Asymmetric Synthesis of α-Aminophosphonates Using the Inexpensive Chiral Catalyst 1,1’-Binaphthol Phosphate

Abstract
:1. Introduction

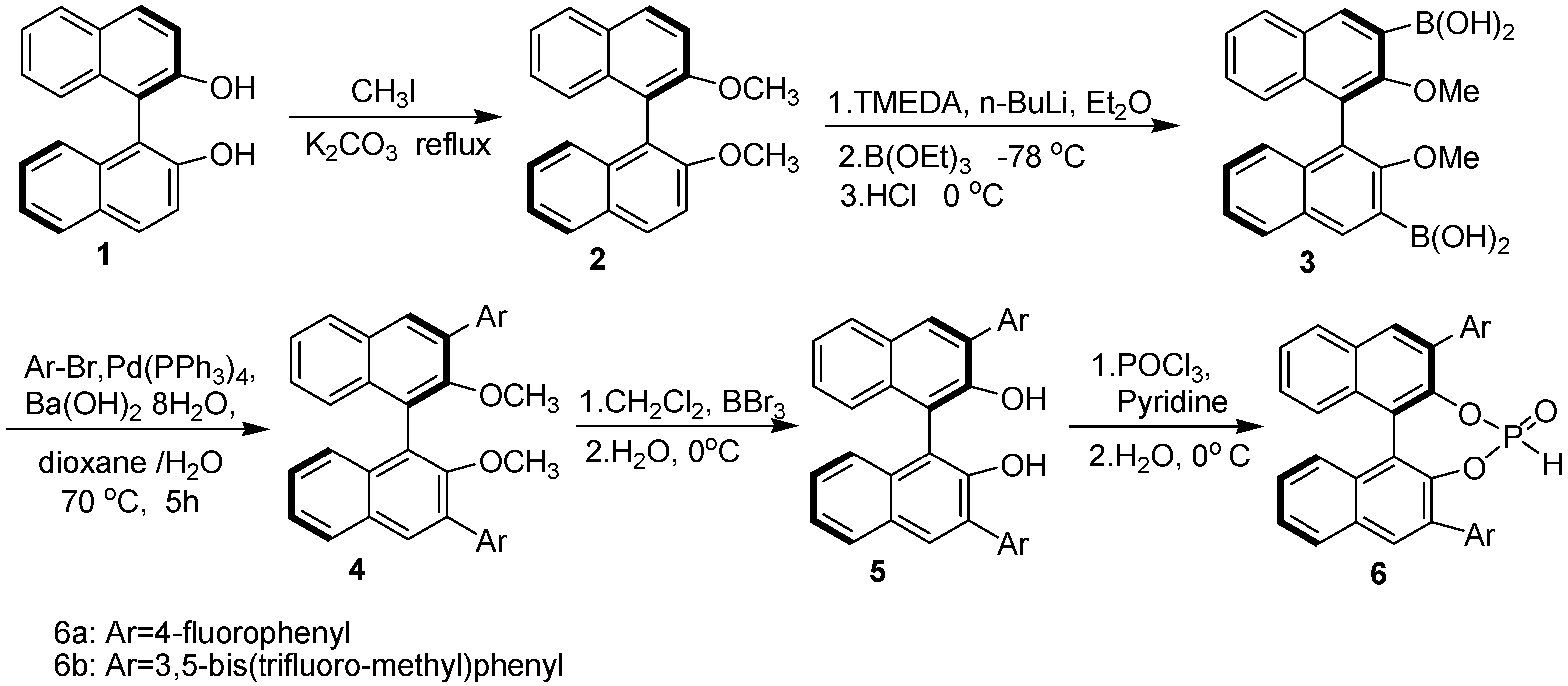
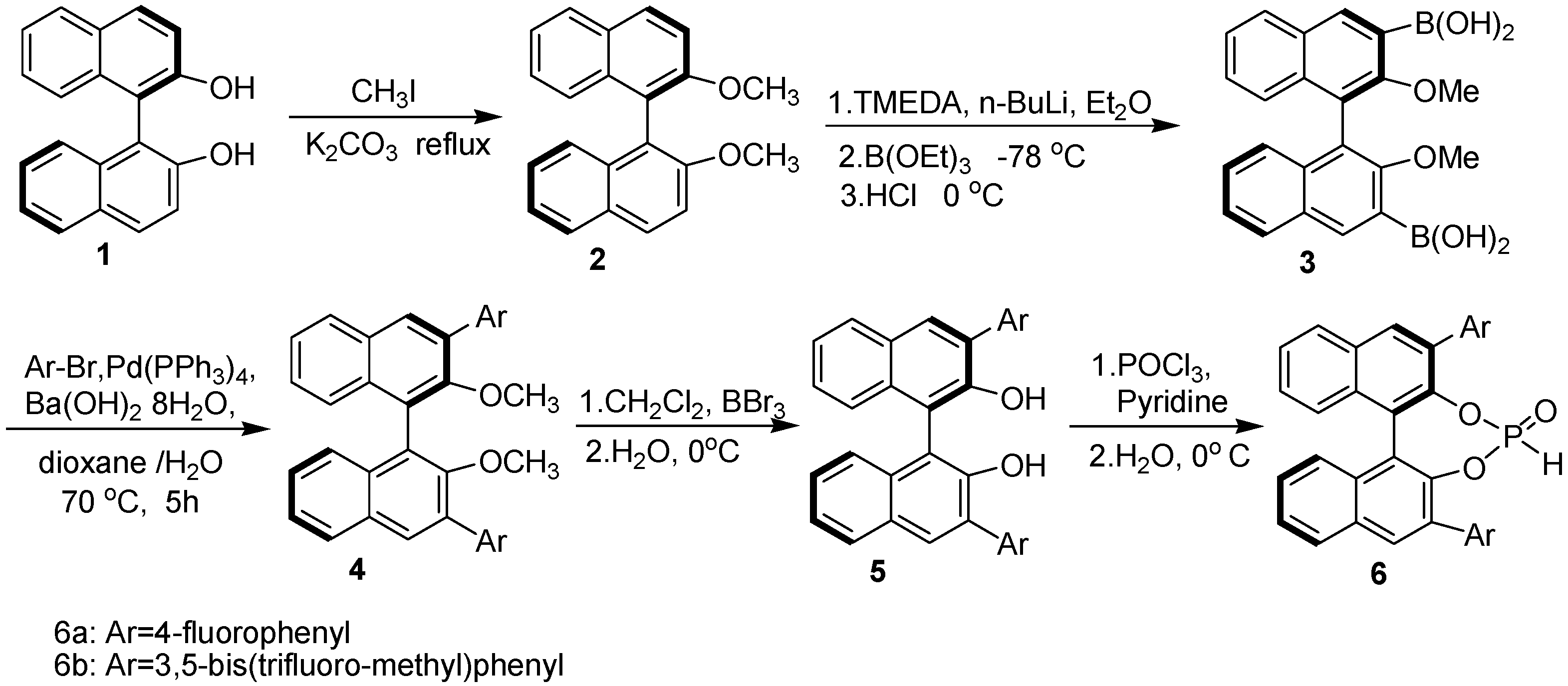
2. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | R3 | R4 | Yield (%) | e.e. (%)b |
|---|---|---|---|---|---|
| 1 | 6a | 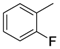 | 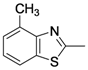 | 20 | 0 |
| 2 | 6b | | | 56 | 10.2 |
| 3 | 6a | 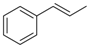 |  | 51 | 31.9 |
| 4 | 6b | | | 45 | 89.5 |
| Entry | 7 | R | Time (h) | Yield (%) |
|---|---|---|---|---|
| 1 | 7a | H | 3 | 85 |
| 2 | 7b | 2-F | 5 | 89 |
| 3 | 7c | 2-CH3 | 8 | 77 |
| 4 | 7d | 4-CH3 | 8 | 65 |
| Entry | Compound | R1 | R2 | Yield (%) | e.e. (%) |
|---|---|---|---|---|---|
| 1 | 9a | H | Et | 41 | 16.5 |
| 2 | 9b | H | Pr | 52 | 17.3 |
| 3 | 9c | H | i-Pr | 33 | 36.9 |
| 4 | 9d | H | Bu | 65 | 16.7 |
| 5 | 9e | 2-F | Et | 60 | 18.8 |
| 6 | 9f | 2-F | Pr | 54 | 8.4 |
| 7 | 9g | 2-F | i-Pr | 31 | 44.2 |
| 8 | 9h | 2-F | Bu | 47 | 27.5 |
| 9 | 9i | 2-CH3 | Et | 51 | 31.9 |
| 10 | 9j | 2-CH3 | Pr | 48 | 16.9 |
| 11 | 9k | 2-CH3 | i-Pr | 32 | 61.9 |
| 12 | 9l | 2-CH3 | Bu | 48 | 30.1 |
| 13 | 9m | 4- CH3 | Et | 50 | 28.3 |
| 14 | 9n | 4- CH3 | Pr | 45 | 15.3 |
| 15 | 9o | 4- CH3 | i-Pr | 30 | 51.5 |
| 16 | 9p | 4- CH3 | Bu | 39 | 22.7 |

| Entry | Solvent | Temp (°C) | Cat 6a (mol%) | ee (%) |
|---|---|---|---|---|
| 1 | acetonitrile | r.t | 10 | 30.1 |
| 2 | methylene chloride | r.t | 10 | 48.0 |
| 3 | toluene | r.t | 10 | 52.8 |
| 4 | xylene | r.t | 10 | 61.9 |
| 5 | xylene | r.t | 20 | 65.1 |
| 6 | xylene | r.t | 5 | 49.9 |
| 7 | xylene | r.t | 2 | 20.9 |
| 8 | xylene | 0 | 10 | 64.9 |
| 9 | xylene | -40 | 10 | 68.3 |
| 10 | xylene | 50 | 10 | 57.9 |
3. Experimental
3.1. General
3.2. Preparation of chiral catalysts 6a-b
3.3. Preparation of intermediate imines 7
3.4. Preparation of title chiral compounds 9a-p
4. Conclusions
Acknowledgements
References and Notes
