Jung, M.E.; Ju, X.; Metzger, D.B.; Simpkins, J.W. Alcohol withdrawal-induced activation of protein kinase P38 targets neurons and a vulnerable age in an estrogen preventable manner. Alcohol Clin. Exp. Res. 2010. 34 Supplement. [Google Scholar]
- Peart, J.N.; Gross, E.R.; Headrick, J.P.; Gross, G.J. Impaired p38 MAPK/HSP27 signaling underlies aging-related failure in opioid-mediated cardioprotection. J. Mol. Cell Cardiol. 2007, 42, 972–980. [Google Scholar] [CrossRef]
- Campos, C.B.; Bedard, P.A.; Linden, R. Requirement of p38 stress-activated MAP kinase for cell death in the developing retina depends on the stage of cell differentiation. Neurochem. Int. 2006, 49, 494–499. [Google Scholar] [CrossRef]
- Zepeda, R.C.; Barrera, I.; Castelan, F.; Soto-Cid, A.; Hernandez-Kelly, L.C.; Lopez-Bayghen, E.; Ortega, A. Glutamate-dependent transcriptional regulation in bergmann glia cells: Involvement of p38 MAP kinase. Neurochem. Res. 2008, 33, 1277–1285. [Google Scholar] [CrossRef]
- Galluzzi, L.; Blomgren, K.; Kroemer, G. Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 2009, 10, 481–494. [Google Scholar] [CrossRef]
- Valles, S.L.; Borras, C.; Gambini, J.; Furriol, J.; Ortega, A.; Sastre, J.; Pallardo, F.V.; Vina, J. Oestradiol or genistein rescues neurons from amyloid beta-induced cell death by inhibiting activation of p38. Aging Cell 2008, 7, 112–118. [Google Scholar] [CrossRef]
- Wu, M.; Han, M.; Li, J.; Xu, X.; Li, T.; Que, L.; Ha, T.; Li, C.; Chen, Q.; Li, Y. 17beta-estradiol inhibits angiotensin II-induced cardiac myofibroblast differentiation. Eur. J. Pharmacol. 2009, 616, 155–159. [Google Scholar] [CrossRef]
- van Eickels, M.; Grohe, C.; Cleutjens, J.P.; Janssen, B.J.; Wellens, H.J.; Doevendans, P.A. 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation 2001, 104, 1419–1423. [Google Scholar] [CrossRef]
- Wang, M.; Tsai, B.M.; Reiger, K.M.; Brown, J.W.; Meldrum, D.R. 17-beta-Estradiol decreases p38 MAPK-mediated myocardial inflammation and dysfunction following acute ischemia. J. Mol. Cell Cardiol. 2006, 40, 205–212. [Google Scholar] [CrossRef]
- Hsu, J.T.; Hsieh, Y.C.; Kan, W.H.; Chen, J.G.; Choudhry, M.A.; Schwacha, M.G.; Bland, K.I.; Chaudry, I.H. Role of p38 mitogen-activated protein kinase pathway in estrogen-mediated cardioprotection following trauma-hemorrhage. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2982–H2987. [Google Scholar] [CrossRef]
- Miyoshi, N.; Oubrahim, H.; Chock, P.B.; Stadtman, E.R. Age-dependent cell death and the role of ATP in hydrogen peroxide-induced apoptosis and necrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 1727–1731. [Google Scholar] [CrossRef]
- Wang, X.; Simpkins, J.W.; Dykens, J.A.; Cammarata, P.R. Oxidative damage to human lens epithelial cells in culture: estrogen protection of mitochondrial potential, ATP, and cell viability. Invest. Ophthalmol. Vis. Sci. 2003, 44, 2067–2075. [Google Scholar] [CrossRef]
- Mansouri, A.; Demeilliers, C.; Amsellem, S.; Pessayre, D.; Fromenty, B. Acute ethanol administration oxidatively damages and depletes mitochondrial dna in mouse liver, brain, heart, and skeletal muscles: Protective effects of antioxidants. J. Pharmacol. Exp. Ther. 2001, 298, 737–743. [Google Scholar]
