2. Results and Discussion
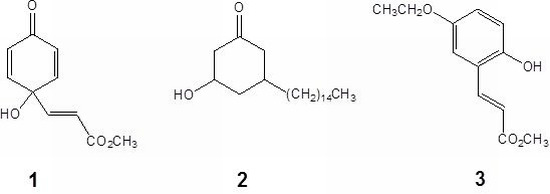
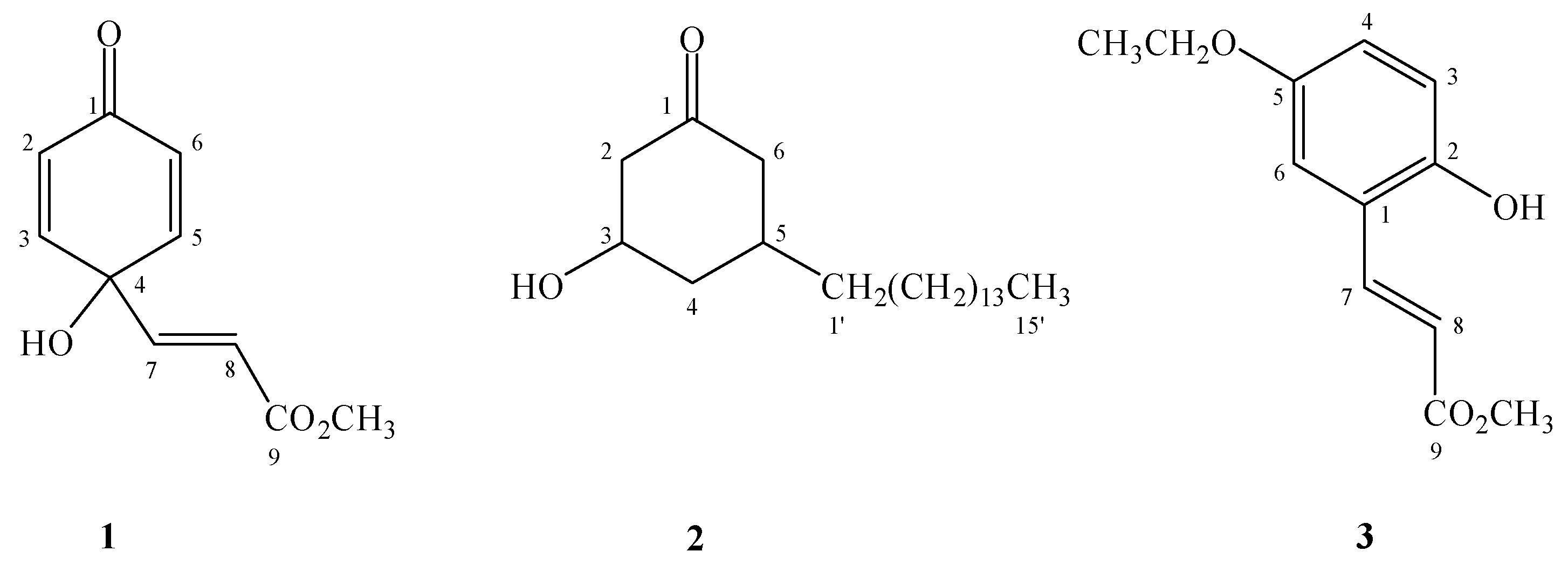
Graviquinone (1) was isolated as a yellowish solid. The molecular formula was determined to be C10H10O4 on the basis of the molecular ion at m/z 194.0577 in the HR-EIMS spectrum. The 1H-NMR and COSY spectra showed the presence of symmetric o-protons at δ 6.13 (2H, d, J = 9.9 Hz, H-2 and -6) and 6.82 (2H, d, J = 9.9 Hz, H-3 and -5) together with an α,β-unsaturated carbonyl unit at δ 6.19 (1H, d, J = 15.7 Hz, H-8) and 6.63 (1H, d, J = 15.7 Hz, H-7). The 13C-NMR, HMQC, and HMBC spectra indicated that both the proton signals at δ 6.13 and 6.82 exhibited 1H-13C long range correlations with a carboxyl carbon at δ 183.6 (C-1) and a quaternary carbon at δ 68.1 (C-4). Moreover, H-7 and H-8 showed HMBC correlations with the quaternary carbon and a carboxylic carbon at δ 164.8 (C-9). Furthermore, this downfield-shifted quaternary carbon correlated with a hydroxyl group (δ 5.49, br s) to which it was attached. A methyl group at δ 3.64 was assigned as forming an ester bond with the carboxylic acid because of the presence of the HMBC correlation between the methyl proton and the carboxylic carbon instead of the quaternary carbon. Consequently, the structure graviquinone (1) was assigned to be 4-hydroxy-4-(3-methoxycarbonyl)ethenylcyclohexadien-1-one.
