An Efficient Procedure Based on a MW-Assisted Horner–Wadsworth-Emmons Reaction for the Synthesis of (Z)-3,3-Trisubstituted-a,b-unsaturated Esters





Abstract
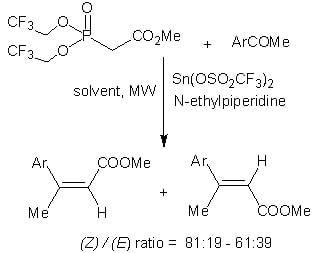
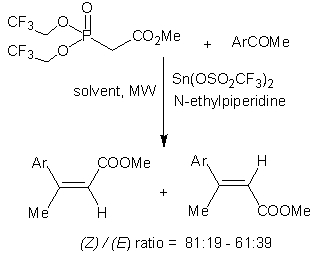
:
1. Introduction


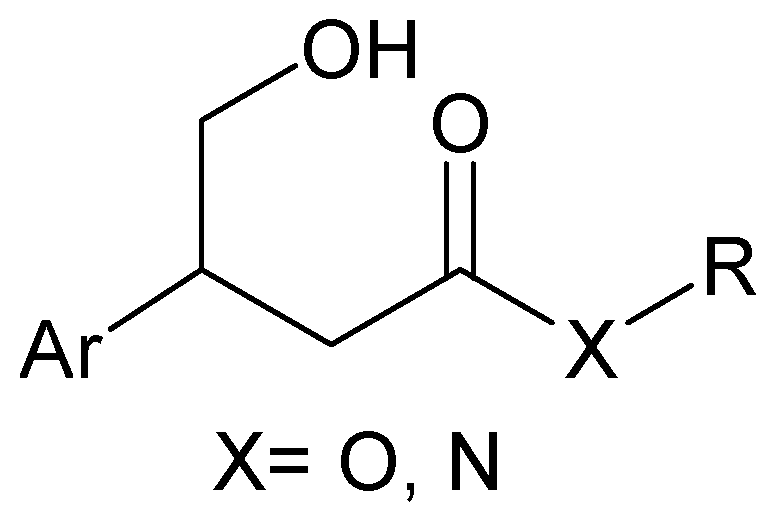
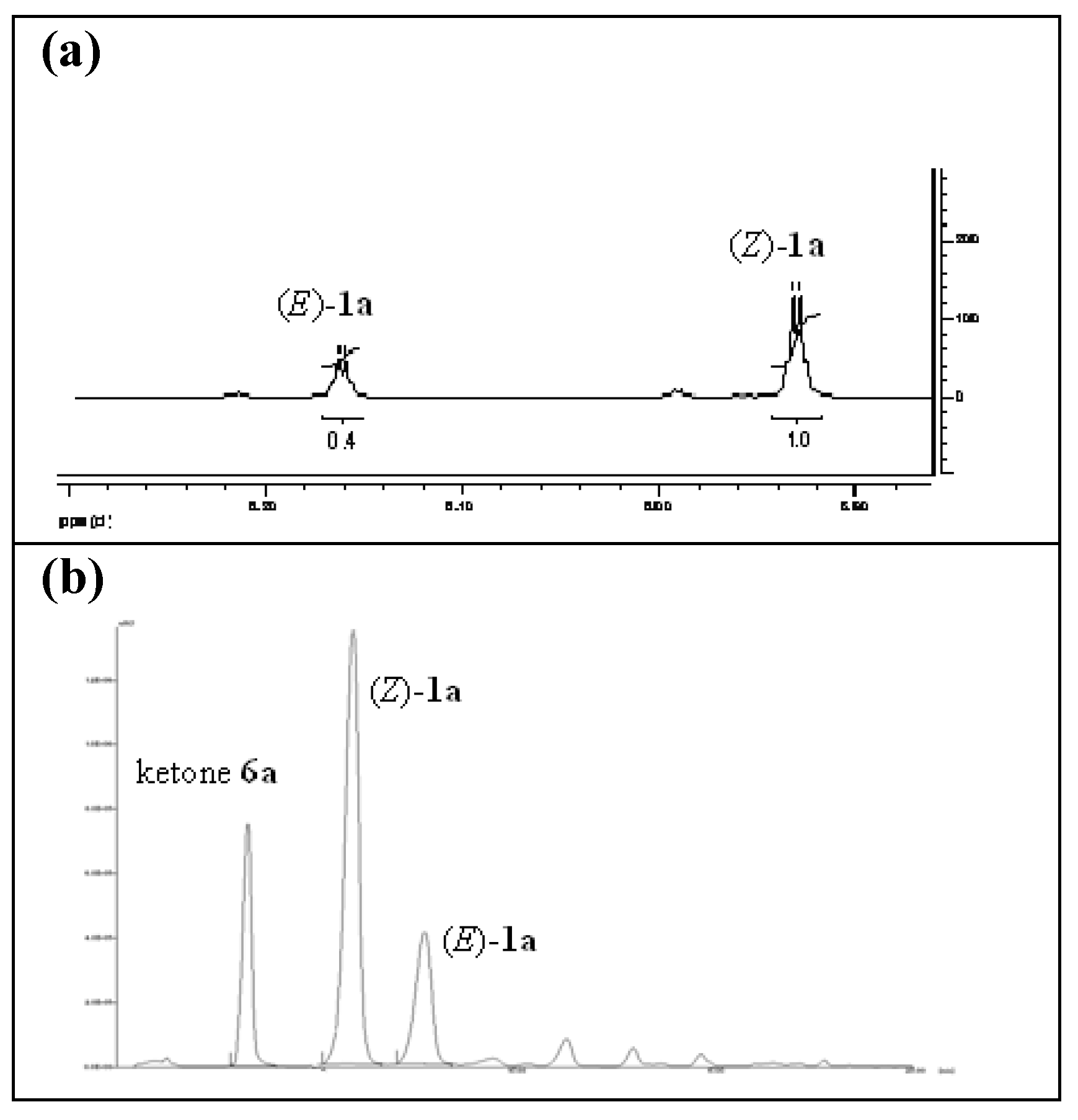
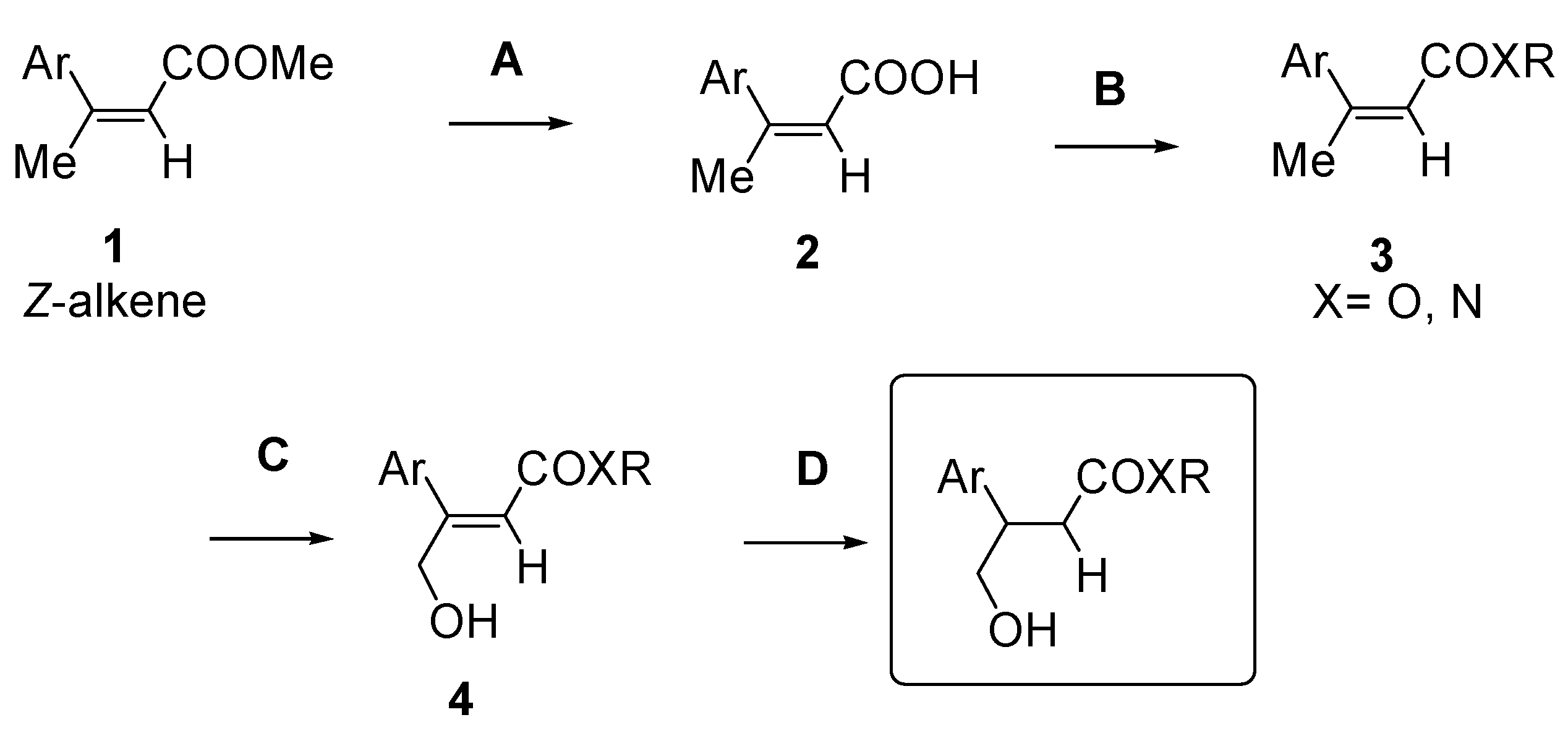
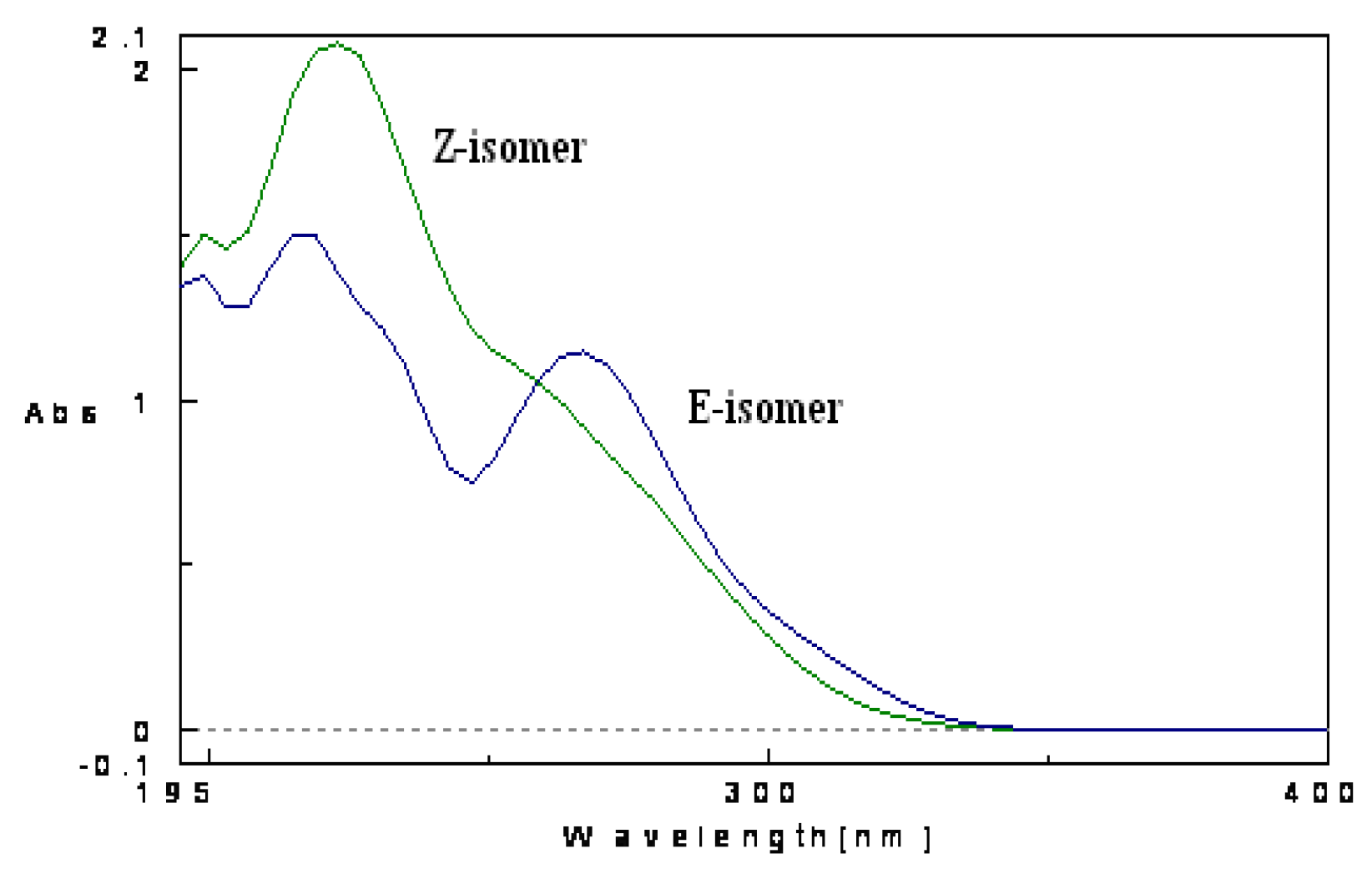
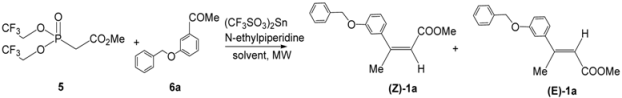
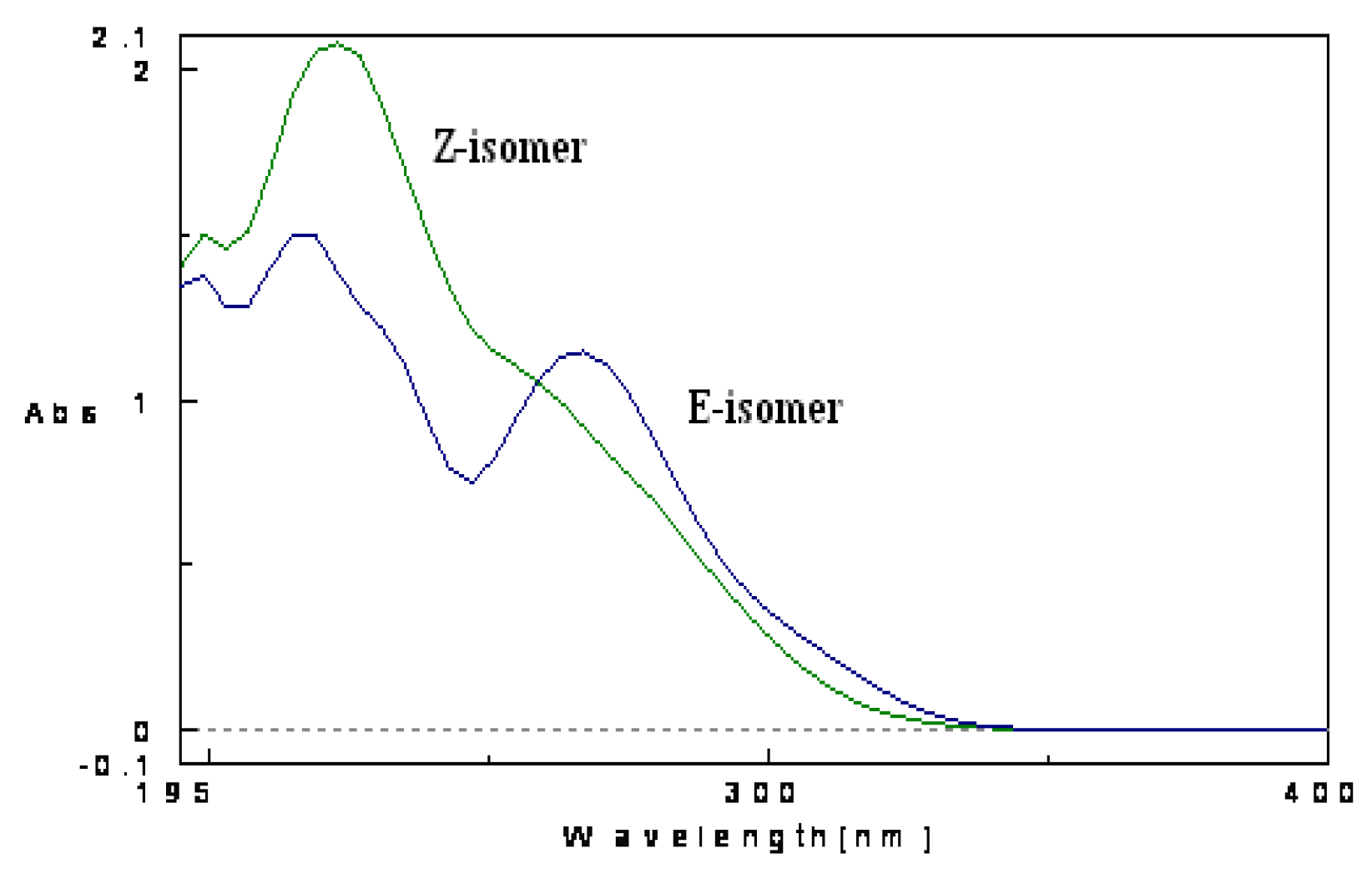
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | 5 equiv | T (°C) | Time (min) | Conversion a (%) | (Z)/(E) ratioa |