- Minana, J.B.; Gomez-Cambronero, L.; Lloret, A.; Pallardo, F.V.; Del Olmo, J.; Escudero, A.; Rodrigo, J.M.; Pelliin, A.; Vina, J.R.; Vina, J.; Sastre, J. Mitochondrial oxidative stress and CD95 ligand: A dual mechanism for hepatocyte apoptosis in chronic alcoholism. Hepatology 2002, 35, 1205–1214. [Google Scholar] [CrossRef]
- Siler-Marsiglio, K.I.; Pan, Q.; Paiva, M.; Madorsky, I.; Khurana, N.C.; Heaton, M.B. Mitochondrially targeted vitamin E and vitamin E mitigate ethanol-mediated effects on cerebellar granule cell antioxidant defense systems. Brain Res. 2005, 1052, 202–211. [Google Scholar] [CrossRef]
- Dolder, M.; Wendt, S.; Wallimann, T. Mitochondrial creatine kinase in contact sites: Interaction with porin and adenine nucleotide translocase, role in permeability transition and sensitivity to oxidative damage. Biol. Signals Recept. 2001, 10, 93–111. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Brennerb, C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef]
- Lipskaya, T.Y. Mitochondrial creatine kinase: Properties and function. Biochemistry (Mosc) 2001, 66, 1098–1111. [Google Scholar] [CrossRef]
- Stuart, R. Insertion of proteins into the inner membrane of mitochondria: The role of the Oxa1 complex. Biochim. Biophys. Acta 2002, 1592, 79–87. [Google Scholar] [CrossRef]
- Norenberg, M.D.; Rao, K.V. The mitochondrial permeability transition in neurologic disease. Neurochem. Int. 2007, 50, 983–997. [Google Scholar] [CrossRef]
- Kessova, I.G.; Cederbaum, A.I. Mitochondrial alterations in livers of Sod1-/- mice fed alcohol. Free Radical Biol. Med. 2007, 42, 1470–1480. [Google Scholar]
- Vyssokikh, M.Y.; Brdiczka, D. The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim. Pol. 2003, 50, 389–404. [Google Scholar]
- Brustovetsky, N.; Brustovetsky, T.; Jemmerson, R.; Dubinsky, J.M. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J. Neurochem. 2002, 80, 207–218. [Google Scholar] [CrossRef]
- Fiskum, G.; Rosenthal, R.E.; Vereczki, V.; Martin, E.; Hoffman, G.E.; Chinopoulos, C.; Kowaltowski, A. Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. J. Bioenerg. Biomembr. 2004, 36, 347–352. [Google Scholar] [CrossRef]
- Halestrap, A.P. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem. Soc. Trans. 2006, 34, 232–237. [Google Scholar] [CrossRef]
- Reynolds, I.J. Mitochondrial membrane potential and the permeability transition in excitotoxicity. Ann. NY Acad. Sci. 1999, 893, 33–41. [Google Scholar] [CrossRef]
- Kratzer, U.; Schmidt, W.J. Caroverine inhibits the conditioned place aversion induced by naloxone-precipitated morphine withdrawal in rats. Neurosci. Lett. 2003, 349, 91–94. [Google Scholar] [CrossRef]
- Gatch, M.B. Ethanol withdrawal and hyperalgesia. Curr. Drug Abuse Rev. 2009, 2, 41–50. [Google Scholar] [CrossRef]
- Davila, J.C.; Olmos, L.; Legaz, I.; Medina, L.; Guirado, S.; Real, M.A. Dynamic patterns of colocalization of calbindin, parvalbumin and GABA in subpopulations of mouse basolateral amygdalar cells during development. J. Chem. Neuroanat. 2008, 35, 67–76. [Google Scholar] [CrossRef]
- Nowak, G.; Bakajsova, D.; Clifton, G.L. Protein kinase C-epsilon modulates mitochondrial function and active Na+ transport after oxidant injury in renal cells. Am. J. Physiol. Renal. Physiol. 2004, 286, F307–F316. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Kusaka, E.; Enokido, Y.; Ikeuchi, T.; Hatanaka, H. Regulation of Bax translocation through phosphorylation at Ser-70 of Bcl-2 by MAP kinase in NO-induced neuronal apoptosis. Mol. Cell Neurosci. 2003, 24, 451–459. [Google Scholar] [CrossRef]