- Hirschmann, R.; Smith, A.B.; Taylor, C.M.; Benkovic, P.A.; Taylor, S.D.; Yager, K.M.; Sprengler, P.A.; Benkovic, S.J. Peptide synthesis catalyzed by an antibody containing a binding site for variable amino acids. Science 1994, 265, 234–237. [Google Scholar]
- Allen, M.C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J.M. Renin inhibitors. Synthesis of transition-state analog inhibitors containing phosphorus acid derivatives at the scissile bond. J. Med. Chem. 1989, 32, 1652–1661. [Google Scholar] [CrossRef]
- Logusch, E.W.; Walker, D.M.; McDonald, J.F.; Leo, G.C.; Franz, J.E. Synthesis of .alpha.- and .gamma.-alkyl-substituted phosphinothricins: Potent new inhibitors of glutamine synthetase. J. Org. Chem. 1988, 53, 4069–4074. [Google Scholar]
- Giannousis, P.P.; Bartlett, P.A. Phosphorus amino acid analogs as inhibitors of leucine aminopeptidase. J. Med. Chem. 1987, 30, 1603–1609. [Google Scholar] [CrossRef]
- Emgenbroich, M.; Wulff, G. A New Enzyme Model For Enantioselective Esterases Based On Molecularly Imprinted Polymers. Chem. Eur. J. 2003, 9, 4106–4177. [Google Scholar] [CrossRef]
- Senten, K.; Danie, L.; Var der Veken, P.; De Meester, I.; Lambeir, A.M.; Scharpe, S.; Haemers, A.; Augustyns, K. Rapid Parallel Synthesis of Dipeptide Diphenyl Phosphonate Esters as Inhibitors of Dipeptidyl Peptidases. J. Comb. Chem. 2003, 5, 336–344. [Google Scholar] [CrossRef]
- Stowasser, B.; Budt, K.H.; Li, J.Q.; Peyman, A.; Ruppert, D. New hybrid transition state analog inhibitors of HIV protease with peripheric C2-symmetry. Tetrahedron Lett. 1992, 33, 6625–6628. [Google Scholar]
- Patel, D.V.; Rielly-Gauvin, K.; Ryono, D.E. Preparation of peptidic α-hydroxy phosphonates a new class of transition state analog renin inhibitors. Tetrahedron Lett. 1990, 31, 5587–5590. [Google Scholar] [CrossRef]
- Beers, S.A.; Schwender, C.F.; Loughney, D.A.; Malloy, E.; Demarest, K.; Jordan, J. Phosphatase inhibitors-III. Benzylaminophosphonic acids as potent inhibitors of human prostatic acid phosphatase. Bioorg. Med. Chem. 1996, 4, 1693–1701. [Google Scholar] [CrossRef]
