Synthesis and Antifungal Activity of Carabrone Derivatives

Abstract
:1. Introduction

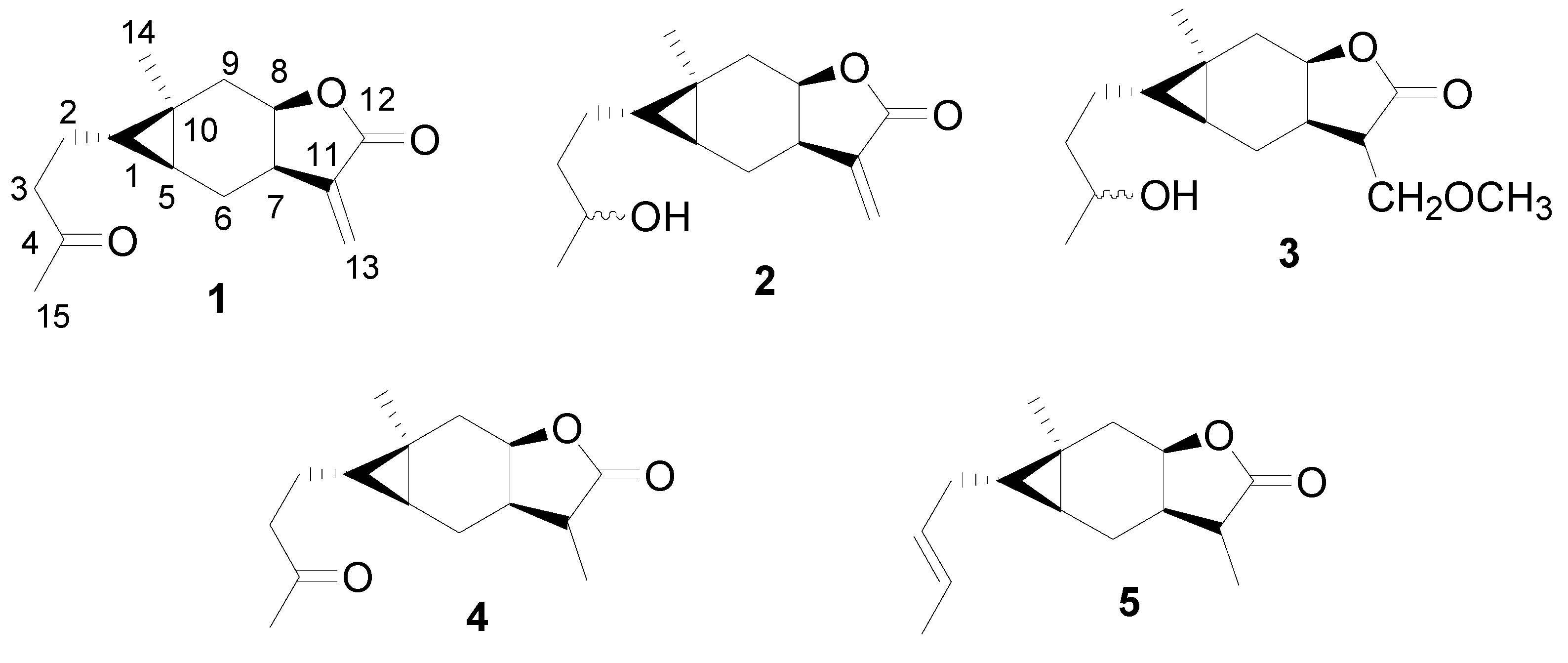
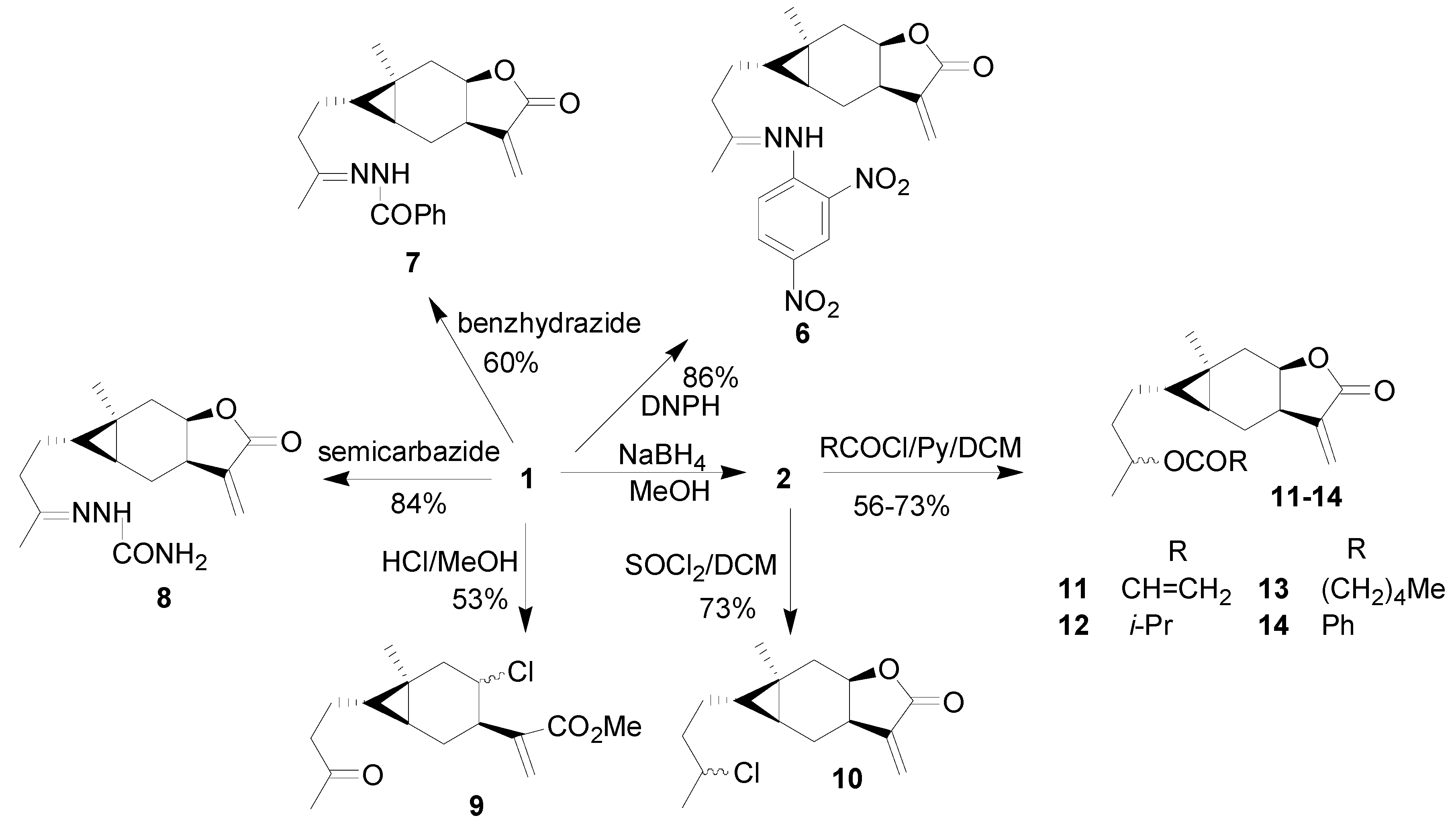
2. Results and Discussion


{kind=link}
{kind=link}
| Compd. | Regression equation (Y = a + bX) | r | EC50b (μg/mL) | EC50 95% CL/(μg/mL) |
|---|---|---|---|---|
| 1 | Y = 3.6090 + 1.6337X | 0.9974 | 7.10 | 6.19~8.02 |
| 6 | Y = 4.5130 + 1.3891X | 0.9923 | 2.24 | 1.97~2.55 |
| 7 | Y = 3.4038 + 2.5118X | 0.9817 | 4.32 | 3.81~4.85 |
| 8 | Y = 4.3577 + 1.3351X | 0.9979 | 3.03 | 2.58~3.55 |
| 9 | Y = 3.1867 + 1.0358X | 0.9942 | 56.30 | 42.95~73.80 |
| 10 | Y = 3.2442 + 1.1736X | 0.9969 | 31.34 | 26.08~37.66 |
| 11 | Y = 4.2780 + 0.6720X | 0.9882 | 10.78 | 9.16~12.68 |
| 12 | Y = 4.3568 + 0.7209X | 0.9920 | 6.39 | 5.33~7.65 |
| 13 | Y = 3.7775 + 0.9365X | 0.9970 | 20.20 | 16.85~24.22 |
| 14 | Y = 3.7676 + 1.0011X | 0.9962 | 17.02 | 14.41~20.11 |
| chlorothalonil c | Y = 5.1247 + 1.0081X | 0.9935 | 0.75 | 0.63~0.90 |
3. Experimental
3.1. General
3.2. Synthesis
General procedure for the synthesis of compounds 11-14 [20]
3.3. Spore Germination Assay
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References
- Minato, H.; Nosaka, S.; Horibe, I. Studies on sesquiterpenoids. Part VIII. The structure of carabrone, a new component of Carpesium abrotanoides Linn. J. Chem. Soc. 1964, 5503–5510. [Google Scholar]
- Holub, M.; Samek, A.; Toman, J. Carabrone from Arnzca foliosa. Phytochemistry 1972, 11, 2627–2628. [Google Scholar] [CrossRef]
- Bohlmann, F.; Mahanta, P.K.; Jakupovicm, J.; Rastogi, R.C.; Natu, A.A. New sesquiterpene lactones from Inula species. Phytochemistry 1978, 17, 1165–1172. [Google Scholar] [CrossRef]
- Bohlmann, F.; Zdero, C.; King, R.M.; Robinson, H. Sesquiterpene lactones from Syncretocarpus sericeus. Phytochemistry 1983, 22, 1288–1290. [Google Scholar] [CrossRef]