- Pan, Z.; Bhat, M.B.; Nieminen, A.L.; Ma, J. Synergistic movements of Ca(2+) and Bax in cells undergoing apoptosis. J. Biol. Chem. 2001, 276, 32257–32263. [Google Scholar]
- Semenova, M.M.; Maki-Hokkonen, A.M.; Cao, J.; Komarovski, V.; Forsberg, K.M.; Koistinaho, M.; Coffey, E.T.; Courtney, M.J. Rho mediates calcium-dependent activation of p38alpha and subsequent excitotoxic cell death. Nat. Neurosci. 2007, 10, 436–443. [Google Scholar]
- Gomez-Lazaro, M.; Galindo, M.F.; Melero-Fernandez de Mera, R.M.; Fernandez-Gomez, F.J.; Concannon, C.G.; Segura, M.F.; Comella, J.X.; Prehn, J.H.; Jordan, J. Reactive oxygen species and p38 mitogen-activated protein kinase activate Bax to induce mitochondrial cytochrome c release and apoptosis in response to malonate. Mol. Pharmacol. 2007, 71, 736–743. [Google Scholar]
- Mancuso, M.; Filosto, M.; Bosetti, F.; Ceravolo, R.; Rocchi, A.; Tognoni, G.; Manca, M.L.; Solaini, G.; Siciliano, G.; Murri, L. Decreased platelet cytochrome c oxidase activity is accompanied by increased blood lactate concentration during exercise in patients with Alzheimer disease. Exp. Neurol. 2003, 182, 421–426. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. Nitric oxide, cytochrome c and mitochondria. Biochem. Soc. Symp. 1999, 66, 17–25. [Google Scholar]
- Jaatinen, P.; Riikonen, J.; Riihioja, P.; Kajander, O.; Hervonen, A. Interaction of aging and intermittent ethanol exposure on brain cytochrome c oxidase activity levels. Alcohol 2003, 29, 91–100. [Google Scholar] [CrossRef]
- Jung, M.E.; Agarwal, R.; Simpkins, J.W. Ethanol withdrawal posttranslationally decreases the activity of cytochrome c oxidase in an estrogen reversible manner. Neurosci. Lett. 2007, 416, 160–164. [Google Scholar] [CrossRef]
- Vina, J.; Lloret, A.; Valles, S.L.; Borras, C.; Badia, M.C.; Pallardo, F.V.; Sastre, J.; Alonso, M.D. Effect of gender on mitochondrial toxicity of Alzheimer's Abeta peptide. Antioxid. Redox Signal. 2007, 9, 1677–1690. [Google Scholar] [CrossRef]
- Borras, C.; Sastre, J.; Garcia-Sala, D.; Lloret, A.; Pallardo, F.V.; Vina, J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radical Biol. Med. 2003, 34, 546–552. [Google Scholar] [CrossRef]
- Wang, J.; Green, P.S.; Simpkins, J.W. Estradiol protects against ATP depletion, mitochondrial membrane potential decline and the generation of reactive oxygen species induced by 3-nitroproprionic acid in SK-N-SH human neuroblastoma cells. J. Neurochem. 2001, 77, 804–811. [Google Scholar] [CrossRef]
- Wang, X.; Dykens, J.A.; Perez, E.; Liu, R.; Yang, S.; Covey, D.F.; Simpkins, J.W. Neuroprotective effects of 17beta-estradiol and nonfeminizing estrogens against H2O2 toxicity in human neuroblastoma SK-N-SH cells. Mol. Pharmacol. 2006, 70, 395–404. [Google Scholar]
- Prokai, L.; Simpkins, J.W. Structure-nongenomic neuroprotection relationship of estrogens and estrogen-derived compounds. Pharmacol. Ther. 2007, 114, 1–12. [Google Scholar] [CrossRef]
- Harms, C.; Lautenschlager, M.; Bergk, A.; Katchanov, J.; Freyer, D.; Kapinya, K.; Herwig, U.; Megow, D.; Dirnagl, U.; Weber, J.R.; Hortnagl, H. Differential mechanisms of neuroprotection by 17 beta-estradiol in apoptotic versus necrotic neurodegeneration. J. Neurosci. 2001, 21, 2600–2609. [Google Scholar]
