Role of Kupffer Cells in Thioacetamide-Induced Cell Cycle Dysfunction

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
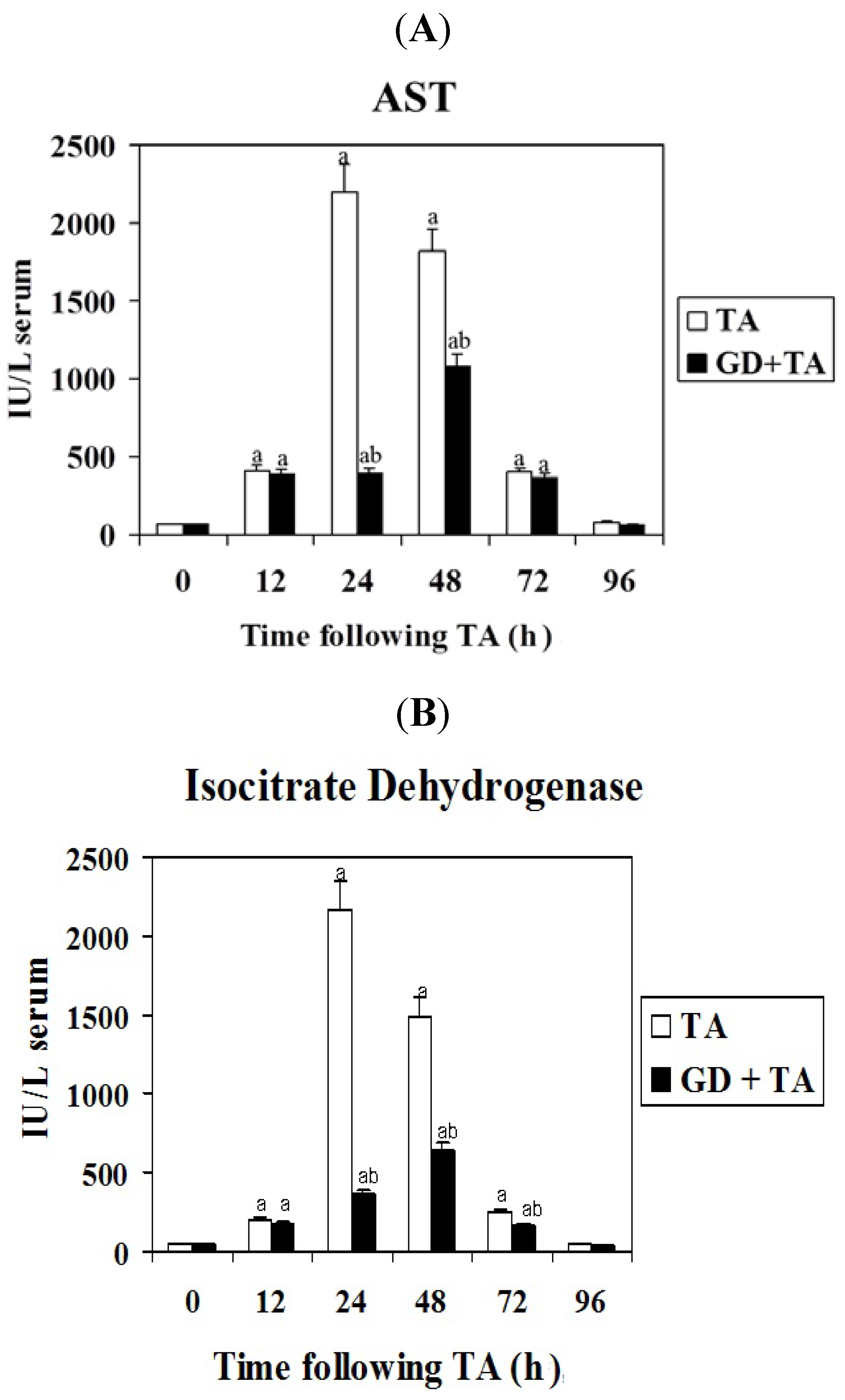
2.1. Effect of GD on Parameters of Liver Necrosis


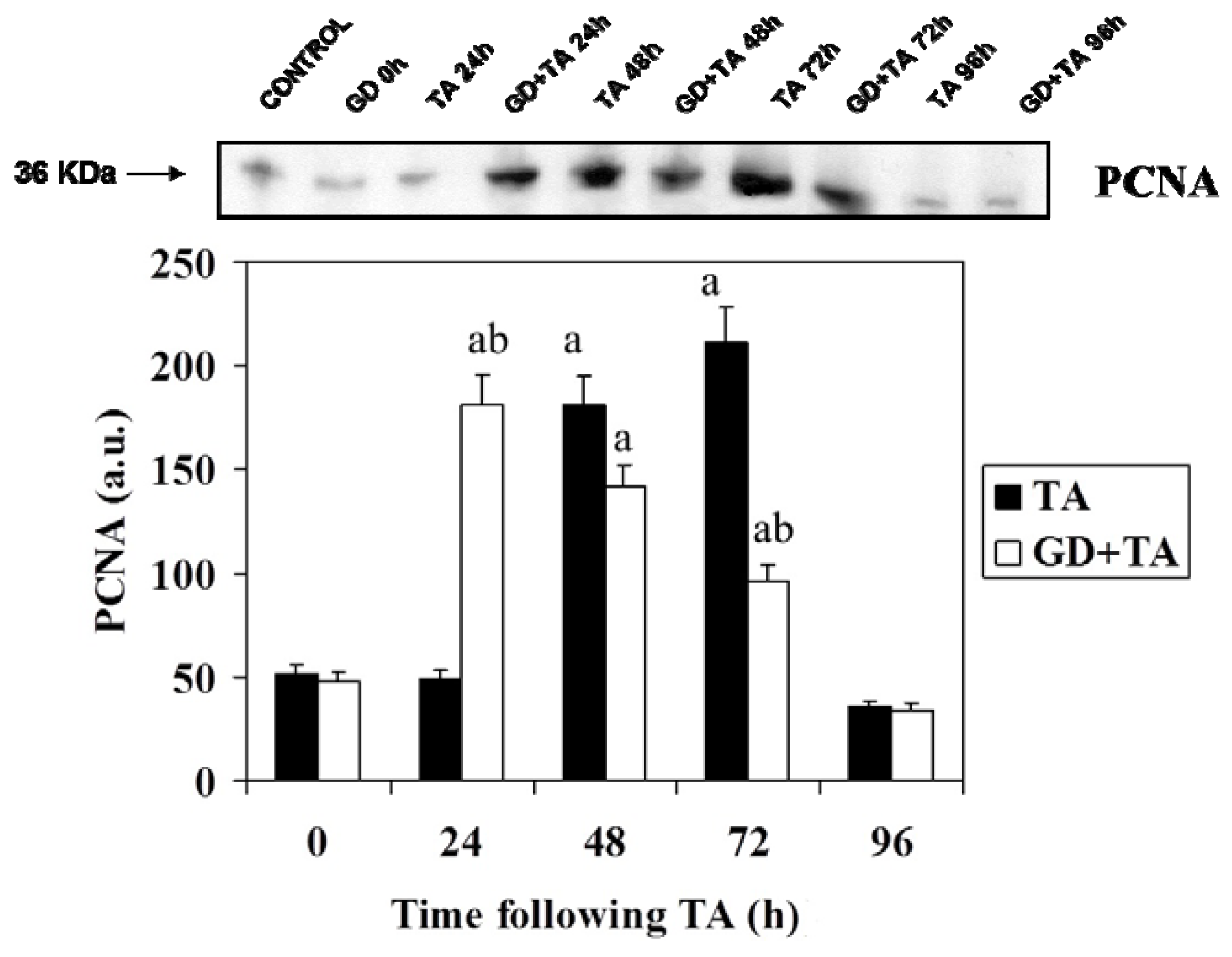
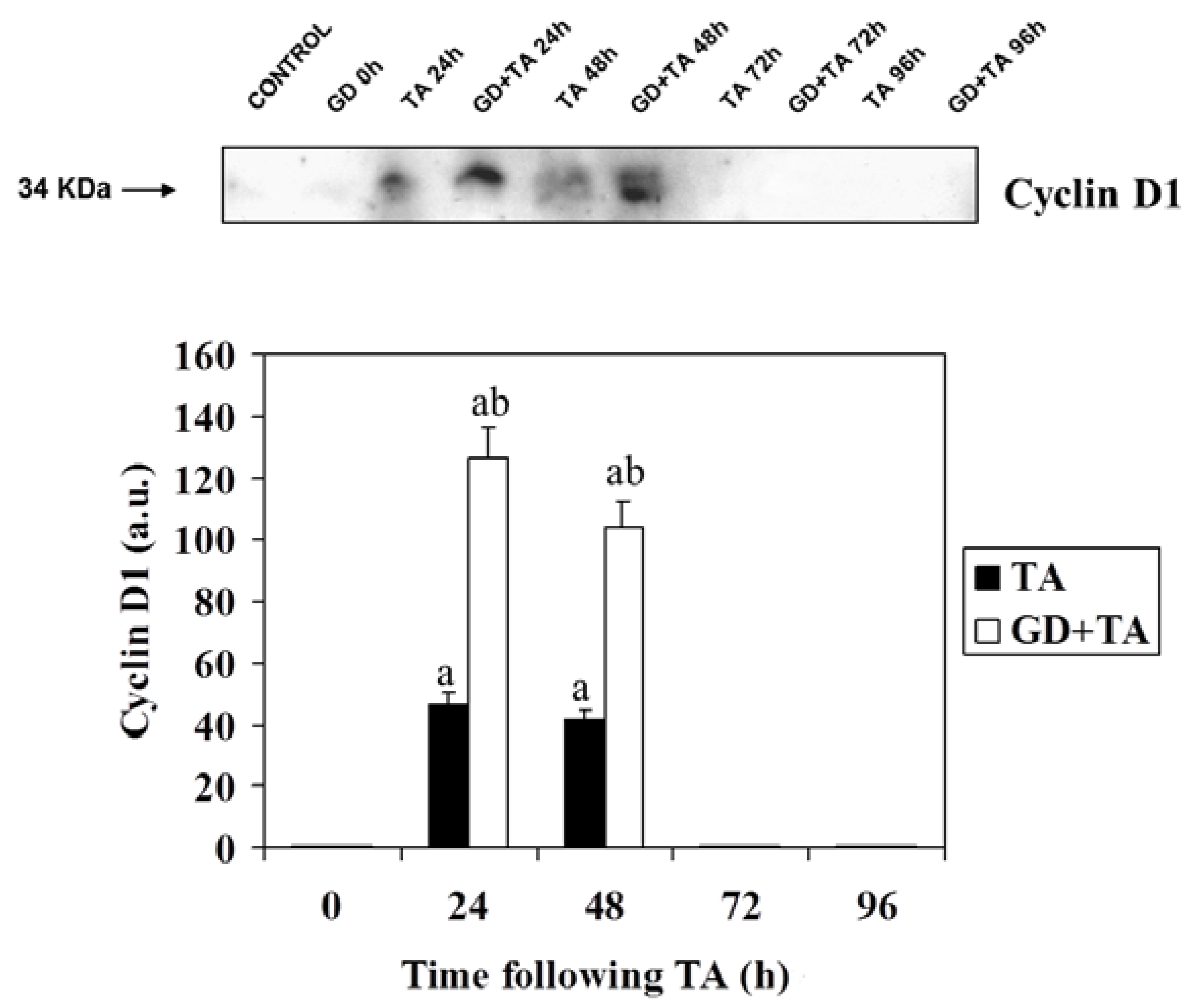
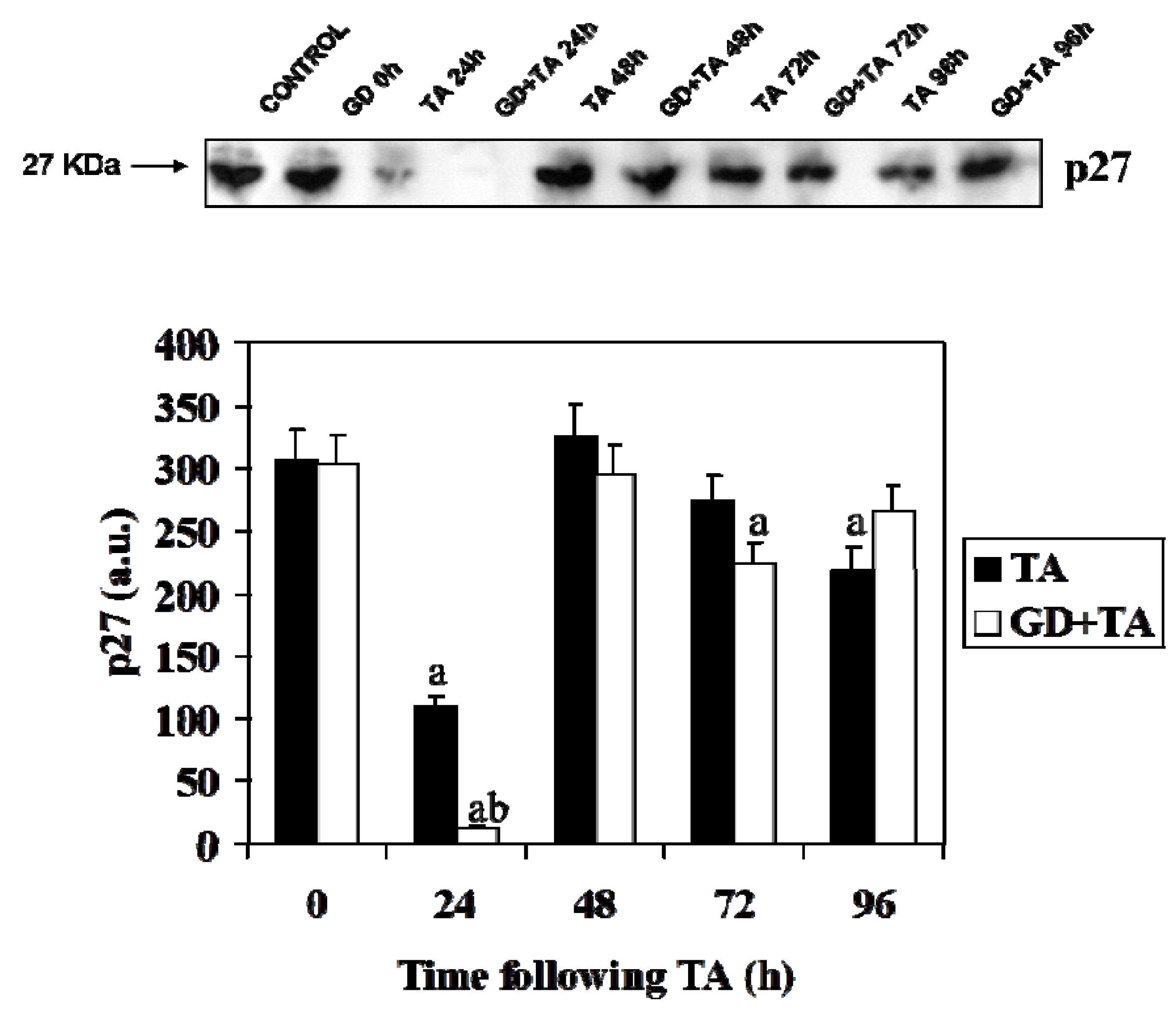
2.2. Effect of GD Pretreatment on Cyclin D1, Cyclin E, p27 and PCNA Level in Liver of Rats Following the Intoxication of TA



3. Discussion
4. Experimental
4.1. Reagents
4.2. Animals and Treatment
4.3. Processing of the Samples
4.4. Determination of Enzymes and Metabolites
4.5. Immunoblotting for Detection of Cyclin D1, Cyclin E, PCNA and p27
4.6. Statistical Analysis
5. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References
- Ishiyama, H.; Sato, M.; Matsumara, K.; Sento, M.; Ogino, K.; Hobara, T. Proliferation of hepatocytes and attenuation from carbon tetrachloride hepatotoxicity by gadolinium chloride. Pharmacol. Toxicol. 1995, 77, 293–298. [Google Scholar]
- Rose, M.L.; Bradford, B.U.; Germolec, D.R.; Lin, M.; Tsukamoto, H.; Thurman, R.G. Gadolinium chloride-induced hepatocytes proliferation is prevented by antibodies to Tumor Necrosis Factor α. Toxicol. Appl. Pharmacol. 2001, 170, 39–45. [Google Scholar]