- Burke, T.R.; Barchi, J.J.; George, C.; Wolf, G.; Shoelson, S.E.; Yan, X. Conformationally constrained phosphotyrosyl mimetics designed as monomeric src homology 2 domain inhibitors. J. Med. Chem. 1995, 38, 1386–1396. [Google Scholar] [CrossRef]
- Atherton, F.R.; Hassall, C.H.; Lambert, R.W. Synthesis and structure-activity relationships of antibacterial phosphonopeptides incorporating (1-aminoethyl)phosphonic acid and (aminomethyl)phosphonic acid. J. Med. Chem. 1986, 29, 29–40. [Google Scholar] [CrossRef]
- Lejczak, B.; Kafarski, P.; Sztajer, H.; Mastalerz, P. Antibacterial activity of phosphono dipeptides related to alafosfalin. J. Med. Chem. 1986, 29, 2212–2217. [Google Scholar] [CrossRef]
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The Most Potent Organophosphorus Inhibitors of Leucine Aminopeptidase. Structure-Based Design, Chemistry, and Activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef]
- Moore, J.D.; Sprott, K.T.; Hanson, P.R. Conformationally Constrained α-Boc-Aminophosphonates via Transition Metal-Catalyzed/Curtius Rearrangement Strategies. J. Org. Chem. 2002, 67, 8123–8129. [Google Scholar] [CrossRef]
- Liu, W.-S; Rogers, C.J.; Fisher, A.J.; Toney, M.D. Aminophosphonate inhibitors of dialkylglycine decarboxylase: Structural basis for slow binding inhibition. Biochemistry. 2002, 41, 12320–12328. [Google Scholar]
- Huang, J.; Chen, R. An overview of recent advances on the synthesis and biological activity of α-aminophosphonic acid derivatives. Heteroatom. Chem. 2000, 11, 480–492. [Google Scholar] [CrossRef]
- Maier, L.; Diel, P.J. Organic phosphorus compounds 941 preparation, physical and biological properties of amino-arylmethylphosphonic-and-phosphonous acids. Phosphor. Sulfur Silicon 1991, 57, 57–64. [Google Scholar] [CrossRef]
- Yager, K.M.; Taylor, C.M.; Smith, A.B., III. Asymmetric synthesis of alpha.-aminophosphonates via diastereoselective addition of lithium diethyl phosphite to chelating imines. J. Am. Chem. Soc. 1994, 116, 9377–9378. [Google Scholar] [CrossRef]