| 1 | DCM | 1.5 | 70 | 30 | 58 | 71:29 |
| 2 | DCM | 3 | 70 | 30 | 58 | 69:31 |
| 3 | DCM | 4.2 | 70 | 30 | 63 | 70:30 |
| 4 | DMF | 4.2 | 70 | 30 | - | - |
| 5 | DMF | 4.2 | 150 | 30 | - | - |
| 6 | THF | 4.2 | 70 | 30 | - | - |
| 7 | THF | 4.2 | 150 | 30 | - | - |
| 8 | DCE | 4.2 | 70 | 30 | 59 | 67:33 |
| 9 | DCE | 4.2 | 120 | 30 | 69 | 64:36 |
| 10 | DCE | 4.2 | 120 | 20 | 72 | 65:35 |
| 11 | DCE | 4.2 | 150 | 30 | 85 | 69:31 |
| 12 | DCE | 4.2 | 150 | 20 | 98 | 81:19 |
| 13 | DCE | 4.2 | 150 | 15 | 84 | 75:25 |
| 14 | DCE | 4.2 | 150 | 10 | 47 | 61:39 |
| 15 | DCE | 3 | 150 | 30 | 77 | 61:39 |
| 16 | DCE | 3 | 150 | 20 | 82 | 65:35 |
| 17 | DCE | 3 | 150 | 15 | 76 | 65:35 |
| 18 | DCE | 3 | 150 | 10 | 61 | 50:50 |

 | ||||
|---|---|---|---|---|
| Entry | Ar | Olefin | Conversion (%)a | (Z)/(E)ratioa |
| 1 |  | 1b | 55 | 66:34 |
| 2 |  | 1c | 99 | 74:26 |
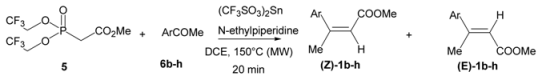
| 3 | 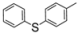 | 1d | 86 | 76:24 |
| 4 |  | 1e | 58 | 79:21 |
| 5 |  | 1f | 92 | 66:34 |
| 6 |  | 1g | 56 | 61:39 |
| 7 |  | 1h | 60 | 74:26 |
3. Experimental
3.1. General
3.2. General Procedure for the Synthesis of 3,3-Trisubstituted-α,β-Unsaturated Methyl Esters
4. Conclusions
- Sample Availability: Samples of the compounds are available from the authors.