- Bautista, A.P.; Skrepnic, N.; Niesman, M.R.; Bagby, G.J. Elimination of macrophages by liposome-encapsulated dichloromethylene diphosphonate suppresses the endotoxin-induced priming of Kupffer cells. J. Leuk. Biol. 1994, 55, 321–327. [Google Scholar]
- Iimuro, Y.; Yamamoto, M.; Kohno, H.; Itakura, J.; Fujii, H.; Matsumoto, Y. Blockade of liver macrophages by gadolinium chloride reduces lethality in endotoxemic rats. Analysis of mechanisms of lethality in endotoxemia. J. Leuk. Biol. 1994, 55, 723–728. [Google Scholar]
- Zhong, Z.; Connor, H.D.; Mason, R.P.; Qu, W.; Lemasters, J.J.; Thurman, R.G. Role of Kupffer cells in reperfusion injury in fat-loaded livers from ethanol-treated rats. J. Pharmacol. Exp. Ther. 1995, 27, 1512–1517. [Google Scholar]
- Andres, D.; Sanchez-Reus, I.; Bautista, M.; Cascales, M. Depletion of Kupffer cell function by gadolinium chloride attenuates thioacetamide-induced hepatotoxicity. Expression of metallothionein and HSP70. Biochem. Pharmacol. 2003, 66, 917–926. [Google Scholar] [CrossRef]
- Cascales, M.; Martín-Sanz, P.; Alvarez, A.; Sanchez-Pérez, M.; Díez-Fernández, C.; Boscá, L. Isoenzymes of carbohydrate metabolism in primary cultures of hepatocytes from thioacetamide-induced rat liver necrosis: responses to growth factors. Hepatology 1992, 16, 232–240. [Google Scholar] [CrossRef]
