New Chiral P-N Ligands for the Regio- and Stereoselective Pd-Catalyzed Dimerization of Styrene

 and
and
Abstract
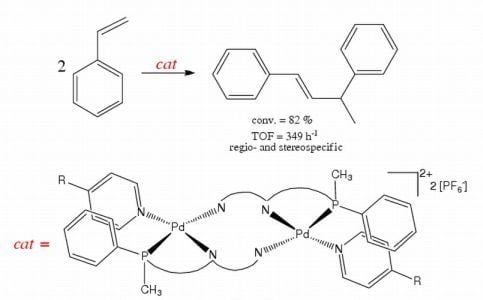
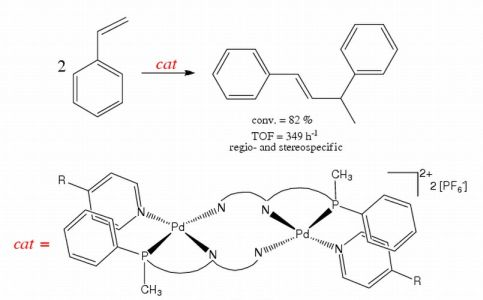
:
1. Introduction
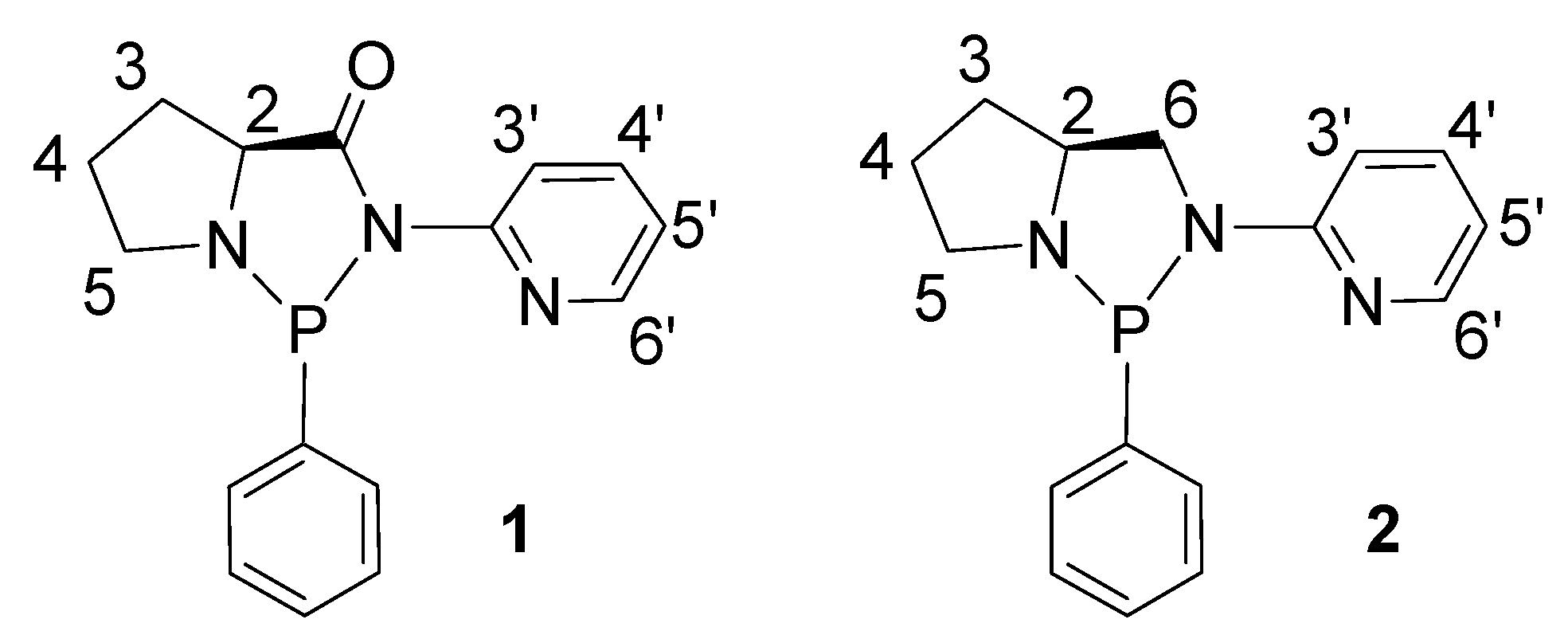
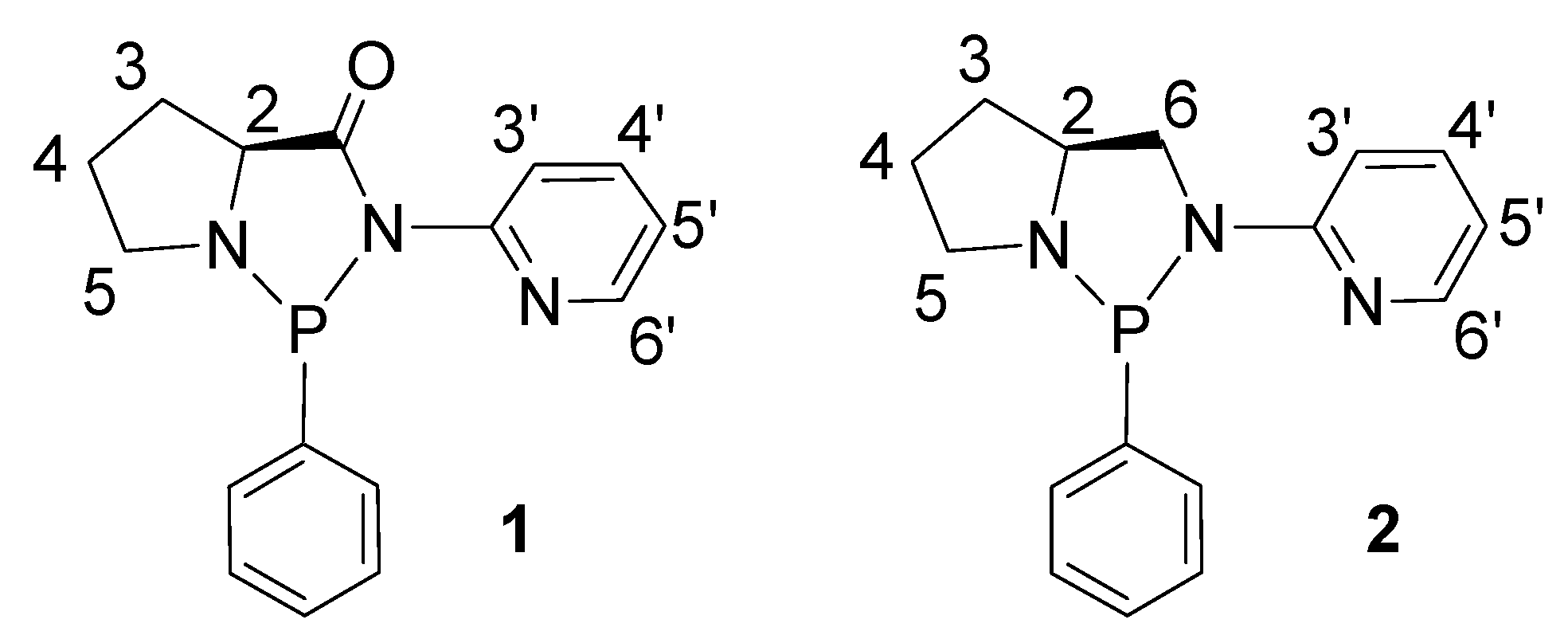
2. Results and Discussion
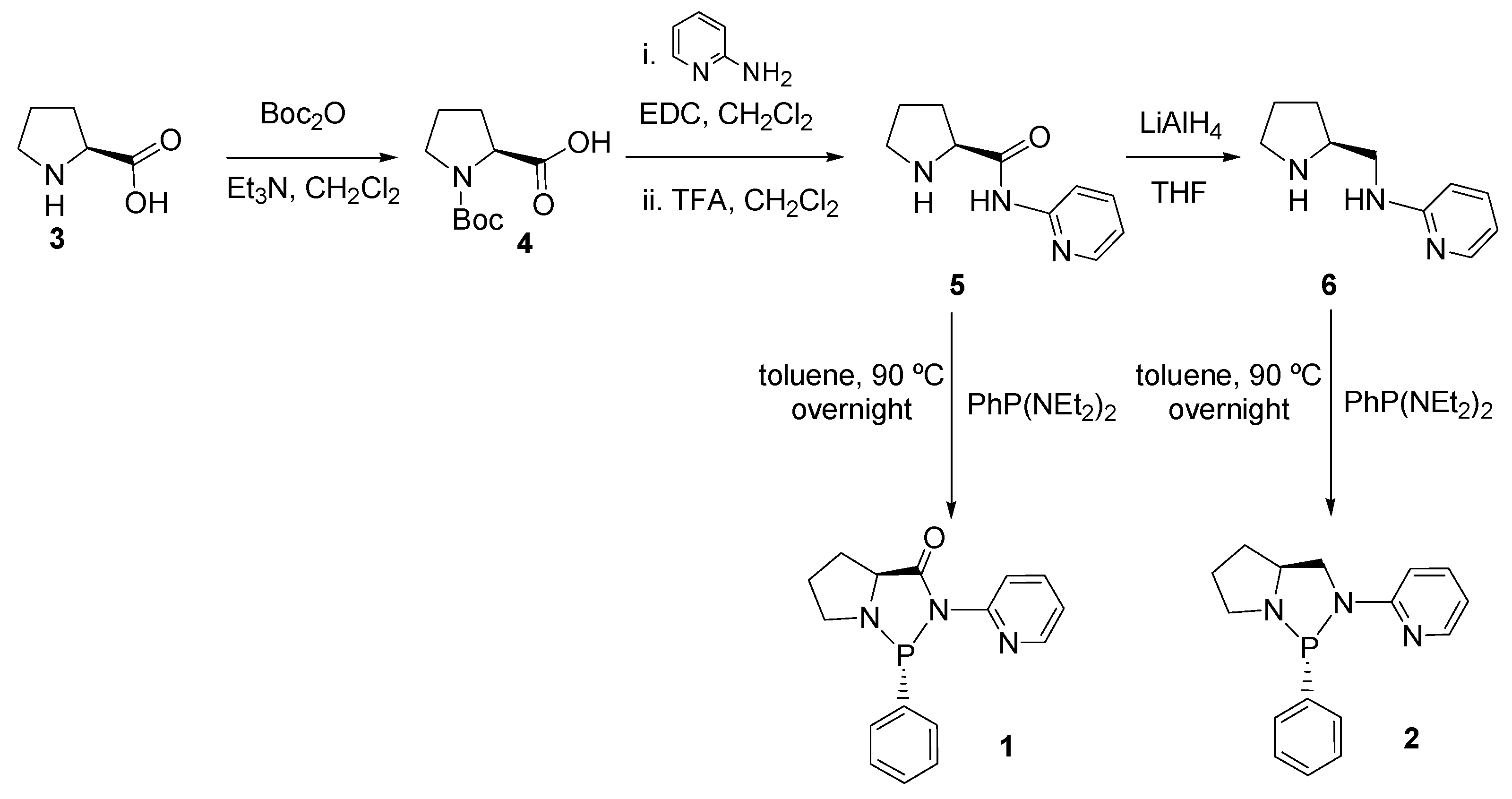
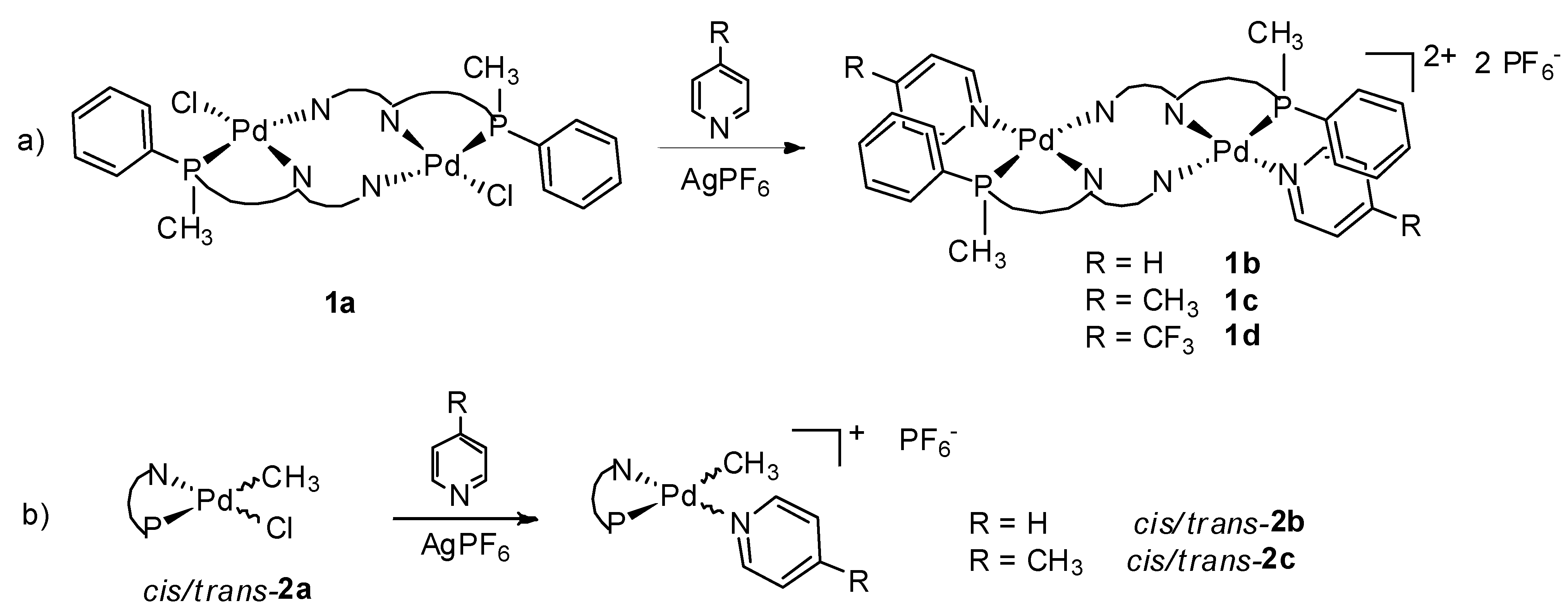
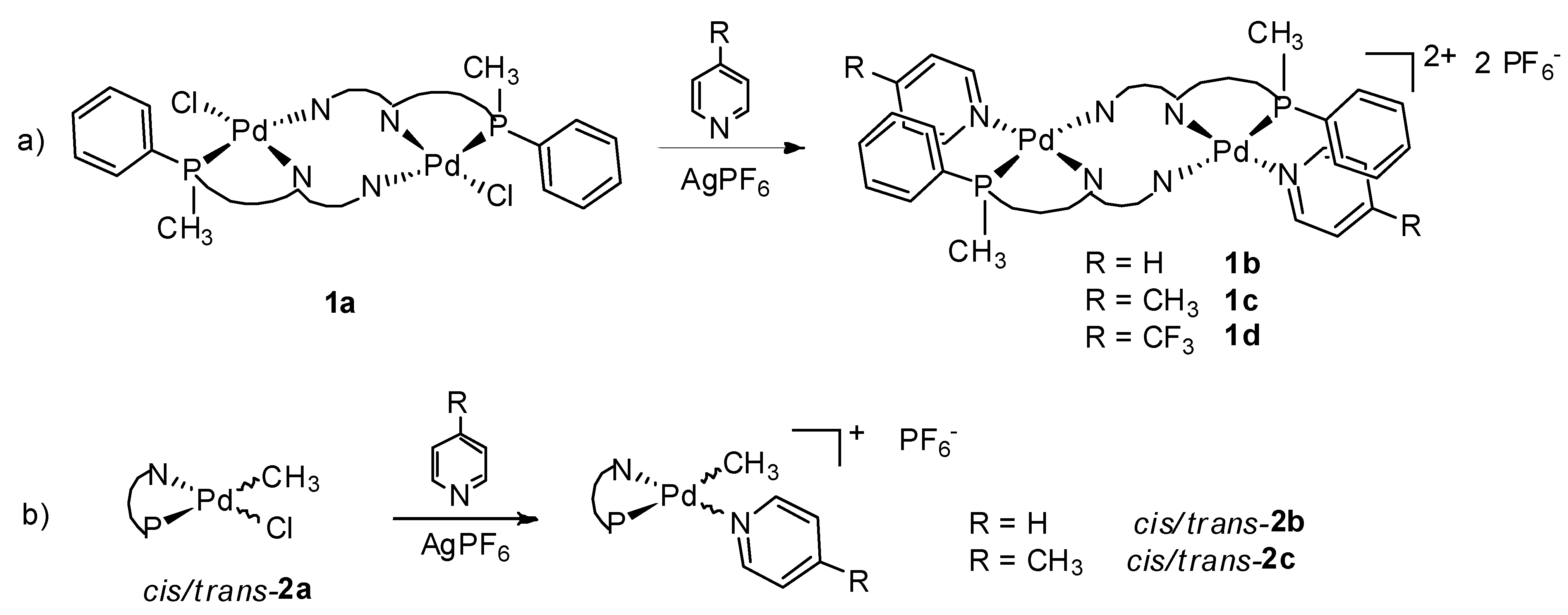
2.1. Synthesis and Characterization of P-N Ligands 1 and 2

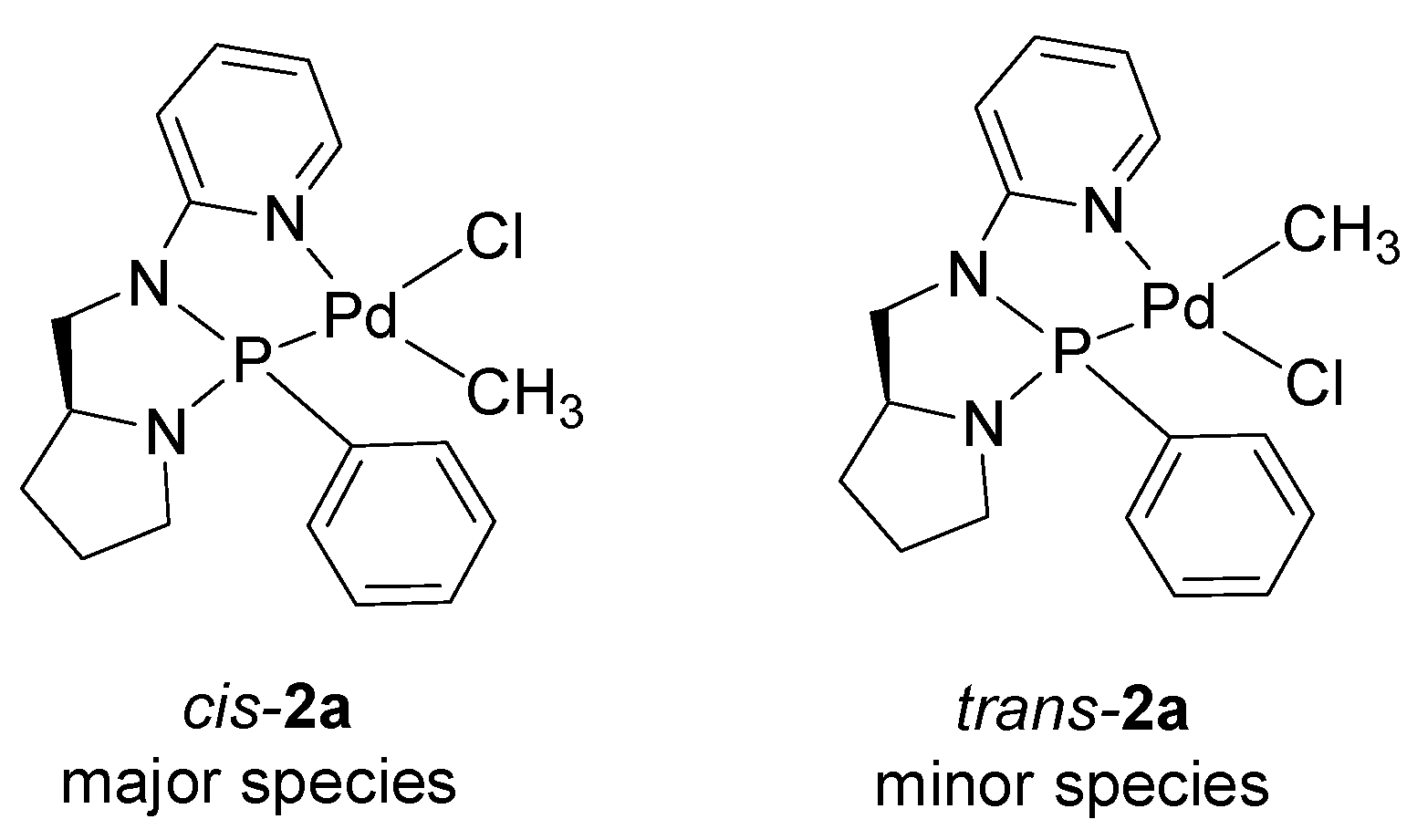
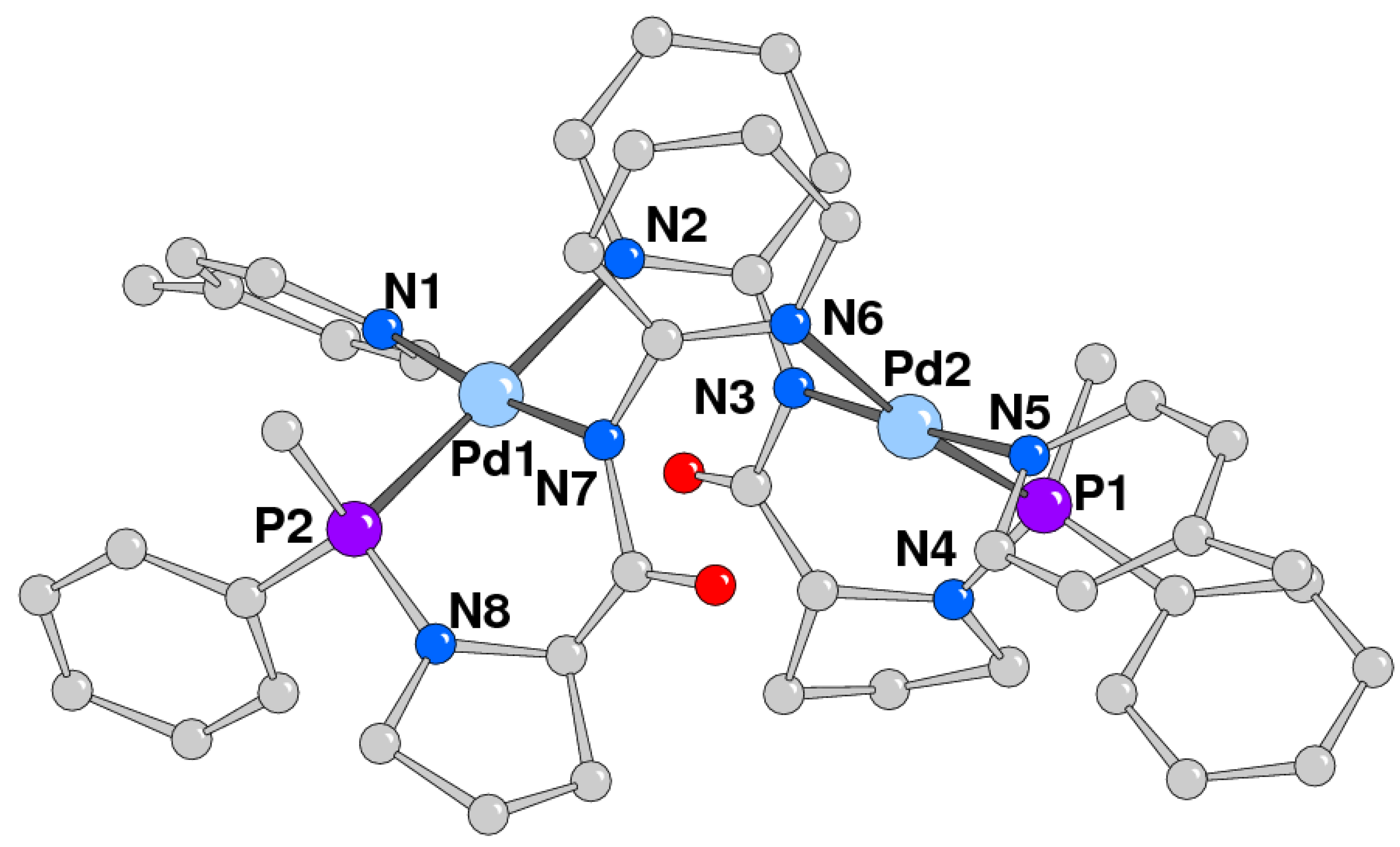
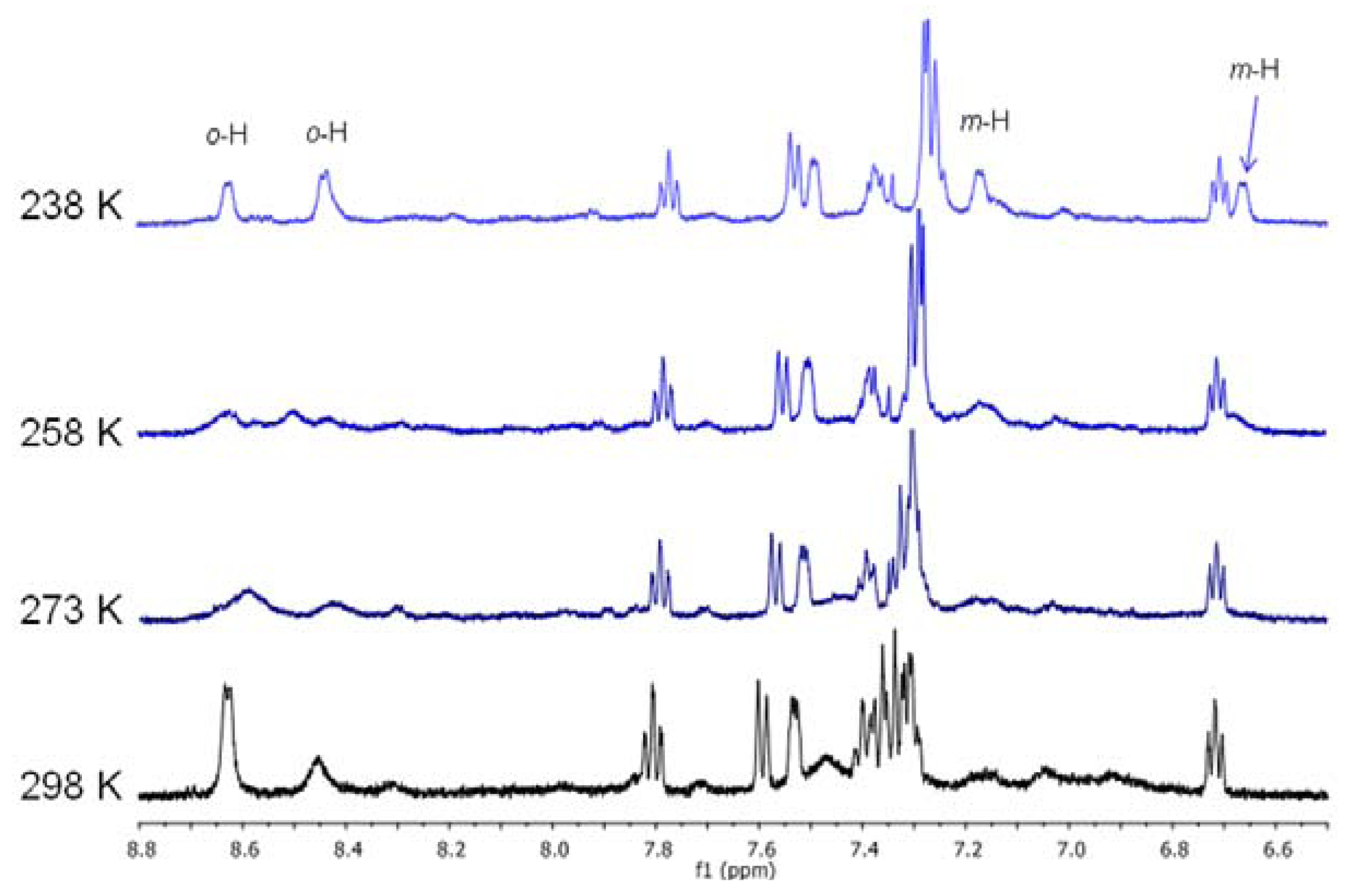
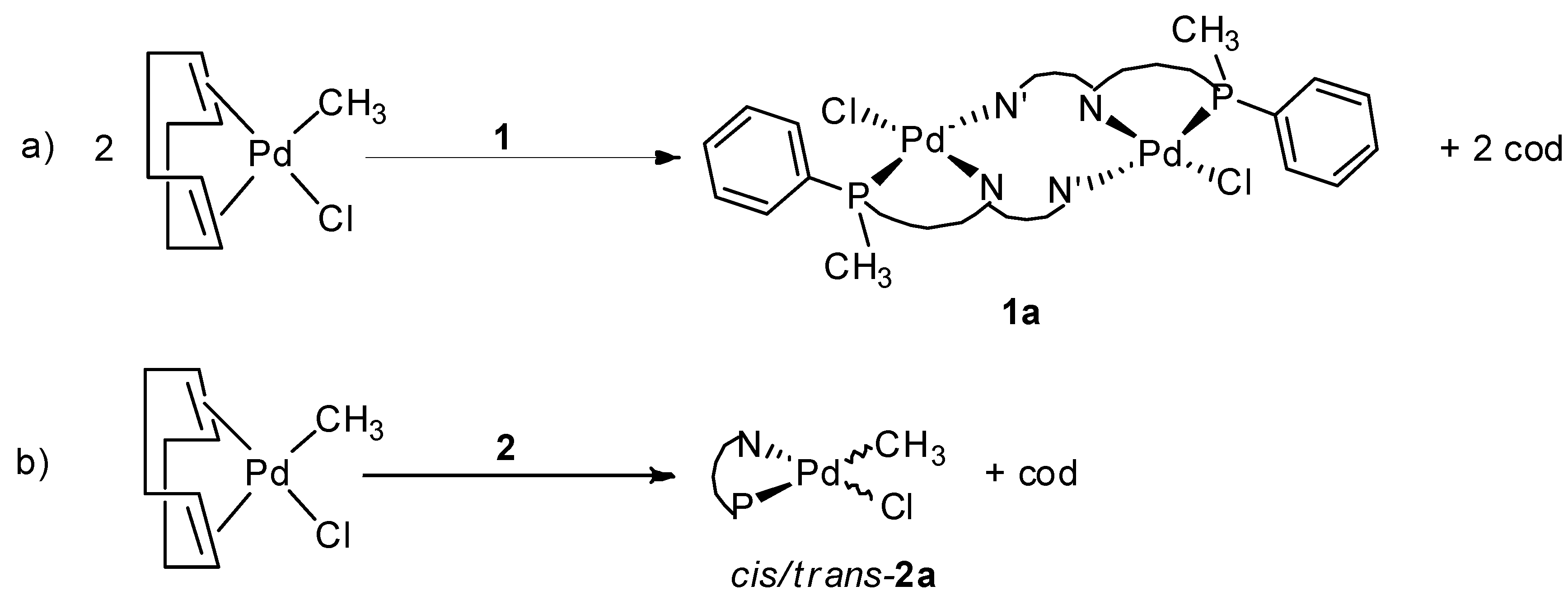
2.2. Study of Palladium Coordination Chemistry


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P-N/ complexes | 31P NMR | CIS[b] (δl-δc) | CH31H NMR | JP-CH3 (Hz) |
|---|---|---|---|---|