- Bird, J.; Rachel, C.D.M.; Gregory, P.H.; David, J.H.; Eric, H.K.; Roger, E.M.; Anette, J.M.; Rahman, S.S.; Ward, R.W. Synthesis of novel N-phosphonoalkyl dipeptide inhibitors of human collagenase. J. Med. Chem. 1994, 37, 158–169. [Google Scholar] [CrossRef]
- Davis, F.A.; Lee, S.; Yan, H.; Titus, D.D. Asymmetric synthesis of quaternary α-amino phosphonates using sulfinimines. Org. Lett. 2001, 3, 1757–1760. [Google Scholar] [CrossRef]
- Brunel, J.M. BINOL: A versatile chiral reagent. Chem. Rev. 2005, 105, 857–897. [Google Scholar] [CrossRef]
- Verkade, J.M.M.; van Hemert, L.J.C.; Quaedflie, P.J.L.M.; Rutjes, F.P.J.T. Organocatalysed asymmetric Mannich reactions. Chem. Soc. Rev. 2008, 37, 29–41. [Google Scholar]
- Bhadury, P.S.; Song, B.A.; Yang, S.; Zhang, Y.; Zhang, S. Some potential chiral catalysts for preparation of asymmetric α-aminophosphonates. Curr. Org. Synth. 2008, 5, 134–150. [Google Scholar] [CrossRef]
- Cheng, X.; Goddard, R.; Buth, G.; List, B. Direct Catalytic asymmetric three-component Kabachnik-Fields reaction. Angew. Chem. Int. Ed. 2008, 47, 5079–5081. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Belokon, Y.N.; Kuzmina, N.A.; Maleev, V.I.; Svistunova, N.Y.; Solodenko, V.A.; Kukhar, V.P. Asymmetric synthesis of phosphorus analogues of dicarboxylic α-amino acids. J. Chem. Soc. Perkin Trans. I 1992, 12, 1525–1529. [Google Scholar]
- Kukhar, V.P.; Soloshonok, V.A.; Solodenko, V.A. Asymmetric Synthesis of Phosphorus Analogs of Amino Acids. Phosphor. Sulfur Silicon 1994, 92, 239–264. [Google Scholar] [CrossRef]
- Akiyama, T.; Morita, H.; Itoh, J.; Fuchibe, K. Chiral brønsted acid catalyzed enantioselective hydrophosphonylation of imines: Asymmetric synthesis of α-amino phosphonates. Org. Lett. 2005, 7, 2583–2585. [Google Scholar] [CrossRef]
- Bhadury, P.S.; Zhang, Y.P.; Zhang, S.; Song, B.A.; Yang, S.; Hu, D.Y.; Chen, Z.; Xue, W.; Jin, L.H. An effective route to fluorine containing asymmetric alpha-aminophosphonates using chiral Bronsted acid catalyst. Chirality 2009, 21, 547–557. [Google Scholar] [CrossRef]