- Cascales, M.; Martin-Sanz, P.; Craciunescu, D.C.; Mayo, I.; Aguilar, A.; Robles-Chillida, E.M.; Cascales, C. Alterations in hepatic peroxidation mechanisms in thioacetamide-induced tumors in rats. Effect of a rhodium complex. Carcinogenesis 1991, 12, 233–240. [Google Scholar] [CrossRef]
- Sanz, N.; Diez-Fernández, C.; Andrés, D.; Cascales, M. Hepatotoxicity and aging: Endogenous antioxidant systems in hepatocytes from 2-, 6-, 12-, 18- and 30-month-old rats following a necrogenic dose of thioacetamide. Biochim. Biophys. Acta 2002, 1587, 12–20. [Google Scholar] [CrossRef]
- Sanz, N.; Díez-Fernández, C.; Alvarez, A.M.; Cascales, M. Age-dependent modifications in rat hepatocyte antioxidant defense systems. J. Hepatol. 1997, 27, 525–534. [Google Scholar]
- Sherr, C.J. Mammalian G1 cyclins. Cell 1993, 73, 1059–1065. [Google Scholar] [CrossRef]
- Sherr, C.J. G1 phase progression: Cyclin on cue. Cell 1994, 79, 551–555. [Google Scholar] [CrossRef]
- Cardoso, M.; Leonhardt, H.; Nadal-Ginard, B. Reversal of terminal differentiation and control of DNA replication. Cell 1993, 74, 979–992. [Google Scholar]
- Nurse, P. Universal control mechanism regulating onset of M-phase. Nature 1990, 344, 503–508. [Google Scholar] [CrossRef]
- Nigg, E.A. Cellular substrates of p34cdc2 and its companion cyclin-dependent kinases. Trends Cell. Biol. 1993, 3, 296–301. [Google Scholar] [CrossRef]