Compound
2 was obtained as an optically active white oil. The HR-EIMS spectrum showed the molecular ion peak at
m/z 324.3206, consistent with the molecular formula C
21H
40O
2. The
1H- and COSY spectra demonstrate that a mutually coupled −CH
2–CH–CH
2–CH–CH
2− unit is present, with a 15-carbon long chain at
δ 0.88 (3H, t,
J = 6.3 Hz, H-15'), 1.25 (24H, m, H-3'−14'), 1.28 (2H, m, H-2'), and 1.31-1.45 (m, H-1'). Methylenes at
δ 2.33 (1H, t,
J = 12.9 Hz, H-2
ax) and 2.72 (1H, br d,
J = 12.9 Hz, H-2
eq) and then at 1.94 (1H, t,
J = 13.2 Hz, H-6
ax) and 2.37 (1H, br d,
J = 13.2 Hz, H-6
eq) exhibited HMBC correlations with a carbonyl carbon (
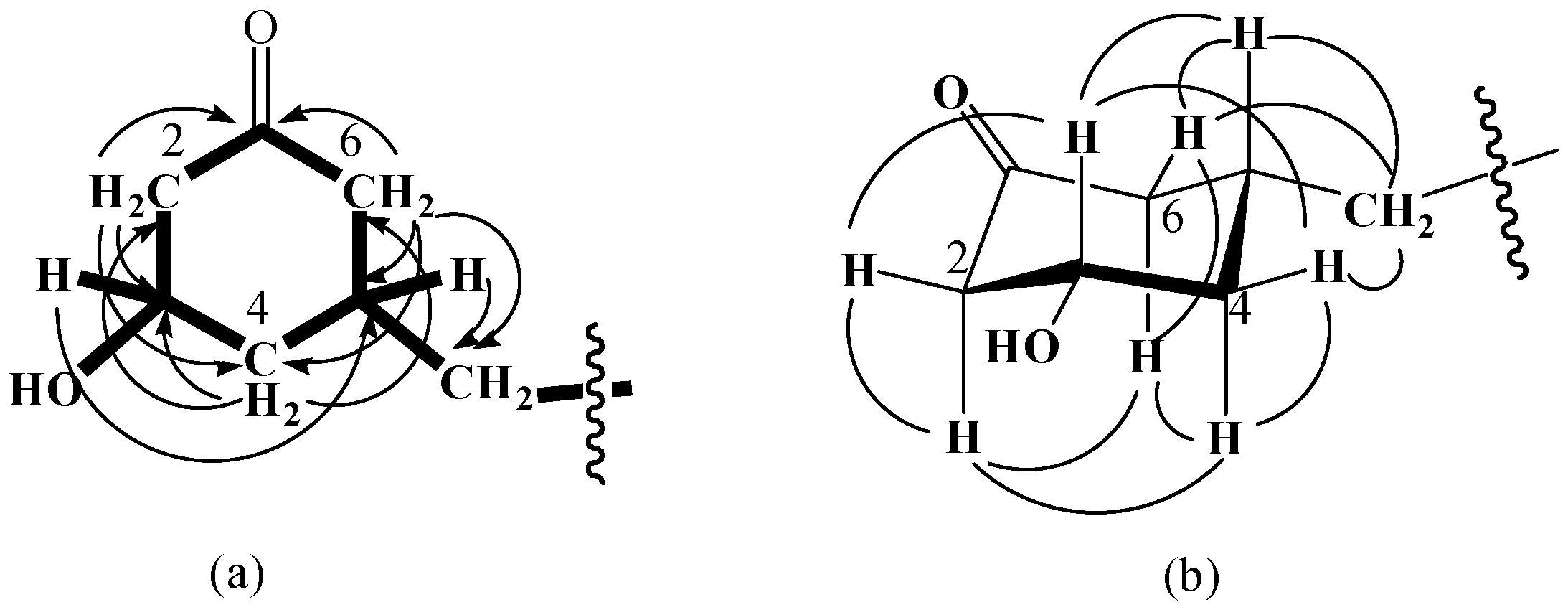
δ 208.8), indicating the presence of a cyclohexanone ring (
Figure 2aδ 3.91 (1H, m)] implied that a hydroxyl group was attached to C-3 (
δ 69.2). The long chain substituent that was apparently attached to C-5 (
δ 33.0) was assigned based on the HMBC correlations of H-5 [
δ 1.60 (1H, m)] with C-1' (
δ 36.5). The NOE correlations between H-3 and H-5 suggested the
cis relative configuration of the two substituents in the cyclohexanone (
Figure 2b). Therefore, compound
2 was assigned as
cis-3-hydroxy-5-pentadecylcyclohexanone.