- Chen, J.Q.; Yager, J.D. Estrogen's effects on mitochondrial gene expression: mechanisms and potential contributions to estrogen carcinogenesis. Ann. NY Acad. Sci. 2004, 1028, 258–272. [Google Scholar] [CrossRef]
- Lee, C.S.; Kim, Y.J.; Ko, H.H.; Han, E.S. Synergistic effects of hydrogen peroxide and ethanol on cell viability loss in PC12 cells by increase in mitochondrial permeability transition. Biochem. Pharmacol. 2005, 70, 317–325. [Google Scholar]
- Sokol, R.J.; Dahl, R.; Devereaux, M.W.; Yerushalmi, B.; Kobak, G.E.; Gumpricht, E. Human hepatic mitochondria generate reactive oxygen species and undergo the permeability transition in response to hydrophobic bile acids. J. Pediatr. Gastroenterol. Nutr. 2005, 41, 235–243. [Google Scholar] [CrossRef]
- Dhandapani, K.M.; Brann, D.W. Protective effects of estrogen and selective estrogen receptor modulators in the brain. Biol. Reprod. 2002, 67, 1379–1385. [Google Scholar] [CrossRef]
- Ha, E.J.; Smith, A.M. Plasma selenium and plasma and erythrocyte glutathione peroxidase activity increase with estrogen during the menstrual cycle. J. Am. Coll. Nutr. 2003, 22, 43–51. [Google Scholar]
- Lapointe, J.; Kimmins, S.; Maclaren, L.A.; Bilodeau, J.F. Estrogen selectively up-regulates the phospholipid hydroperoxide glutathione peroxidase in the oviducts. Endocrinology 2005, 146, 2583–2592. [Google Scholar] [CrossRef]
- Nilsen, J.; Diaz Brinton, R. Mechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expression. Proc. Natl. Acad. Sci. USA 2003, 100, 2842–2847. [Google Scholar] [CrossRef]
- Culberson, J.W. Alcohol use in the elderly: Beyond the CAGE. Part 1 of 2: prevalence and patterns of problem drinking. Geriatrics 2006, 61, 23–27. [Google Scholar]
- Culberson, J.W. Alcohol use in the elderly: beyond the CAGE. Part 2: Screening instruments and treatment strategies. Geriatrics 2006, 61, 20–26. [Google Scholar]
- Meier, P.; Seitz, H.K. Age, alcohol metabolism and liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 21–26. [Google Scholar] [CrossRef]
- Tarter, R.E.; Edwards, K.L. Multifactorial etiology of neuropsychological impairment in alcoholics. Alcohol Clin. Exp. Res. 1986, 10, 128–135. [Google Scholar] [CrossRef]
- Riihioja, P.; Jaatinen, P.; Haapalinna, A.; Kiianmaa, K.; Hervonen, A. Effects of ageing and intermittent ethanol exposure on rat locus coeruleus and ethanol-withdrawal symptoms. Alcohol Alcohol. 1999, 34, 706–717. [Google Scholar]
- Cocuzza, M.; Athayde, K.S.; Agarwal, A.; Sharma, R.; Pagani, R.; Lucon, A.M.; Srougi, M.; Hallak, J. Age-related increase of reactive oxygen species in neat semen in healthy fertile men. Urology 2008, 71, 490–494. [Google Scholar] [CrossRef]
- Judge, S.; Jang, Y.M.; Smith, A.; Hagen, T.; Leeuwenburgh, C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005, 19, 419–421. [Google Scholar]
- Mehta, R.; Shangari, N.; O'Brien, P.J. Preventing cell death induced by carbonyl stress, oxidative stress or mitochondrial toxins with vitamin B anti-AGE agents. Mol. Nutr. Food Res. 2008, 52, 379–385. [Google Scholar] [CrossRef]
- Abdel, S.E.; Abdel-Meguid, I.; Korraa, S. Markers of oxidative stress and aging in Duchene muscular dystrophy patients and the possible ameliorating effect of He:Ne laser. Acta Myol. 2007, 26, 14–21. [Google Scholar]
- Navarro, A.; Lopez-Cepero, J.M.; Bandez, M.J.; Sanchez-Pino, M.J.; Gomez, C.; Cadenas, E.; Boveris, A. Hippocampal mitochondrial dysfunction in rat aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R501–R509. [Google Scholar]