Appendix I
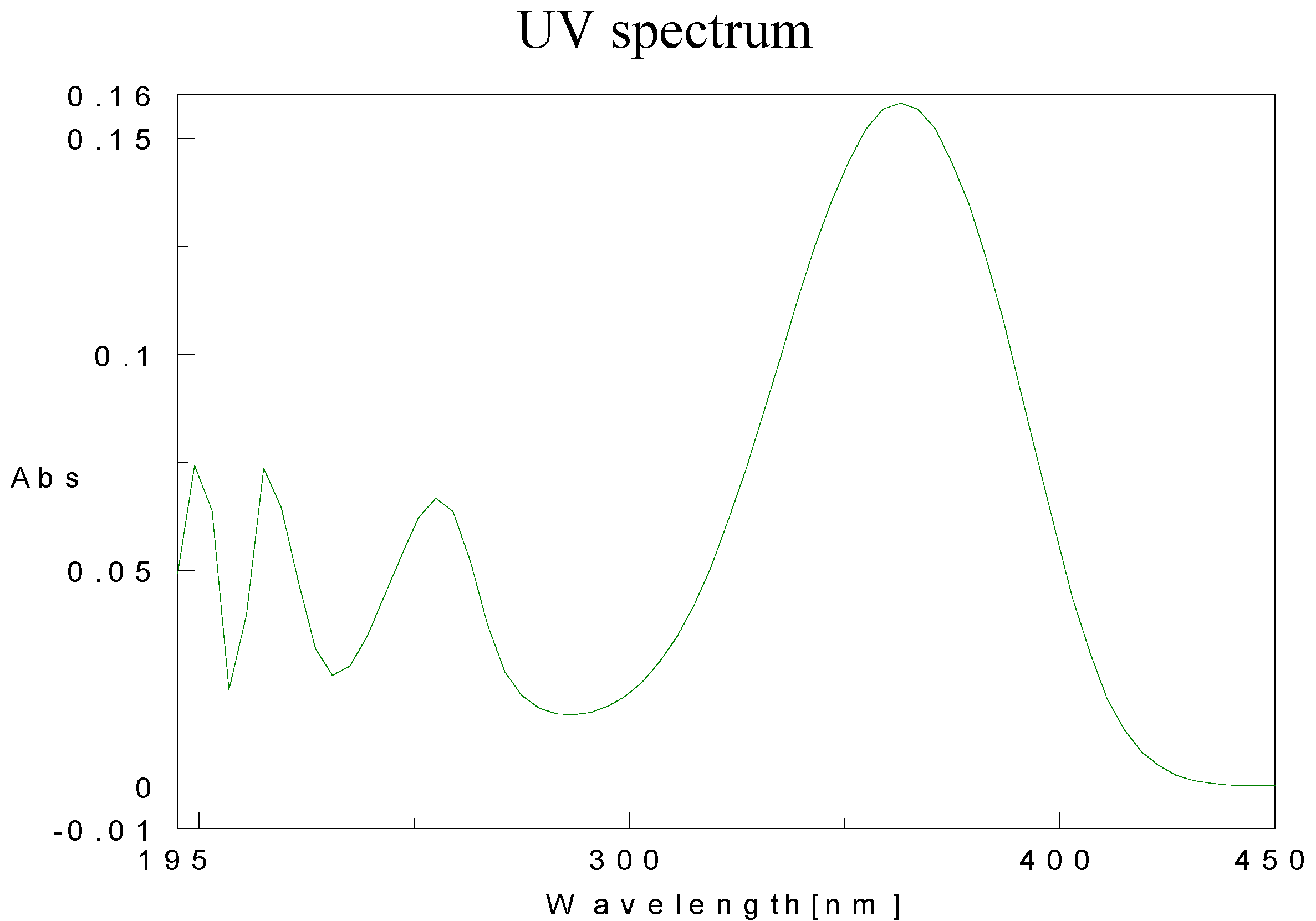
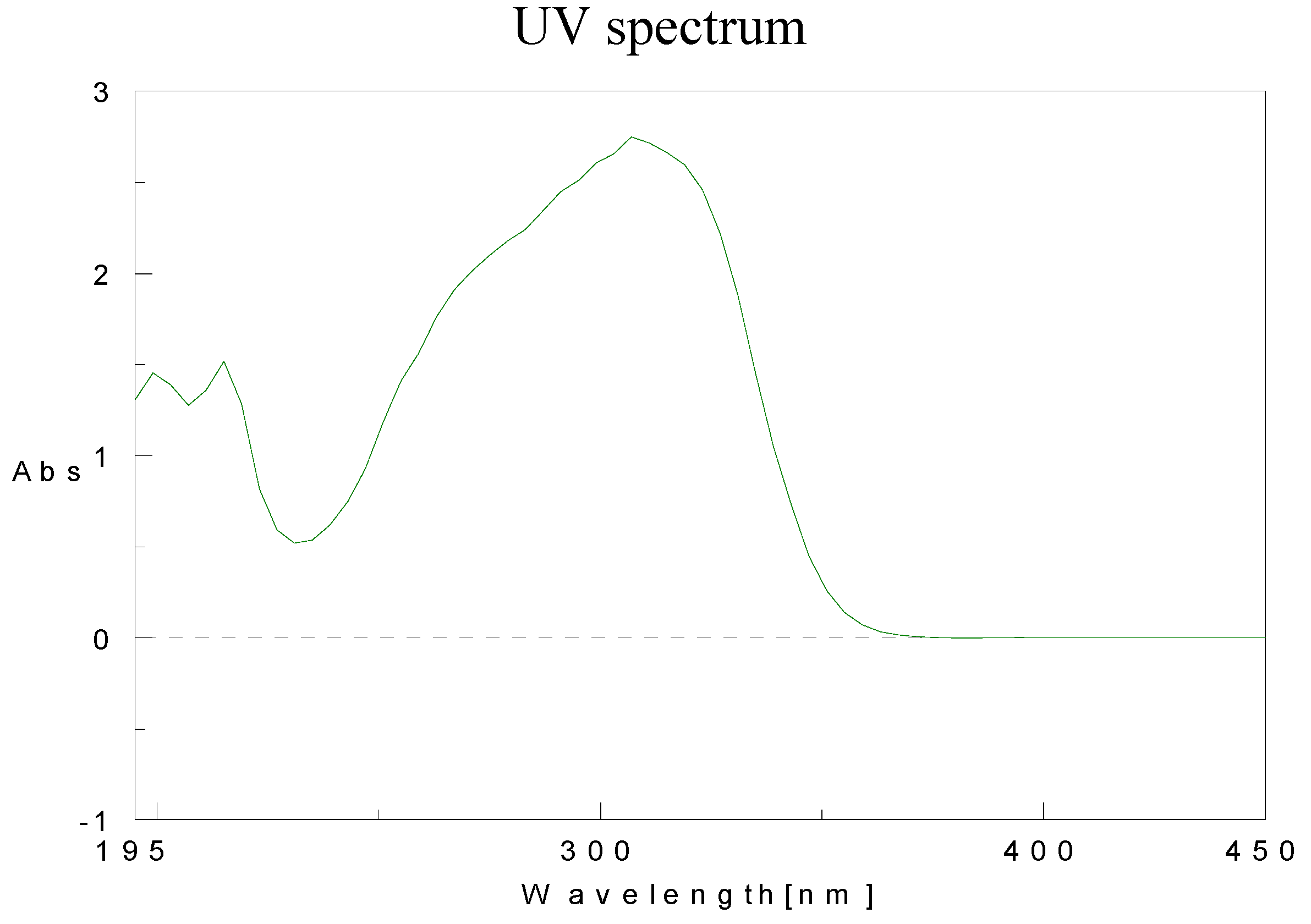
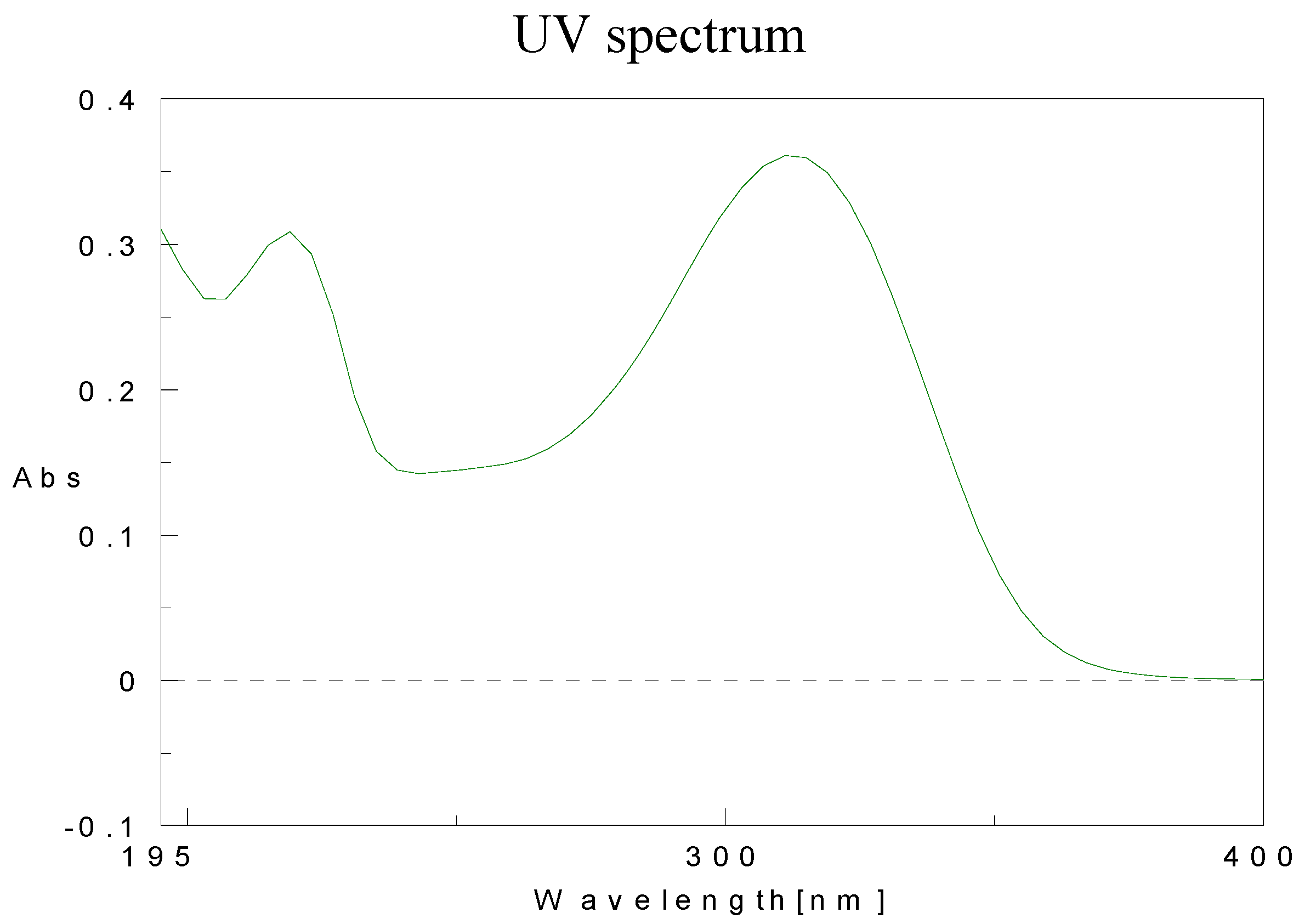
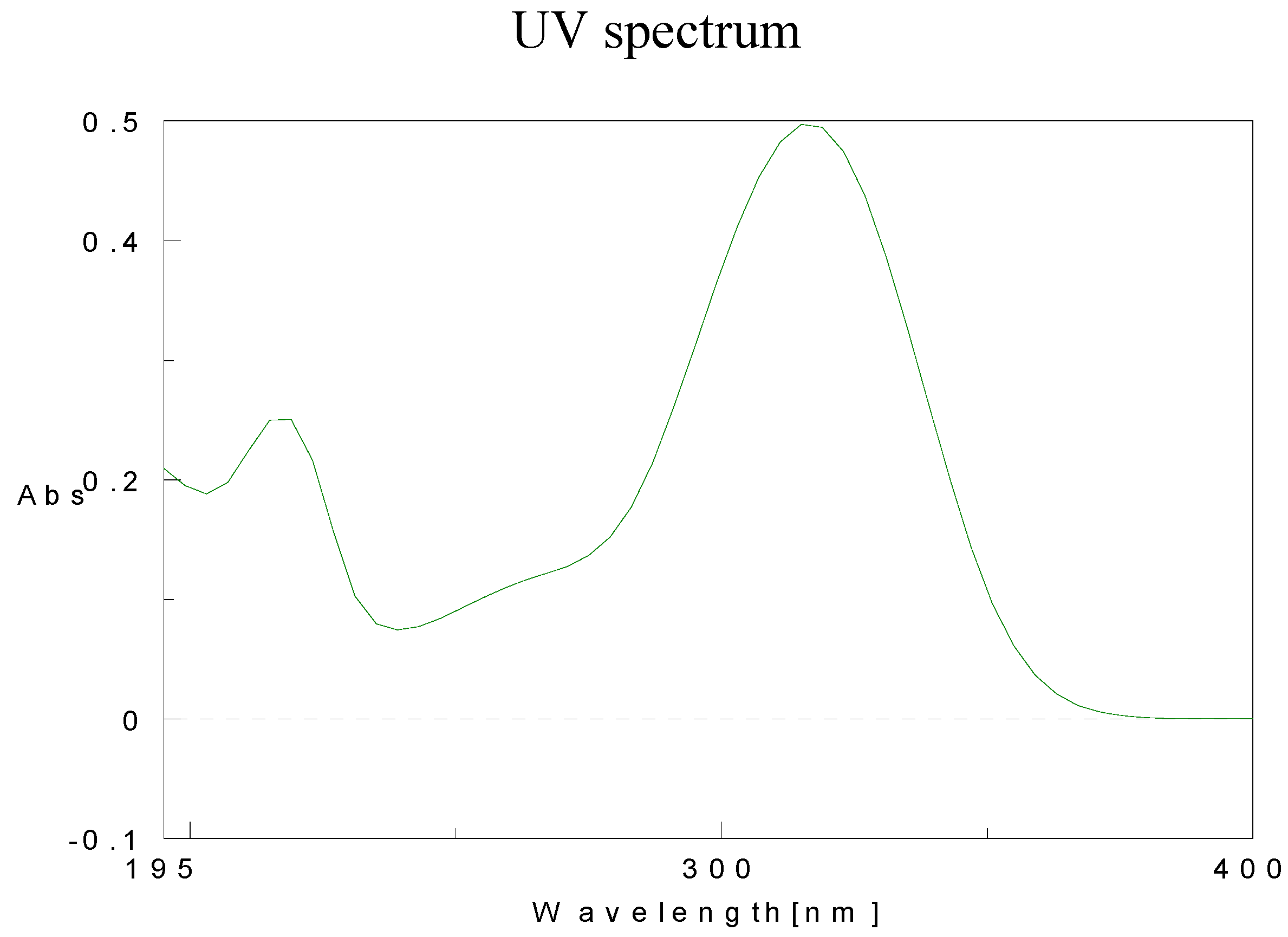
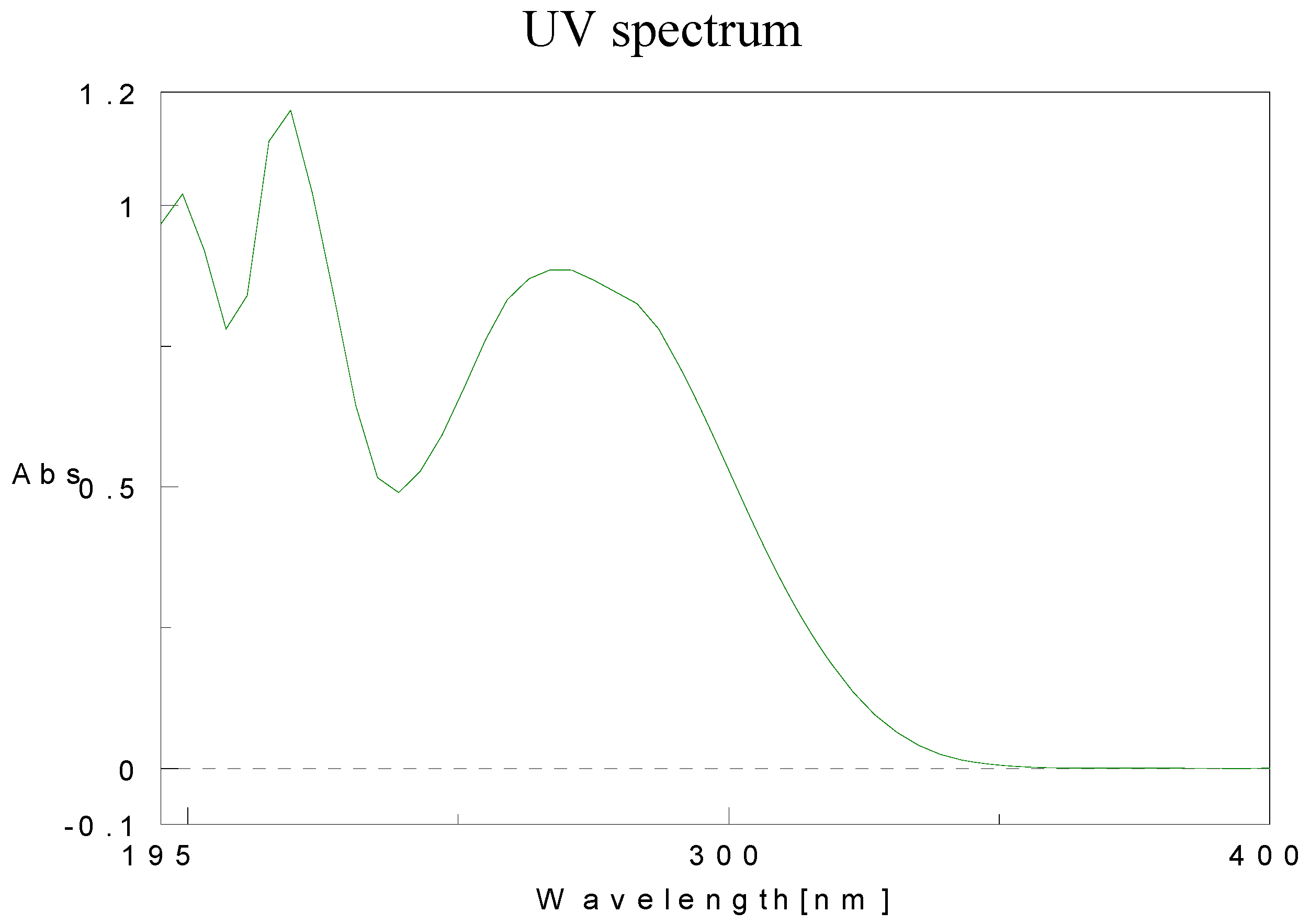
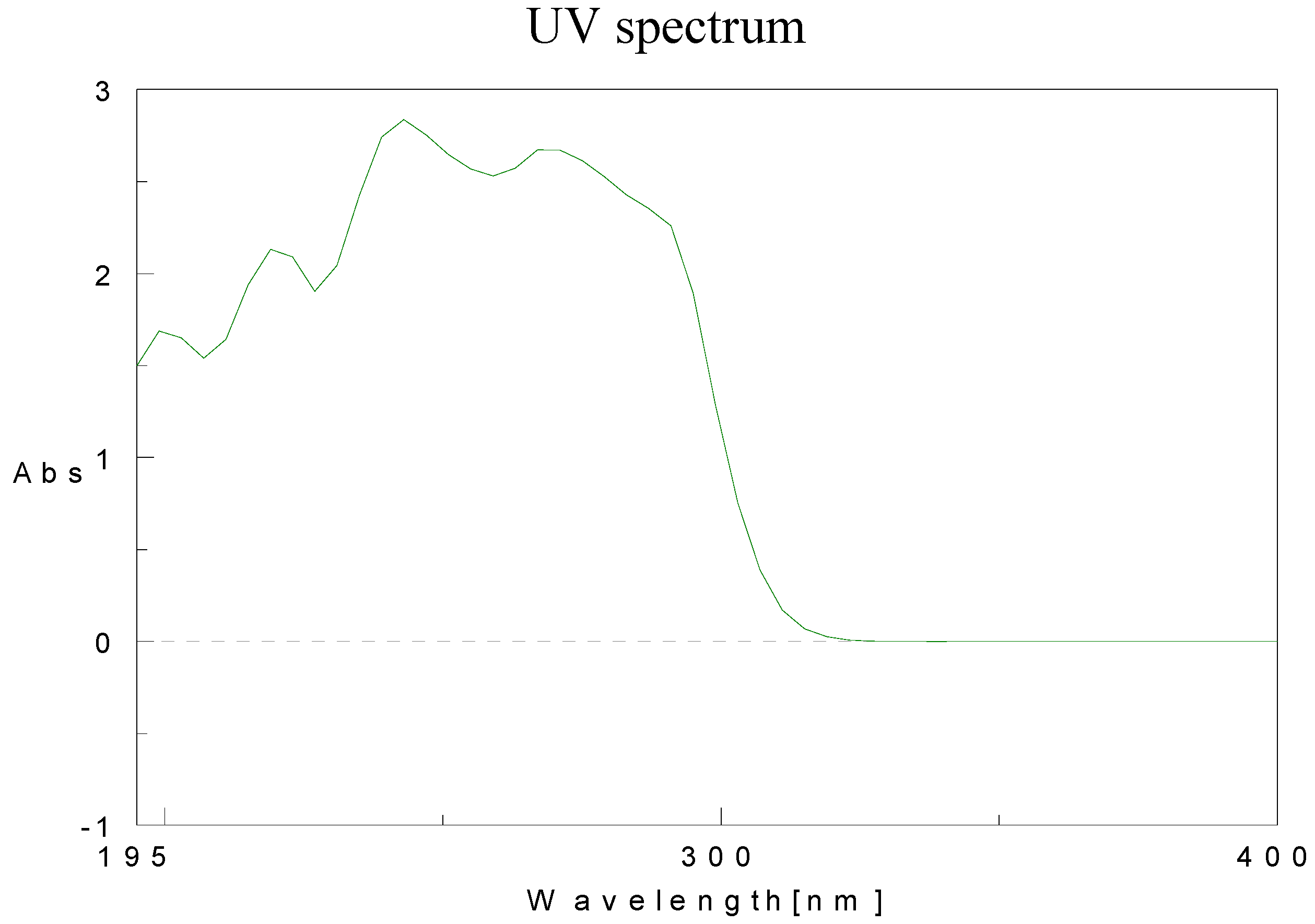
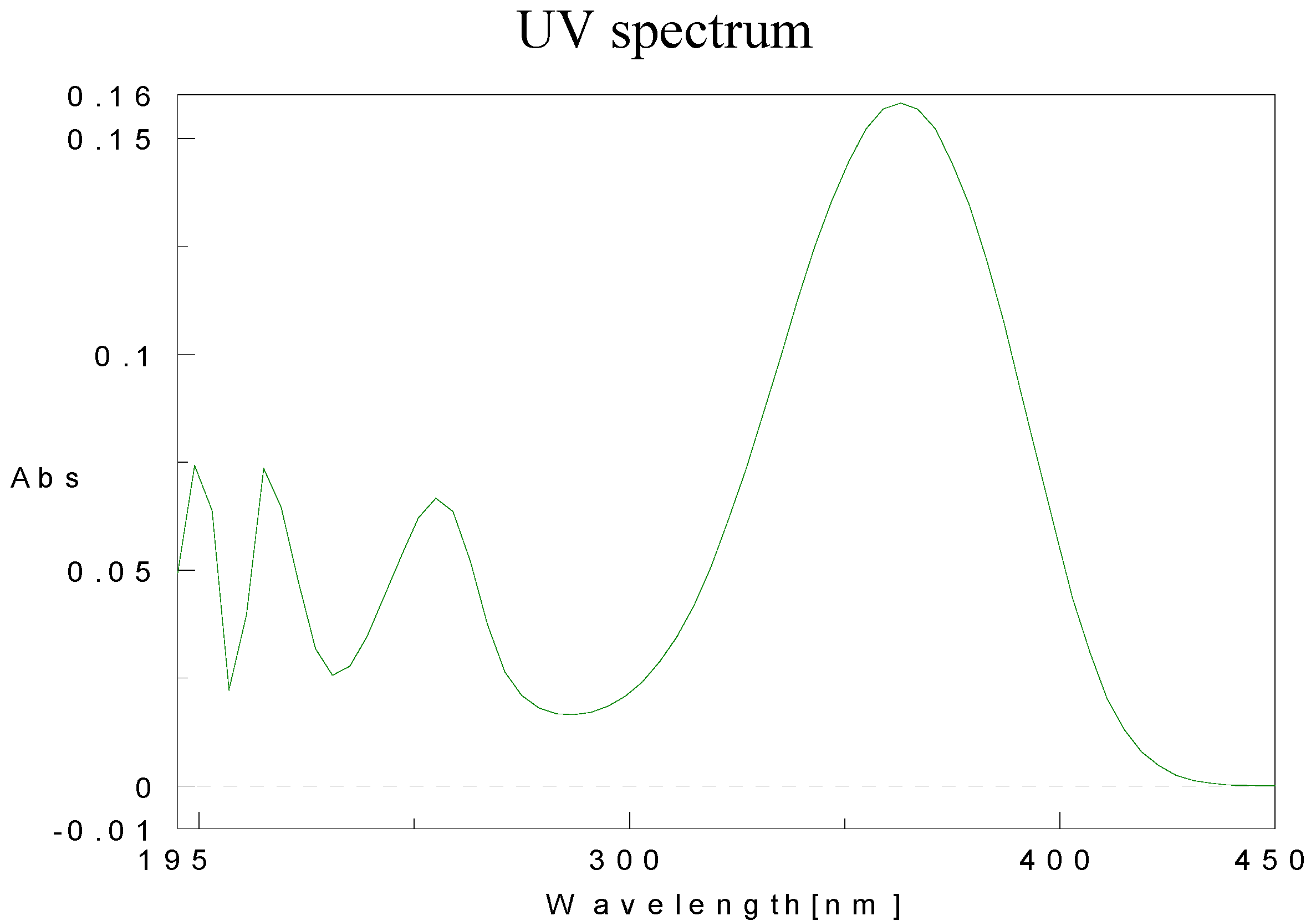
Physical data for compounds 1b-h











References
- Baraglia, C.A.; Rossi, D.; Amadio, M.; Pascale, A.; Laggner, C.; Yli-Kauhaluoma, J.; Tuominen, R.K.; Azzolina, O.; Collina, S. Novel PKCα ligands: Rational design, preparation and biological investigation. Drugs Fut. 2009, 34, 184. [Google Scholar]
- Paz, J.L.; Rodrigues, J.A.R. Preparation of aromatic geraniol analogues via a Cu(I)-mediated Grignard coupling. J. Braz. Chem. Soc. 2003, 14, 975–981. [Google Scholar] [CrossRef]