Figure 2.
The key HMBC (a) and NOE (b) correlations of compound 2.
Figure 2.
The key HMBC (a) and NOE (b) correlations of compound 2.
Compound
3 was determined to have the molecular formula C
12H
14O
4 from the molecular ion peak at
m/z 222.0891 in the HR-EIMS spectrum. The
1H-NMR signals at
δ 3.81 (3H, s, 9-OCH
3), 6.53 (1H, d,
J = 16.0 Hz, H-8), 6.75 (1H, d,
J = 8.6 Hz, H-3), 6.82 (1H, dd,
J = 8.6, 2.5 Hz, H-4), 6.98 (1H, d,
J = 2.5 Hz, H-6), and 7.96 (1H, d,
J = 16.0 Hz, H-7) were very closely related to methyl 2,5-dihydroxycinnamate
4 [
3], which implied that compound
3 could likewise be a 2,5-disubstituted cinnamic acid methyl ester derivative. The HMBC correlations between -OCH
3, H-7, H-8 and C-9 (
δ 168.1); H-7 and C-1 (
δ 122.1), C-2 (
δ 149.2), C-6 (
δ 113.5), as well as the NOE correlation between H-6 and H-7, H-8, confirm this deduction. An ethoxy group at
δ 1.39 (3H, t,
J = 6.9 Hz) and 3.98 (2H, q,
J = 6.9 Hz) was assigned as a substituent of C-5 owing to the signal at
δ 3.98 (CH
2). Moreover, this group demonstrated a HMBC correlation with C-5 (
δ 152.9) and NOE correlations with H-4 and H-6. The hydroxyl group at
δ 5.68 (1H, br s, 2-OH) appeared to be attached to C-2. Hence, the structure of
3 was determined to be methyl 5-ethoxy-2-hydroxycinnamate.
In addition to these three new compounds, thirty-eight known compounds were isolated from the MeOH extract of the leaves from
G. robust. These known compounds could be divided into three types: benzenoid compounds, including methyl 2,5-dihydroxycinnamate (
4), cinnamic acid, methyl coumarate,
p-coumaric acid, pentacosyl dihydro-
p-coumarate, methyl 3,4-dihydroxybenzoate, 4-hydroxybenzaldehyde,
p-nitrophenol, methyl
p-hydroxybenzoate, 4-hydroxyacetophenone, hydro- quinone, and
p-hydroxybenzoic acid; alkylresorcinols, including gravicycle (
5) [
4], dehydrogravicycle (
6) [
4], robustol (
7) [
4], dehydrobustol-A (
8) [
4], dehydrobustol-B (
9) [
5], gravirobustol C (
10) [
5], methylgraviphane (
11) [
4], methyldehydrograviphane (
12) [
4], graviphane (
13) [
4], dehydrograviphane (
14) [
4], bisgravillol (
15) [
4], dehydrobisgravillol (
16) [
4], bis-norstriatol (
17) [
4], 5-[14'-(3'',5''-dihydroxyphenyl)-
cis-tetradec-6'-en-1'-yl]benzene-1,3-diol (
18) [
4], gravirobustol A (
19) [
5],
cis-5-
n-pentadecylresorcinol (
20) [
4], and
cis-5-
n-pentadec-8'-enylresorcinol (
21) [
4]; and flavonoids, including rhamnocitrin [
6], quercetin [
7], kaempferol [
8], rhamnetin [
9], 7-
O-methylrutin [
10], eriodictyol-7-methyl ether [
11], and sakranetin [
11]. Two other compounds, grasshopperketone [
12] and itaconic acid 4-methyl ester [
13], were also identified by the comparison of their spectral data with those reported in the literature.