- Bautista, M.; Andres, D.; Cascales, M.; Morales-González, J.A.; Sánchez-Reus, M.I. Effect of gadolinium chloride on liver regeneration following thioacetamide-induced necrosis in rats. Int. J. Mol. Sci. 2010, 11, 4426–4440. [Google Scholar] [CrossRef]
- Chung, H.; Kim, J.A.; Song, B.C.; Song, I.H.; Koh, M.S.; Lee, H.C.; Yu, E.; Lee, Y.S.; Suh, D.J. Isocitrate dehydrogenase as a marker ofcentrilobular hepatic necrosis in the experimental model of rats. J. Gastroenterol. Hepatol. 2001, 16, 328–332. [Google Scholar] [CrossRef]
- Decker, K. Biologically active products of stimulated liver macrophages (Kupffer cells). Eur. J. Biochem. 1990, 192, 245–261. [Google Scholar]
- Jaeschke, H.; Smith, C.W. Mechanisms of neutrophil-induced parenchymal cell injury. J. Leuk. Biol. 1997, 61, 647–653. [Google Scholar]
- Essani, N.A.; Fisher, M.A.; Farhood, A.; Manning, A.M.; Smith, C.W.; Jaeschke, H. Cytokine-induced upregulation of hepatic intercellular adhesion molecule-1 messenger RNA expression and its role in the pathophysiology of murine endotoxin shock and acute liver failure. Hepatology 1995, 21, 1632–1639. [Google Scholar]
- Matsushime, H.; Roussel, M.F.; Ashmun, R.A.; Sherr, C.J. Colony-stimulating factor I regulates novel cyclins during the G1 phase of the cell cycle. Cell 1991, 65, 701–713. [Google Scholar] [CrossRef]