- Yu, J.S.; Wiemer, F. Temperature effects on stereocontrol in the Horner-Wadsworth-Emmonds condensation of α-phosphono lactones. J. Org. Chem. 2007, 72, 6263–6265. [Google Scholar] [CrossRef]
- Dakdouki, S.C.; Villemin, D.; Bar, N. Solid-phase reactive chromatography (SPRC): A new methodology for Wittig and Horner–Emmons reactions on a column under microwave irradiation. Eur. J. Org. Chem. 2010, 2, 333–337. [Google Scholar]
- Sano, S.; Takehisa, T.; Ogawa, S.; Yokoyama, K.; Nagao, Y. Stereoselective synthesis of tetrasubstituted (Z)-alkenes from aryl alkyl ketones utilizing the Horner-Wadsworth-Emmonds reaction. Chem. Pharm. Bull. 2002, 50, 1300–1302. [Google Scholar] [CrossRef]
- Wang, G.T.; Li, S.; Wideburg, N.; Krafft, G.A.; Kempf, D.J. Synthetic chemical diversity: Solid phase synthesis of libraries of C2 symmetric inhibitors of HIV protease containing diamino diol and diamino alcohol cores. J. Med. Chem. 1995, 38, 2995–3002. [Google Scholar] [CrossRef]