Compounds
1−8,
11−18,
20, and
21 were subjected to cytotoxic evaluation. The clinically applied anticancer agent, actinomycin D, was used as a positive control for the cytotoxicity assays. Among these compounds, graviquinone (
1) showed the strongest cytotoxicity against MCF-7, NCI-H460, and SF-268 cell lines, with IC
50 values of 15.0, 10.8, and 5.9 μM, respectively (
Table 1).
cis-3-Hydroxy-5-pentadecyl-cyclohexanone (
2) also showed moderate activity, with IC
50 values of 26.7, 42.6, and 20.5 μM. The alkylresorcinols
5−8,
11−18,
20, and
21 showed marginal cytotoxicity, with IC
50 values of 39.8−22.8 μM [
4]. The similar IC
50 values of these compounds indicated that the alkyl chain structures, either cyclic or straight chain, had no impact on the anti-cancer activity of these alkylresorcinols.
Table 1.
Cytotoxicity of compounds 1–8, 11–18, 20, and 21 toward some cancer lines.
Table 1.
Cytotoxicity of compounds 1–8, 11–18, 20, and 21 toward some cancer lines.
| Compounds | IC50 (μM) a |
|---|
| MCF-7 | NCI-H460 | SF-268 |
|---|
| 1 | 15.0 ± 3.0 | 10.8 ± 2.3 | 5.9 ± 0.1 |
| 2 | 26.7 ± 1.9 | 42.6 ± 3.6 | 20.5 ± 0.6 |
| 3 | >50 | >50 | >50 |
| 4 | >50 | >50 | >50 |
| Actinomycin Db | 0.103 | 0.008 | 0.016 |
Subsequently, compounds
1,
4,
5,
7,
8,
11−18, and
20 (
Figure 3) were examined for their antioxidant properties based on the scavenging of the α,α-diphenyl-β-picrylhydrazyl free radical (DPPH) (
Table 2). Amoung these compounds, methyl 2,5-dihydroxycinnamate (
4), containing the
p-dihydroxy functionality, together with graviphane (
13) and dehydrograviphane (
14), containing tetrahydroxycyclophane moieties, showed very potent activity, with IC
50 values of 0.53, 3.96, and 2.05 μM, respectively. The results were compared with α-tocopherol, which is commonly used in the food industry as an antioxidant (IC50, 3.10 μM). The above compounds were also examined for their tyrosinase inhibition activities (
Table 2). Methyl 2,5-dihydroxycinnamate (
4) and bis-norstriatol (
17) strongly inhibited L-DOPA oxidation by mushroom tyrosinase with IC
50 values of 69.22 and 65.54 μM, respectively, and was compared with kojic acid (IC
50, 114.54 μM) as a reference.
Figure 3.
Structures of compounds 4, 5, 7, 8, 11–18, and 20.
Figure 3.
Structures of compounds 4, 5, 7, 8, 11–18, and 20.
Table 2.
DPPH inhibition activity and tyrosinase inhibition activity of compounds 1, 4, 5, 7, 8, 11–18, and 20.
Table 2.