| 1 | 106.3 | - | - | - |
| 1a | 67.0 (M), 75.6 (m) | 39.3 (M), 30.7 (m) | 2.12 (M), 2.45 (m) | 13.0 (M), 11.5 (m) |
| 1b | 50.7 | 55.6 | 2.38 | 11.5 |
| 1c | 50.7 | 55.6 | 2.36 | 11.5 |
| 1d | 50.7 | 55.6 | 2.38 | 11.5 |
| 2 | 99.7 | - | - | - |
| 2a | 119.9 (m), 133.8 (M) | -20.2 (m), -34.0 (M) | 0.69 (M), 1.65 (m) | 2.0 (M), 8.5 (m) |
| 2bc | 120.5 (M), 122.9, 134.2 | -20.8 (M), -23.2, -34.5 | 0.58,d 1.61 (M), 1.71 | 8.0 (M), 7.8 |
| 2c | 122.2 (m), 135.5 (M) | -22.5 (m), -35.8 (M) | 0.58 (M), 1.77 (m) | 1.8 (M), 8.6 (m) |



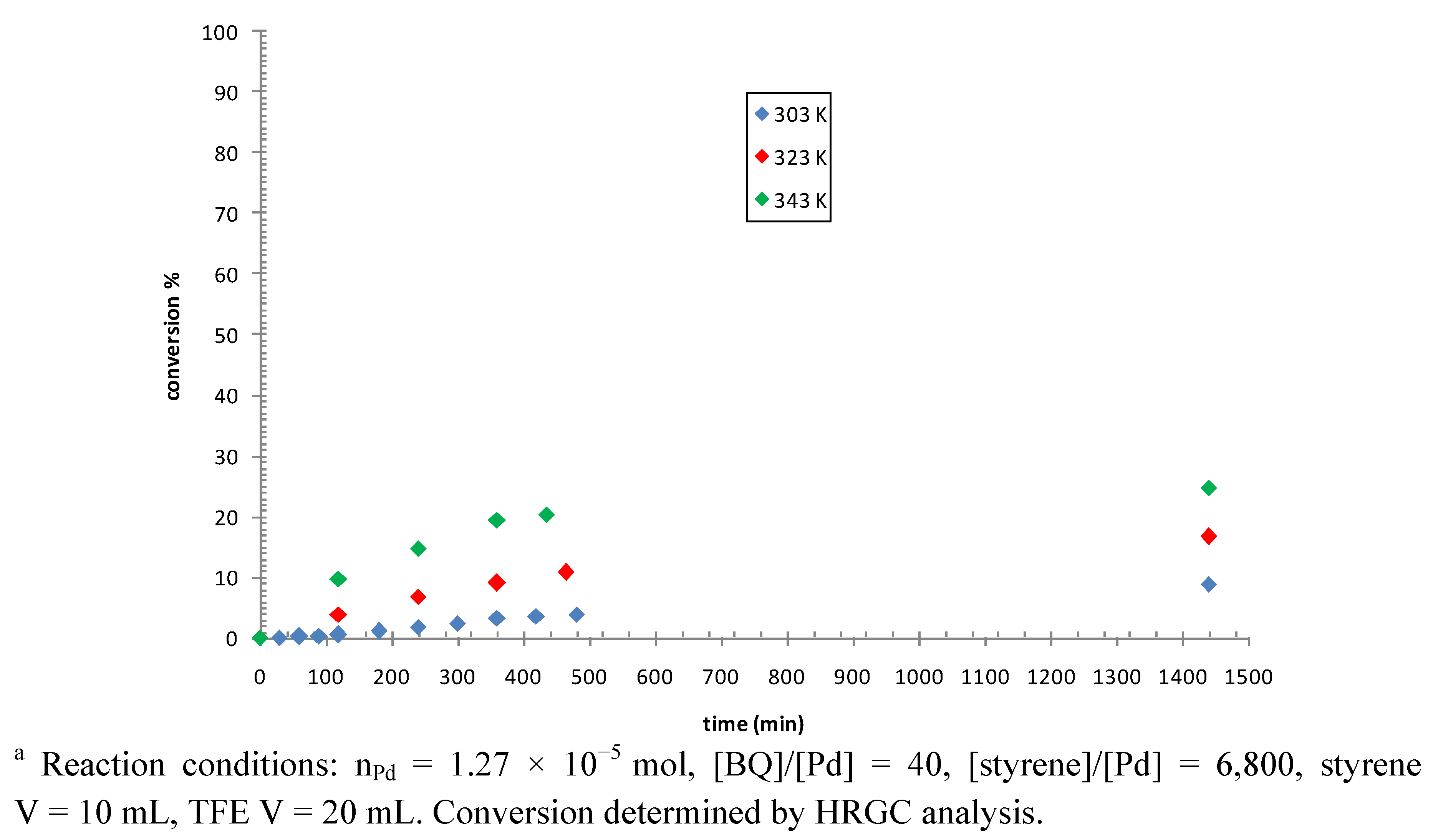
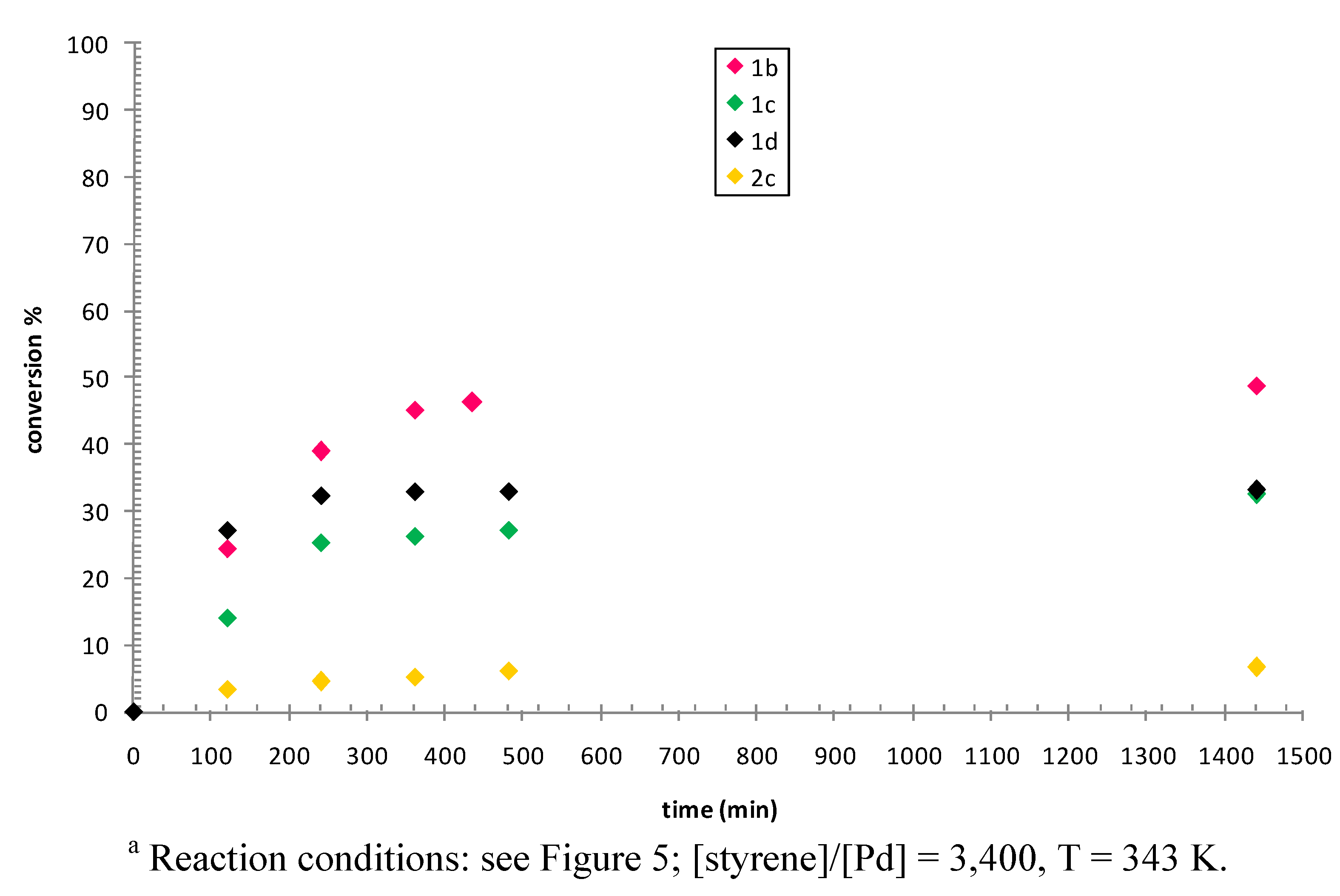
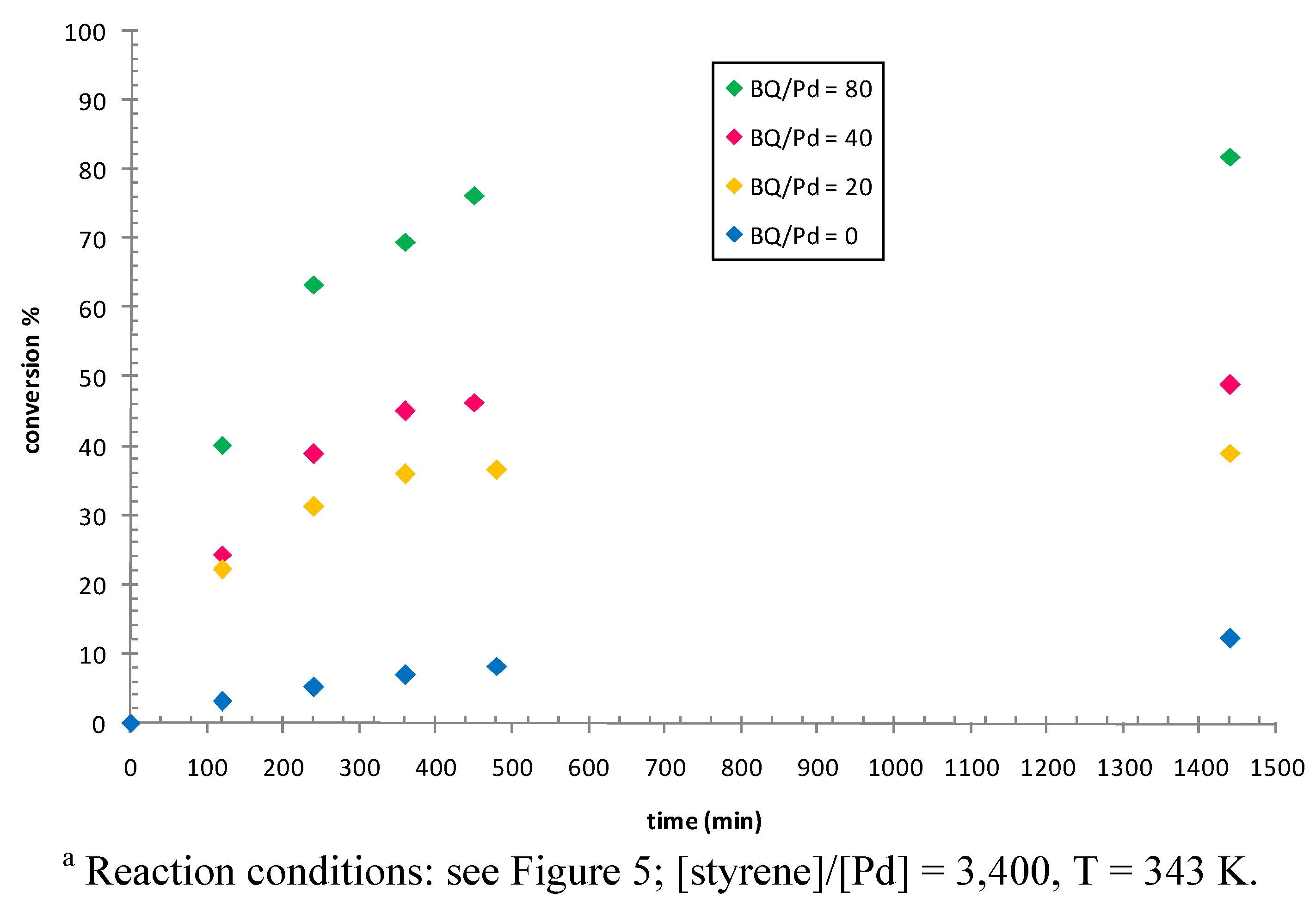
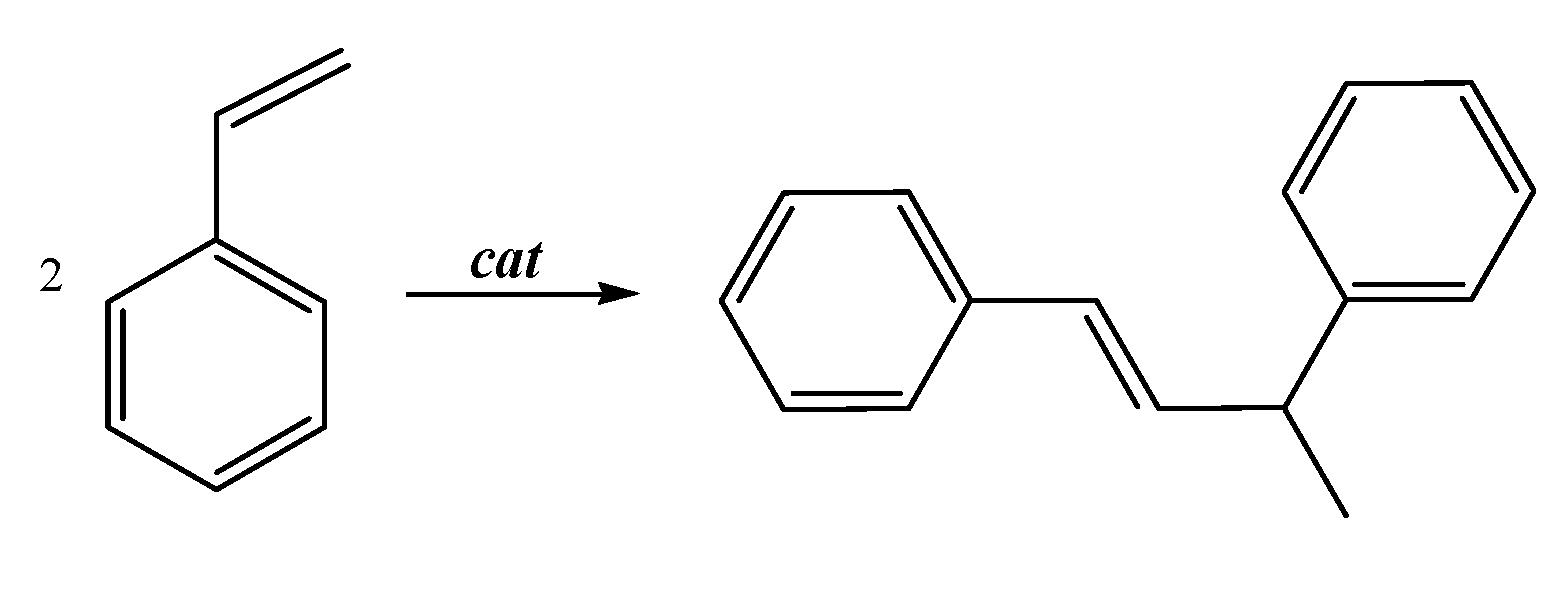
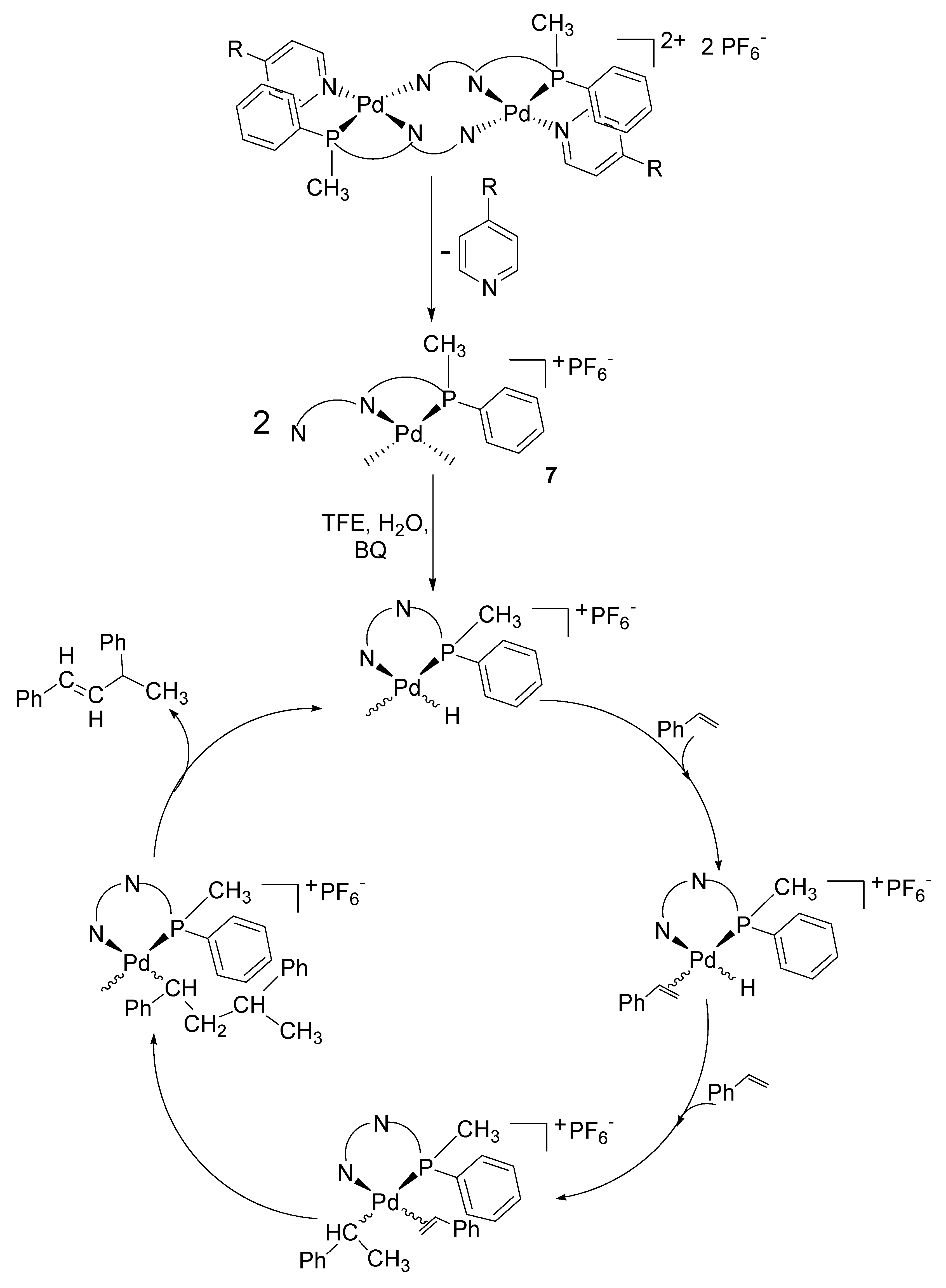
2.3. Catalytic Activity of Cationic Complexes 1b-d, 2b-c




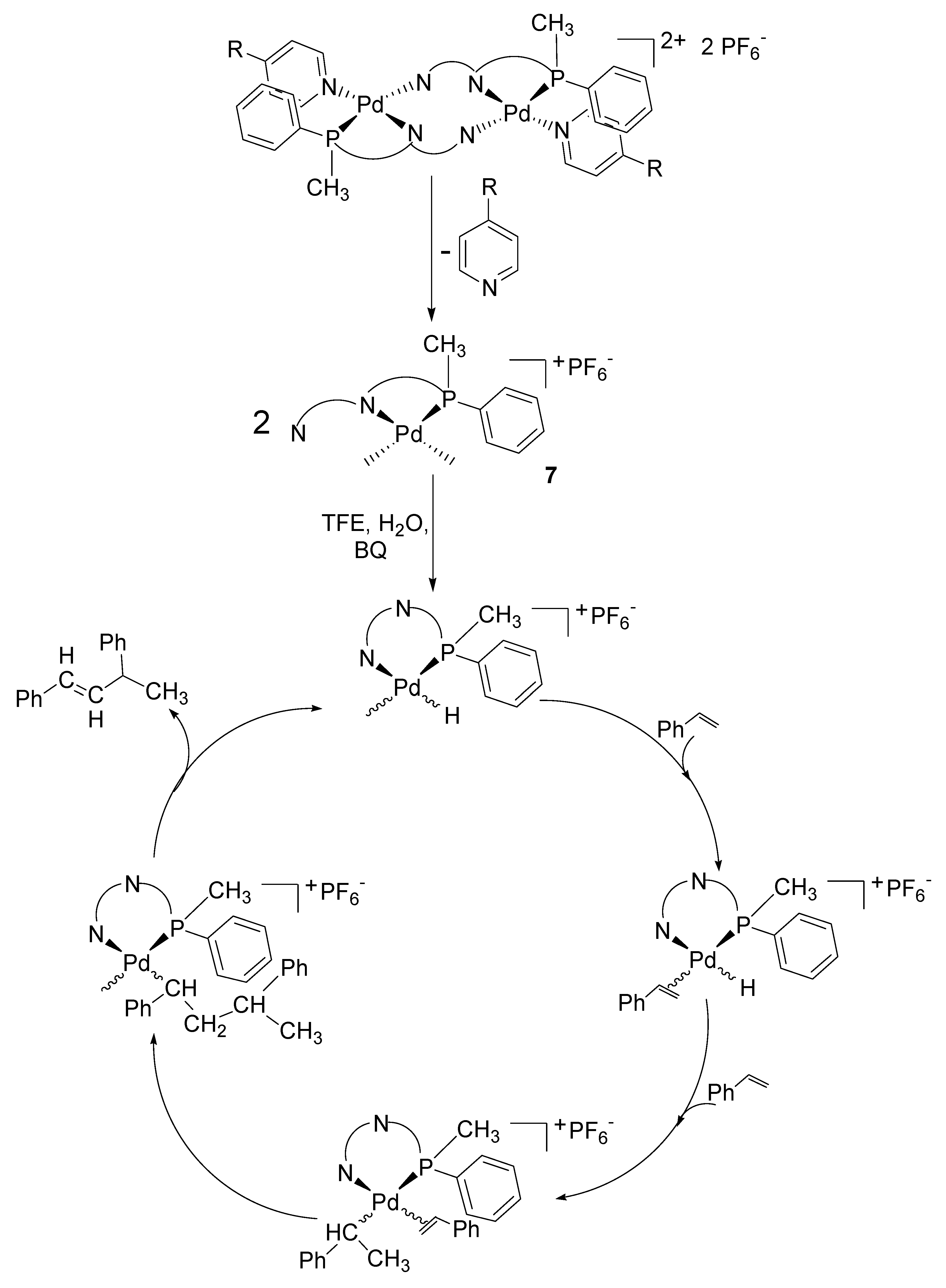
3. Experimental
3.1. General