- Opii, W.O.; Joshi, G.; Head, E.; Milgram, N.W.; Muggenburg, B.A.; Klein, J.B.; Pierce, W.M.; Cotman, C.W.; Butterfield, D.A. Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer's disease. Neurobiol. Aging 2008, 29, 51–70. [Google Scholar] [CrossRef]
- Safciuc, F.; Constantin, A.; Manea, A.; Nicolae, M.; Popov, D.; Raicu, M.; Alexandru, D.; Constantinescu, E. Advanced glycation end products, oxidative stress and metalloproteinases are altered in the cerebral microvasculature during aging. Curr. Neurovasc. Res. 2007, 4, 228–234. [Google Scholar] [CrossRef]
- Bugalho, P.; Viana-Baptista, M.; Jordao, C.; Secca, M.F.; Ferro, J.M. Age-related white matter lesions are associated with reduction of the apparent diffusion coefficient in the cerebellum. Eur. J. Neurol. 2007, 14, 1063–1066. [Google Scholar] [CrossRef]
- Giusto, N.M.; Salvador, G.A.; Castagnet, P.I.; Pasquare, S.J.; Ilincheta de Boschero, M.G. Age-associated changes in central nervous system glycerolipid composition and metabolism. Neurochem. Res. 2002, 27, 1513–1523. [Google Scholar] [CrossRef]
- Wojnar, M.; Wasilewski, D.; Zmigrodzka, I.; Grobel, I. Age-related differences in the course of alcohol withdrawal in hospitalized patients. Alcohol Alcohol. 2001, 36, 577–583. [Google Scholar]
- Davies, B.T.; Bowen, C.K. Total body water and peak alcohol concentration: A comparative study of young, middle-age, and older females. Alcohol Clin. Exp. Res. 1999, 23, 969–975. [Google Scholar]
- Young, C.; Klocke, B.J.; Tenkova, T.; Choi, J.; Labruyere, J.; Qin, Y.Q.; Holtzman, D.M.; Roth, K.A.; Olney, J.W. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ. 2003, 10, 1148–1155. [Google Scholar] [CrossRef]
- Kim, Y.C.; Kim, S.Y.; Sohn, Y.R. Effect of age increase on metabolism and toxicity of ethanol in female rats. Life Sci. 2003, 74, 509–519. [Google Scholar]
- Williamson, D.; Gallagher, P.; Harber, M.; Hollon, C.; Trappe, S. Mitogen-activated protein kinase (MAPK) pathway activation: Effects of age and acute exercise on human skeletal muscle. J. Physiol. 2003, 547, 977–987. [Google Scholar] [CrossRef]
- Kelleher, I.; Garwood, C.; Hanger, D.P.; Anderton, B.H.; Noble, W. Kinase activities increase during the development of tauopathy in htau mice. J. Neurochem. 2007, 103, 2256–2267. [Google Scholar] [CrossRef]
- Hensley, K.; Floyd, R.A.; Zheng, N.Y.; Nael, R.; Robinson, K.A.; Nguyen, X.; Pye, Q.N.; Stewart, C.A.; Geddes, J.; Markesbery, W.R.; Patel, E.; Johnson, G.V.; Bing, G. p38 kinase is activated in the Alzheimer's disease brain. J. Neurochem. 1999, 72, 2053–2058. [Google Scholar]
- Vereker, E.; O'Donnell, E.; Lynch, M.A. The inhibitory effect of interleukin-1beta on long-term potentiation is coupled with increased activity of stress-activated protein kinases. J. Neurosci. 2000, 20, 6811–6819. [Google Scholar]
- Watanabe, M. Molecular mechanisms governing competitive synaptic wiring in cerebellar Purkinje cells. Tohoku J. Exp. Med. 2008, 214, 175–190. [Google Scholar] [CrossRef]
- Becker, E.B.; Oliver, P.L.; Glitsch, M.D.; Banks, G.T.; Achilli, F.; Hardy, A.; Nolan, P.M.; Fisher, E.M.; Davies, K.E. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6706–6711. [Google Scholar]
- Levin, S.I.; Khaliq, Z.M.; Aman, T.K.; Grieco, T.M.; Kearney, J.A.; Raman, I.M.; Meisler, M.H. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a (NaV1.6) in cerebellar purkinje neurons and granule cells. J. Neurophysiol. 2006, 96, 785–793. [Google Scholar] [CrossRef]