DPPH inhibition activity and tyrosinase inhibition activity of compounds 1, 4, 5, 7, 8, 11–18, and 20.
| Compounds | IC50 (μM) or (Inhibition %) a |
|---|
| DPPH inhibition activity | Tyrosinase inhibition activity |
|---|
| 1 | 23.01 ± 0.53 | (32.78 ± 0.54) |
| 4 | 0.53 ± 0.09 | 69.22 ± 1.27 |
| 5 | 25.68 ± 0.32 | 210.35 ± 2.13 |
| 7 | 28.78 ± 0.42 | (30.87 ± 0.98) |
| 8 | 11.78 ± 0.88 | (37.92 ± 0.67) |
| 11 | (28.21 ± 0.23) | 178.41 ± 2.23 |
| 12 | 15.67 ± 0.31 | 250.53 ± 3.26 |
| 13 | 3.96 ± 0.55 | (23.66 ± 0.76) |
| 14 | 2.05 ± 0.22 | (42.76 ± 0.53) |
| 15 | 36.73 ± 0.43 | 296.65 ± 3.32 |
| 16 | 28.93 ± 0.67 | (48.22 ± 0.43) |
| 17 | 31.31 ± 0.34 | 65.54 ± 0.87 |
| 18 | 35.22 ± 0.48 | 245.14 ± 2.21 |
| 20 | (21.32 ± 0.21) | 233.23 ± 1.02 |
| α-tocopherol | 3.10 ± 0.05 | - |
| Kojic acid | - | 114.54 ± 1.21 |
3. Experimental
3.1. General
Melting points were recorded on a Yanaco MP-3 melting point apparatus and were not corrected. Optical rotations were measured on a Jasco DIP-370 digital polarimeter. UV spectra were recorded on an Agilent 8453 spectrophotometer. IR spectra were recorded on a Nicolet Magna FT-IR spectrophotometer. NMR spectra were recorded on Bruker Avance 300 and AMX 400 FT-NMR spectrometers; all chemical shifts were given in ppm from tetramethylsilane as an internal standard. Mass spectra were obtained on a VG 70-250S spectrometer by a direct inlet system.
3.2. Plant Material
The leaves of G. robusta were collected on the campus of National Cheng Kung University, Tainan, Taiwan, in September of 2003. The samples were authenticated by Professor C.S. Kuoh of the Department of Life Sciences, National Cheng Kung University. A voucher specimen (No: PLW-0303) was deposited in the Herbarium of the same school.
3.3. Extraction and Isolation
The dry leaves of G. robusta (7.7 kg) were extracted with MeOH (6 × 10 L) for 6 h under reflux conditions. The filtrate was concentrated under reduced pressure to obtain a dark green syrup. This syrup was re-suspended in H2O (1.5 L) and then partitioned with n-hexane (6 × 1 L), CHCl3 (6 × 1 L) and EtOAc (6 × 1 L) to give n-hexane- (150 g), CHCl3- (15 g), EtOAc- (125 g) and H2O-soluble portions, respectively.
The n-hexane extract was subjected to silica gel column chromatography (CC), eluting with n-hexane−Me2CO (4:1) in a step gradient that gradually increased the polarity of the solution with pure Me2CO to afford eight fractions. Fractions 4–7 contained a large amount of resorcinols, which were difficult to separate. Purification of fraction 4 was accomplished using silica gel CC and eluting with a gradient of CHCl3-MeOH (50:1 to pure MeOH) to gave a mixture of methylgraviphane (11) and methyldehydrograviphane (12) (105 mg), a mixture of cis-5-n-pentadecylresorcinol (20), cis-5-n-pentadec-8'-enylresorcinol (21) (2.15 g), cis-3-hydroxy-5-pentadecylcyclohexanone (2) (3 mg), and pentacosyl dihydro-p-coumarate (12 mg), and a mixture of robustol (7), dehydrobustol-A (8), dehydrobustol-B (9), and gravirobustol C (10) (825 mg). Unfortunately, compounds 9, 10 and 21 could not be obtained in pure samples. Fraction 5 was separated by using silica gel CC and by eluting with a gradient of CHCl3-MeOH (30:1 to pure MeOH) to give a mixture of bisgravillol (15) and dehydrobisgravillol (16) (35 mg) and a mixture of graviphane (13) and dehydrograviphane (14) (180 mg). Fraction 6 was separated by using silica gel CC and by eluting with a gradient of CHCl3-MeOH (20:1 to pure MeOH) to gave a mixture of bis-norstriatol (17), 5-[14'-(3'',5''-dihydroxyphenyl)-cis- tetradec-6'-en-1'-yl]benzene-1,3-diol (18), and gravirobustol A (19) (545 mg), which unfortunately could not be obtained as a pure compound. Using the same elution conditions as for fraction 6, fraction 7 produced a mixture of gravicycle (5) and dehydrogravicycle (6) (52 mg). However, extensive efforts were made through repeated CC to purify these components for identification.