- Spring, O.; Vargas, D.; Fischer, N.H. Sesquiterpene lactones and benzofurans in glandular trichomes of three Pappobolus species. Phytochemistry 1991, 30, 1861–1867. [Google Scholar] [CrossRef]
- ksüz, S.; Topar, G. A eudesmanolide and other constituents fromInula graveolens. Phytochemistry 1992, 31, 195–197. [Google Scholar]
- Mossa, J.S.; El-Feraly, F.S.; Muhammad, I.; Zaw, K.; Mbwambo, Z.H.; Pezzuto, J.M.; Fong, H.H.S. Sesquiterpene lactones and tymol esters from Vicoa pentanema. J. Nat. Prod. 1997, 60, 550–555. [Google Scholar] [CrossRef]
- Kim, M.R.; Lee, S.K.; Kim, C.S.; Kim, K.S.; Moon, D.C. Phytochemical constituents of Carpesium macrocephalum FR. et SAV. Arch. Pharm. Res. 2004, 27, 1029–1033. [Google Scholar] [CrossRef]
- Wang, F.; Yang, K.; Ren, F.C.; Liu, J.K. Sesquiterpene lactones from Carpesium abrotanoides. Fitoterapia 2009, 80, 21–24. [Google Scholar] [CrossRef]
- Lee, J.S.; Min, B.S.; Lee, S.; Na, M.; Kwon, B.; Lee, C.; Kim, Y.; Bae, K. Cytotoxic sesquiterpene lactones from carpesium abrotanoides. Planta Med. 2002, 68, 745–747. [Google Scholar] [CrossRef]
- Maruyama, M.; Omura, S. Carpesiolin from Carpesium abrotanoides. Phytochemistry 1977, 16, 782–783. [Google Scholar] [CrossRef]
- Yang, C.; Shi, Y. P.; Jia, Z.J. Sesquiterpene lactone glycosides, eudesmanolides, and other constituents from Carpesium macrocephalu. Planta Med. 2002, 68, 626–630. [Google Scholar] [CrossRef]
- Jiang, J.W. Manuscript of Active Ingredients of Vegetable Drug; People's Health Press: Beijing, China, 1986; pp. 832–833. [Google Scholar]
- Feng, J.T.; Zhu, M.J.; Yu, P.R.; Li, Y.P.; Han, J.H.; Shao, H.J.; Ding, H.X.; Zhang, X. Screening on the resources of botanical fungicides in Northwest China. Acta Agric. Boreali-occidentalis Sinica 2002, 30, 129–133. [Google Scholar]