3.2. Synthesis of P-N Ligands
General procedure for preparation of P-N ligands 1 and 2
3.3. Synthesis of Pd Complexes
3.4. Synthesis of the Cationic Complexes 1b-d, 2b-c
3.5. Dimerization Reaction
3.6. Codimerization Reaction
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Espinet, P.; Soulantica, K. Phosphine-pyridyl and related ligands in synthesis and catalysis. Coord. Chem. Rev. 1999, 193-195, 499–556. [Google Scholar] [CrossRef]
- Maggini, S. Classification of P,N-binucleating ligands for hetero- and homobimetallic complexes. Coord. Chem. Rev. 2009, 253, 1793–1832. [Google Scholar] [CrossRef]
- Schoenleber, M.; Hilgraf, R.; Pfaltz, A. Chiral Bis(N-arylamino)phosphine-oxazolines: Synthesis and Application in Asymmetric Catalysis. Adv. Synth. Catal. 2008, 350, 2033–2038. [Google Scholar] [CrossRef]
- Mothes, E.; Sentets, S.; Luquin, M.A.; Mathieu, M.; Lugan, N.; Lavigne, G. New insight into the reactivity of pyridine-functionalized phosphine complexes of ruthenium(II) with respect to olefin metathesis and transfer hydrogenation. Organometallics 2008, 27, 1197–1206. [Google Scholar]
- Roseblade, S.J.; Pfaltz, A. Iridium-Catalyzed Asymmetric Hydrogenation of Olefins. Acc. Chem. Res. 2007, 40, 1402–1411. [Google Scholar] [CrossRef]
- Cipot, J.; McDonald, R.; Ferguson, M.J.; Schatte, G.; Stradiotto, G. Cationic and formally zwitterionic rhodium(I) and iridium(I) derivatives of a P,N-substituted indene: A comparative synthetic, structural and catalytic investigation. Organometallics 2007, 26, 594–608. [Google Scholar]
- Kimura, M.; Uozomi, Y. Development of New P-Chiral Phosphorodiamidite Ligands Having a Pyrrolo[1,2-c]diazaphosphol-1-one Unit and Their Application to Regio- and Enantioselective Iridium-Catalyzed Allylic Etherification. J. Org. Chem. 2007, 72, 707–714. [Google Scholar] [CrossRef]