- Nevins, J.R. E2F: A link between the Rb tumor supresor protein and viral oncoproteins. Science 1992, 258, 424–429. [Google Scholar]
- Matsushime, H.; Ewen, M.E.; Strom, D.K.; Kato, J.; Hanks, S.K.; Roussel, M.F.; Sherr, C.J. Identification and properties of an atypical catalytic subunit (p34PSKJ3) for mammalian D-type G1 cyclin. Cell 1992, 71, 323–334. [Google Scholar] [CrossRef]
- Linden, M.D.; Torres, F.X.; Kubus, J.; Zarbo, R.J. Clinical application of morphologic and immunocytochemical assessments of cell proliferation. J. Clin. Pathol. 1992, 97, S4–S13. [Google Scholar]
- Pellicciari, C.; Mangiarotti, R.; Bottone, M.G.; Danova, M.; Wang, E. Identification of resting cells by dual-parameter flow cytometry of statin expression and DNA content. Cytometry 1995, 21, 329–327. [Google Scholar]
- Sawtell, R.M.; Rew, D.A.; Stradling, R.N.; Wilson, G.D. Pan cycle expression of proliferating cell nuclear antigen in human colorectal cancer and its proliferative correlations. Cytometry 1995, 22, 190–199. [Google Scholar] [CrossRef]
- Bravo, R.; McDonald-Bravo, H. Existence of two populations of cyclin-proliferating cell nuclear antigen during the cell cycle: Association with DNA replication sites. J. Cell Biol. 1987, 105, 1549–1554. [Google Scholar]