The CHCl3 extract was subjected to silica gel CC, eluting with CHCl3-MeOH (20:1) in a step gradient that gradually increased the polarity with pure MeOH to afford five fractions. Fraction 2 was separated using silica gel CC, eluting with a gradient of (i-Pr)2O-MeOH (50:1 to pure MeOH), to give p-nitrophenol (6 mg), methyl p-hydroxybenzoate (4 mg), 4-hydroxybenzaldehyde (6 mg), cinnamic acid (1 mg), and graviquinone (1) (1.8 g), successively. Fraction 3 was purified by silica gel CC, eluting with (i-Pr)2O-MeOH (50:1 to pure MeOH), to obtain itaconic acid 4-methyl ester (2 mg), 4-hydroxyacetophenone (2 mg), methyl 3,4-dihydroxybenzoate (2 mg), and rhamnocitrin (4 mg). Fraction 4 was separated using silica gel CC, eluting with (i-Pr)2O-Me2CO (5:1 to pure Me2CO), to afford methyl coumarate (4 mg) and 5-ethoxy-2-hydroxycinnamate (3) (5 mg). Using the same elution conditions as for fraction 4, fraction 5 produced hydroquinone (2 mg), p-hydroxybenzoic acid (3 mg), and grasshopperketone (5 mg).
The EtOAc extract was subjected to silica gel CC, eluting with a gradient of CHCl3-MeOH (20:1 to pure MeOH) to yield twelve fractions. Fractions 1 and 2 were further purified by silica gel CC, eluting with a gradient of CHCl3-MeOH (6:1 to pure MeOH), to give quercetin (30 mg) and kaempferol (4 mg), respectively. Fraction 3 was separated using silica gel CC, eluting with CHCl3-Me2CO (10:1 to pure Me2CO), to afford methyl 2,5-dihydroxycinnamate (4) (1.1 g), eriodictyol-7-methyl ether (24 mg) and p-coumaric acid (18 mg). Fraction 5 was chromatographed using the same elution conditions as for fraction 3 to give sakranetin (10 mg), rhamnetin (18 mg), and 7-O-methylrutin (5 mg).
3.4. Cytotoxicity Assay
The cytotoxicity assay was carried out according to procedures that have been previously described in the literature [
14].
3.5. Free Radical Scavenging Activity Assay
The free radical scavenging activity of compounds
1,
4,
5,
7,
8,
11−18, and
20 was measured with DPPH
• using the method of Chiu
et al. [
15]. Briefly, 0.1 mM solution of DPPH
• in ethanol was prepared, and sample (20 μL) was added. The sample was thoroughly mixed and kept in the dark for 30 min. The absorbance was measured at 517 nm on a Quant universal microplate spectrophotometer. α-Tocopherol (Sigma Chemical Co.) was used as a standard agent. A lower absorbance of the reaction mixture indicated a higher free radical scavenging activity. The sample concentration that was used to provide the IC
50 data was calculated from a graph plotting inhibition percentage against sample concentration. Tests were performed in triplicate.
3.6. Tyrosinase Inhibitory Activity
The tyrosinase assay was carried out according to a procedure previously described in the literature [
16]. The test substance was dissolved in 10% DMSO-H
2O solution (0.1 mL) and then incubated with mushroom tyrosinase (0.1 mL, 135 U/mL, PBS pH 6.8) at 25 °C for 10 min. Next, L-dopa phosphate buffer solution (0.1 mL, 0.5 mM, pH 6.8) was added. The reaction mixture was incubated for 5 min. The amount of dopachrome in the mixture was determined by measuring the optical density (OD) at 475 nm using a Quant universal microplate spectrophotometer. Kojic acid (Sigma Chemical Co.) was used as a standard agent. The inhibitory percentage of tyrosinase was calculated as follows:
Where:
A: OD at 475 nm without test substance;
B: OD at 475 nm without test substance and tyrosinase;
C: OD at 475 nm with test substance.