- Urbano, M.; Collina, S.; Rossi, D.; Carnevale Baraglia, A.; Alcaro, S.; Artese, A.; Azzolina, O. Design and synthesis of a (N-alkylaminoalkyl-substituted)arylalkenylamide drug discovery library. Lett. Drug Design Disc. 2007, 4, 605–610. [Google Scholar] [CrossRef]
- Kappe, C.O. High-speed combinatorial synthesis utilizing microwave irradiation. Curr. Opin. Chem. Biol. 2002, 6, 314–320. [Google Scholar] [CrossRef]
- Kappe, C.O. Microwave dielectric heating in synthetic organic chemistry. Chem. Soc. Rev. 2008, 37, 1127–1139. [Google Scholar] [CrossRef]
- Mazzucato, U.; Spalletti, A. Competition between photoisomerization and photocyclization of the cis isomers of n-styrylnaphthalenes and –phenantrenes. J. Phys. Chem. A 2009, 113, 14521–14529. [Google Scholar] [CrossRef]
- Stariat, J.; Kovarikova, P.; Klimes, J.; Kalinowski, D.S.; Richardson, D.R. Development of an LC-MS/MS method for analysis of interconvertible Z/E isomers of the novel anticancer agent, Bp4eT. Anal. Bioanal. Chem. 2010, 397, 161–171. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals; Pergamon: Oxford, UK, 1988. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rossi, D.; Baraglia, A.C.; Serra, M.; Azzolina, O.; Collina, S. An Efficient Procedure Based on a MW-Assisted Horner–Wadsworth-Emmons Reaction for the Synthesis of (Z)-3,3-Trisubstituted-a,b-unsaturated Esters. Molecules 2010, 15, 5928-5942. https://doi.org/10.3390/molecules15095928
Rossi D, Baraglia AC, Serra M, Azzolina O, Collina S. An Efficient Procedure Based on a MW-Assisted Horner–Wadsworth-Emmons Reaction for the Synthesis of (Z)-3,3-Trisubstituted-a,b-unsaturated Esters. Molecules. 2010; 15(9):5928-5942. https://doi.org/10.3390/molecules15095928
Chicago/Turabian StyleRossi, Daniela, Anna Carnevale Baraglia, Massimo Serra, Ornella Azzolina, and Simona Collina. 2010. "An Efficient Procedure Based on a MW-Assisted Horner–Wadsworth-Emmons Reaction for the Synthesis of (Z)-3,3-Trisubstituted-a,b-unsaturated Esters" Molecules 15, no. 9: 5928-5942. https://doi.org/10.3390/molecules15095928
APA StyleRossi, D., Baraglia, A. C., Serra, M., Azzolina, O., & Collina, S. (2010). An Efficient Procedure Based on a MW-Assisted Horner–Wadsworth-Emmons Reaction for the Synthesis of (Z)-3,3-Trisubstituted-a,b-unsaturated Esters. Molecules, 15(9), 5928-5942. https://doi.org/10.3390/molecules15095928