- Arrayas, R.G.; Adrio, J.; Carrettero, J.C. Recent applications of chiral ferrocene ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef]
- Giuri, P.J.; Saunders, C.P. The development of bidentate P,N ligands for asymmetric catalysis. Adv. Synth. Catal. 2004, 346, 497–537. [Google Scholar] [CrossRef]
- Franciò, G.; Drommi, D.; Graiff, C.; Faraone, F.; Tiripicchio, A. Synthesis of phosphonito,N and phosphito,N ligands based on quinolines and (R)-binaphthol or substituted biphenol and their rhodium(I), palladium(II) and platinum(II) complexes. Inorg. Chim. Acta 2002, 338, 59–69. [Google Scholar] [CrossRef]
- Brumel, J.M.; Legrand, O.; Reymond, S.; Buono, G. First Iminodiazaphospholidines with a Stereogenic Phosphorous Center. Application to Asymmetric Copper-Catalyzed Cyclopropanation. J. Am. Chem. Soc. 1999, 121, 5807–5808. [Google Scholar] [CrossRef]
- Bluhm, M.E.; Folli, C.; Walter, O.; Doering, M. Nitrogen- and phosphorus-coordinated nickel(II) complexes as catalysts for the oligomerization of ethylene. J. Mol. Cat. A Chem. 2005, 229, 177–181. [Google Scholar]
- Cheng, H.-P.; Liu, Y.-H.; Peng, S.-M.; Liu, S.-T. New bulky phosphino-pyridine ligands. Palladium and nickel complexes for the catalytic polymerization and oligomerization of ethylene . Organometallics 2003, 22, 4893–4899. [Google Scholar]
- Sun, W.-H.; Li, Z.; Hu, H.; Wu, B.; Yang, H.; Zhu, N.; Leng, X.; Wang, H. Synthesis and characterization of novel nickel(II) complexes bearing N,P ligands and their catalytic activity in ethylene oligomerization. New J. Chem. 2002, 26, 1474–1478. [Google Scholar] [CrossRef]