- Feng, J.T.; Zhang, Y.M.; Wang, J.R.; Zhang, X. Synthesis and antifugal activities of carabrone derivatives. Chin. J. Pestic. Sci. 2007, 9, 185–188. [Google Scholar]
- Feng, J.T.; Ma, Z.Q.; Wang, J.R.; Wang, Z.H.; Su, Z.S.; Li, G.Z.; Zhang, X. Preparation of calacane-type sesquiterpenoids separated from Carpesium macrocephalum and application as agricultural fungicide. China Patent ZL200610104867.7, 3 June 2009. [Google Scholar]
- Hitoshi, M.; Satoko, N.; Isao, H. Sesquiterpenoids. VIII. The structure of carabrone, a new component of carpesium abrotanoides. J. Chem. Soc., Suppl. 1964, 1, 5503–5510. [Google Scholar]
- Li, J.; Huang, W.L.; Zhang, H.B. Synthesis of amide derivatives of andrographolide and their inhibitory activities on COX-2 expression. J. China Pharm. Univ. 2007, 38, 299–304. [Google Scholar]
- Peng, H.; Gong, Y.F. Facile snthesis of ethyl 2-(4-hydroxyphenyl)-3,3,3-trifluoropropionate. Chin. J. Org. Chem. 2004, 24, 516–520. [Google Scholar]
- Pamela, J.R.; Joel, R.C. Insecticidal properties of monoterpenoid derivatives to the housefly and red flour beetle. Pestic. Sci. 1994, 41, 195–202. [Google Scholar] [CrossRef]
- Mu, L.Y.; Wu, W.J.; Wang, K.Y. Methods of plant protection with chemicals; China Agriculture Press: Beijing, China, 1994; pp. 71–78. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Feng, J.-T.; Ma, Z.-Q.; Li, J.-H.; He, J.; Xu, H.; Zhang, X. Synthesis and Antifungal Activity of Carabrone Derivatives. Molecules 2010, 15, 6485-6492. https://doi.org/10.3390/molecules15096485
Feng J-T, Ma Z-Q, Li J-H, He J, Xu H, Zhang X. Synthesis and Antifungal Activity of Carabrone Derivatives. Molecules. 2010; 15(9):6485-6492. https://doi.org/10.3390/molecules15096485
Chicago/Turabian StyleFeng, Jun-Tao, Zhi-Qing Ma, Jiang-Hua Li, Jun He, Hui Xu, and Xing Zhang. 2010. "Synthesis and Antifungal Activity of Carabrone Derivatives" Molecules 15, no. 9: 6485-6492. https://doi.org/10.3390/molecules15096485