- Kang, Q.; Zhao, Z.A.; You, S.L. Highly Enantioselective Friedel-Crafts Reaction of Indoles with Imines by a Chiral Phosphoric Acid. J. Am. Chem. Soc. 2007, 129, 1484–1485. [Google Scholar]
- Bhadury, P.S.; Song, B.A.; Yang, S.; Hu, D.Y.; Xue, W. Bifunctional Chiral Organocatalysts in Organic Transformations. Curr. Org. Synth. 2009, 6, 380–399. [Google Scholar] [CrossRef]
- Shi, F.Q.; Song, B.A. Origins of enantioselectivity in the chiral Brønsted acid catalyzed hydrophosphonylation of imines. Org. Biomol. Chem. 2009, 7, 1292–1298. [Google Scholar] [CrossRef]
- Wipf, P.; Jung, J.-K. Formal total synthesis of (+)-diepoxin σ. J. Org. Chem. 2000, 65, 6319–6337. [Google Scholar] [CrossRef]
- Soloshonok, V.A. Remarkable Amplification of the Self-Disproportionation of Enantiomers on Achiral-Phase Chromatography Columns. Angew. Chem. In. Ed. Engl. 2006, 45, 766–769. [Google Scholar]
- Soloshonok, V.A.; Ueki, H.; Yasumoto, M.; Mekala, S.; Hirschi, J.S.; Singleton, D.A. Phenomenon of Optical Self-Purification of Chiral Non-Racemic Compounds. J. Am. Chem. Soc. 2007, 129, 12112–12113. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, W.; Zhang, S.; Yang, S.; Jin, L.-H.; Bhadury, P.S.; Hu, D.-Y.; Zhang, Y. Asymmetric Synthesis of α-Aminophosphonates Using the Inexpensive Chiral Catalyst 1,1’-Binaphthol Phosphate. Molecules 2010, 15, 5782-5796. https://doi.org/10.3390/molecules15085782
Xu W, Zhang S, Yang S, Jin L-H, Bhadury PS, Hu D-Y, Zhang Y. Asymmetric Synthesis of α-Aminophosphonates Using the Inexpensive Chiral Catalyst 1,1’-Binaphthol Phosphate. Molecules. 2010; 15(8):5782-5796. https://doi.org/10.3390/molecules15085782
Chicago/Turabian StyleXu, Weiming, Sha Zhang, Song Yang, Lin-Hong Jin, Pinaki S. Bhadury, De-Yu Hu, and Yuping Zhang. 2010. "Asymmetric Synthesis of α-Aminophosphonates Using the Inexpensive Chiral Catalyst 1,1’-Binaphthol Phosphate" Molecules 15, no. 8: 5782-5796. https://doi.org/10.3390/molecules15085782