- Carta, M.; Mameli, M.; Valenzuela, C.F. Alcohol potently modulates climbing fiber-->Purkinje neuron synapses: role of metabotropic glutamate receptors. J. Neurosci. 2006, 26, 1906–1912. [Google Scholar] [CrossRef]
- Markowska, A.L. Sex dimorphisms in the rate of age-related decline in spatial memory: Relevance to alterations in the estrous cycle. J. Neurosci. 1999, 19, 8122–8133. [Google Scholar]
- Kok, E.; Haikonen, S.; Luoto, T.; Huhtala, H.; Goebeler, S.; Haapasalo, H.; Karhunen, P.J. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann. Neurol. 2009, 65, 650–657. [Google Scholar] [CrossRef]
- Finch, C.E. The neurobiology of middle-age has arrived. Neurobiol. Aging 2009, 30, 515–520, discussion 530-533. [Google Scholar] [CrossRef]
- Li, L.; Shou, Y.; Borowitz, J.L.; Isom, G.E. Reactive oxygen species mediate pyridostigmine-induced neuronal apoptosis: Involvement of muscarinic and NMDA receptors. Toxicol. Appl. Pharmacol. 2001, 177, 17–25. [Google Scholar] [CrossRef]
- Guarnieri, C.; Muscari, C.; Caldarera, C.M. Mitochondrial production of oxygen free radicals in the heart muscle during the life span of the rat: Peak at middle age. EXS 1992, 62, 73–77. [Google Scholar]
- Joseph, J.A.; Kochman, K.; Roth, G.S. Reduction of motor behavioural deficits in senescence via chronic prolactin or estrogen administration: time course and putative mechanisms of action. Brain Res. 1989, 505, 195–202. [Google Scholar] [CrossRef]
- Rapp, S.R.; Espeland, M.A.; Shumaker, S.A.; Henderson, V.W.; Brunner, R.L.; Manson, J.E.; Gass, M.L.; Stefanick, M.L.; Lane, D.S.; Hays, J.; Johnson, K.C.; Coker, L.H.; Dailey, M.; Bowen, D. Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women's Health Initiative Memory Study: A randomized controlled trial. JAMA 2003, 289, 2663–2672. [Google Scholar]
- Ziegler, D.R.; Gallagher, M. Spatial memory in middle-aged female rats: assessment of estrogen replacement after ovariectomy. Brain Res. 2005, 1052, 163–173. [Google Scholar] [CrossRef]
- Gibbs, R.B. Long-term treatment with estrogen and progesterone enhances acquisition of a spatial memory task by ovariectomized aged rats. Neurobiol. Aging 2000, 21, 107–116. [Google Scholar] [CrossRef]
- Lacreuse, A.; Wilson, M.E.; Herndon, J.G. Estradiol, but not raloxifene, improves aspects of spatial working memory in aged ovariectomized rhesus monkeys. Neurobiol. Aging 2002, 23, 589–600. [Google Scholar] [CrossRef]
- Zandi, P.P.; Carlson, M.C.; Plassman, B.L.; Welsh-Bohmer, K.A.; Mayer, L.S.; Steffens, D.C.; Breitner, J.C. Hormone replacement therapy and incidence of Alzheimer disease in older women: The Cache County Study. JAMA 2002, 288, 2123–2129. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jung, M.E.; Metzger, D.B. Alcohol Withdrawal and Brain Injuries: Beyond Classical Mechanisms. Molecules 2010, 15, 4984-5011. https://doi.org/10.3390/molecules15074984
Jung ME, Metzger DB. Alcohol Withdrawal and Brain Injuries: Beyond Classical Mechanisms. Molecules. 2010; 15(7):4984-5011. https://doi.org/10.3390/molecules15074984
Chicago/Turabian StyleJung, Marianna E., and Daniel B. Metzger. 2010. "Alcohol Withdrawal and Brain Injuries: Beyond Classical Mechanisms" Molecules 15, no. 7: 4984-5011. https://doi.org/10.3390/molecules15074984