- Allegranza, A.; Girlando, S.; Arrigoni, G.L.; Veronese, S.; Mauri, F.A.; Gambacorta, M.; Pollo, B.; Dalla-Palma, P.; Barbareschi, M. Proliferating cell nuclear antigen expression in central nervous system neoplasms. Virchows Arch. 1991, 419, 417–423. [Google Scholar]
- Sanz, N.; Díez-Fernández, C.; Alvarez, A.M.; Cascales, M. Age-dependent modifications in rat hepatocyte antioxidant defense systems. J. Hepatol. 1997, 27, 525–534. [Google Scholar]
- Sanz, N.; Díez-Fernández, C.; Fernández-Simón, L.; Alvarez, A.; Cascales, M. Necrogenic and regenerative responses of liver of newly weaned rats. Biochem. Biophys. Acta 1998, 1384, 66–78. [Google Scholar]
- Rej, R.; Horder, M. Aspartate aminotransferase. L-aspartate: 2-Oxoglutarate aminotranferase, EC 2.6.2.1. Routine U.V. method. In Methods of Enzymatic Analysis, 3rd; Bergmeyer, H.U., Ed.; Verlag Chemie: Weinheim, Germany, 1984; Volume III, pp. 416–424. [Google Scholar]
- Goldberg, D.M.; Ellis, G. Isocitrate dehydrogenase. In Methods of Enzymatic Analysis, 3rd; Bergmeyer, H.U., Ed.; Verlag Chemie: Weinheim, Germany, 1986; Volume 3, pp. 183–189. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantification of micrograms quantities of protein utilizing the principle of protein-dye-binding. Anal. Biochem. 1975, 72, 248–254. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bautista, M.; Andres, D.; Cascales, M.; Morales-González, J.A.; Sánchez-Reus, M.I.; Madrigal-Santillán, E.; Valadez-Vega, C.; Fregoso-Aguilar, T.; Mendoza-Pérez, J.A.; Gutiérrez-Salinas, J.; et al. Role of Kupffer Cells in Thioacetamide-Induced Cell Cycle Dysfunction. Molecules 2011, 16, 8319-8331. https://doi.org/10.3390/molecules16108319
Bautista M, Andres D, Cascales M, Morales-González JA, Sánchez-Reus MI, Madrigal-Santillán E, Valadez-Vega C, Fregoso-Aguilar T, Mendoza-Pérez JA, Gutiérrez-Salinas J, et al. Role of Kupffer Cells in Thioacetamide-Induced Cell Cycle Dysfunction. Molecules. 2011; 16(10):8319-8331. https://doi.org/10.3390/molecules16108319
Chicago/Turabian StyleBautista, Mirandeli, David Andres, María Cascales, José A. Morales-González, María Isabel Sánchez-Reus, Eduardo Madrigal-Santillán, Carmen Valadez-Vega, Tomas Fregoso-Aguilar, Jorge Alberto Mendoza-Pérez, José Gutiérrez-Salinas, and et al. 2011. "Role of Kupffer Cells in Thioacetamide-Induced Cell Cycle Dysfunction" Molecules 16, no. 10: 8319-8331. https://doi.org/10.3390/molecules16108319
APA StyleBautista, M., Andres, D., Cascales, M., Morales-González, J. A., Sánchez-Reus, M. I., Madrigal-Santillán, E., Valadez-Vega, C., Fregoso-Aguilar, T., Mendoza-Pérez, J. A., Gutiérrez-Salinas, J., & Esquivel-Soto, J. (2011). Role of Kupffer Cells in Thioacetamide-Induced Cell Cycle Dysfunction. Molecules, 16(10), 8319-8331. https://doi.org/10.3390/molecules16108319