Graviquinone (1). Yellow amorphous powder. mp 79−80 °C. UV (MeOH) λmax (log ε) 213 (4.0), 372 (1.6) nm. IR (KBr) νmax 3423, 1715, 1664 cm−1. EIMS m/z (rel. int.) 194 (43, M+), 162 (100), 134 (83), 107 (36), 77 (33), 55 (49); HR-EIMS m/z 194.0577 [M]+ (calcd for C10H10O4 194.0579). 1H-NMR (acetone-d6) δ 3.64 (3H, s, 9-OCH3), 5.49 (1H, br s, 4-OH), 6.13 (2H, d, J = 9.9 Hz, H-2 and -6), 6.19 (1H, d, J = 15.7 Hz, H-8), 6.63 (1H, d, J = 15.7 Hz, H-7), 6.82 (2H, d, J = 9.9 Hz, H-3 and -5); 13C NMR (acetone-d6) δ 50.2 (9-OCH3), 68.1 (C-4), 120.3 (C-8), 126.5 (C-2 and -6), 145.5 (C-7), 147.9 (C-3 and -5), 164.8 (C-9), 183.6 (C-1).
cis-3-Hydroxy-5-pentadecylcyclohexanone (2). White amorphous powder mp 62−64 °C [α]D −101° (c 0.10, CHCl3) UV (CHCl3) λmax (log ε) 241 (3.1), 280 (2.9) nm IR (KBr) νmax 3424, 1709, 1595, 1469 cm−1 EIMS m/z (rel. int.) 324 (3, M+), 306 (6), 113 (100), 95 (19); HR-EIMS m/z 324.3206 [M]+ (calcd for C21H40O2 324.3208). 1H NMR (CDCl3) δ 0.88 (3H, t, J = 6.3 Hz, H-15'), 1.25 (24H, m, H-3'−14'), 1.28 (2H, m, H-2'), 1.31–1.45 (3H, m, H-4ax and H-1'), 1.60 (1H, m, H-5ax), 1.94 (1H, t, J = 13.2 Hz, H-6ax), 2.21 (1H, br d, J = 12.3 Hz, H-4eq), 2.33 (1H, t, J = 12.9 Hz, H-2ax), 2.37 (1H, br d, J = 13.2 Hz, H-6eq), 2.72 (1H, br d, J = 12.9 Hz, H-2eq), 3.91 (1H, m, H-3ax)H, H; 13C-NMR (CDCl3) δ 14.1 (C-15'), 22.7 (C-14'), 26.6 (C-2'), 29.3–29.7 (C-3'−12'), 31.9 (C-13'), 33.0 (C-5), 36.5 (C-1'), 41.0 (C-4), 47.0 (C-6), 50.9 (C-2), 69.2 (C-3), 208.8 (C-1).
Methyl 5-ethoxy-2-hydroxycinnamate (3). Yellow amorphous powder. mp 98−100 °C. UV (MeOH) λmax (log ε) 249 (3.65), 278 (3.77), 351 (3.41) nm. IR (KBr) νmax 3404, 1705, 1630, 1455 cm−1. EIMS m/z (rel. int.) 222 (8, M+), 162 (32), 149 (24), 134 (100), 107 (33), 105 (38), 77 (81); HR-EIMS m/z 222.0891 [M]+ (calcd for C12H14O4 222.0892). 1H NMR (CDCl3) δ 1.39 (3H, t, J = 6.9 Hz, 5-OCH2CH3), 3.81 (3H, s, 9-OCH3), 3.98 (2H, q, J = 6.9 Hz, 5-OCH2CH3), 5.68 (1H, br s, 2-OH), 6.53 (1H, d, J = 16.0 Hz, H-8), 6.75 (1H, d, J = 8.6 Hz, H-3), 6.82 (1H, dd, J = 8.6, 2.5 Hz, H-4), 6.98 (1H, d, J = 2.5 Hz, H-6), 7.96 (1H, d, J = 16.0 Hz, H-7); 13C NMR (CDCl3) δ 14.9 (5-OCH2CH3), 51.7 (9-OCH3), 64.2 (5-OCH2CH3), 113.5 (C-6), 117.3 (C-3), 118.4 (C-8), 118.5 (C-4), 122.1 (C-1), 140.1 (C-7), 149.2 (C-2), 152.9 (C-5), 168.1 (C-9).





{kind=link}
{kind=link}
{kind=link}
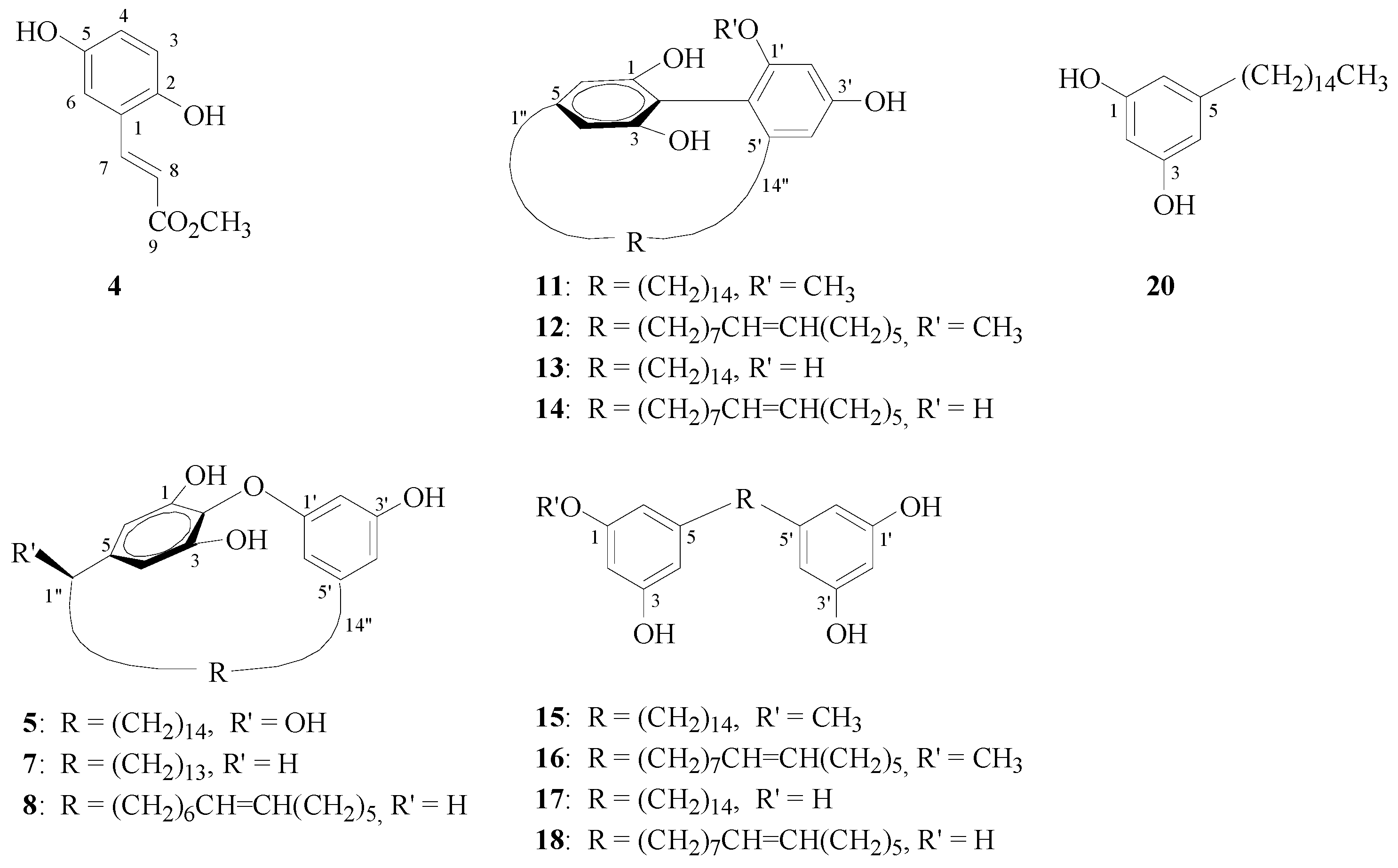{kind=link}