- Daugulis, O.; Brookhart, M. Polymerization of ethylene with cationic palladium and nickel catalysts containing bulky nonenolizable imine-phosphine ligands. Organometallics 2002, 21, 5926–5934. [Google Scholar]
- Keim, W.; Killat, S.; Nobile, C.F.; Suranna, G.P.; Englert, U.; Wang, R.; Mecking, S.; Schroeder, D.L. Synthesis, characterization and catalytic activity of Pd(II) and Ni(II) complexes with new cyclic alpha-diphenylphosphino-ketoimines. Crystal structure of 2,6-diisopropyl-N-(2-diphenylphosphino-cyclopropylidene)aniline and of 2,6-diisopropyl-N-(2-diphenylphosphino-cyclohexylidene)aniline. J. Organomet. Chem. 2002, 662, 150–171. [Google Scholar]
- Sirbu, D.; Consiglio, G.; Gischig, S. Palladium and nickel complexes of (P,N)-ligands based on quinolines: Catalytic activity for polymerization and oligomerization. J. Organomet. Chem. 2006, 691, 1143–1150. [Google Scholar] [CrossRef]
- Aeby, A.; Consiglio, G. P^N versus N^N ligands for the palladium-catalyzed alternating copolymerization of styrene and carbon monoxide. Inorg. Chim. Acta 1999, 296, 45–51. [Google Scholar] [CrossRef]
- Gsponer, A.; Schmid, T.M.; Consiglio, G. Ligand-dependent diastereoselectivity in the palladium-catalyzed copolymerization of styrene with carbon monoxide. Helv. Chim. Acta 2001, 84, 2986–2995. [Google Scholar] [CrossRef]
- Braunstein, P.; Fryzuk, M.D.; Le Dall, M.; Naud, F.; Rettig, S.J.; Speiser, F. Synthesis and structure of Pd(II) complexes containing chelating (phosphinomethyl)oxazoline P,N-type ligands. copolymerisation of ethylene/CO. J. Chem. Soc., Dalton Trans. 2000, 1067–1074. [Google Scholar]
- Agostinho, M.; Braunstein, P. Palladium(II) complexes with the new P,N-type ligand (2-oxazoline-2-ylmethyl)di-isopropylphosphine as intermediates in CO/ethylene or CO/methyl acrylate insertion. C.R. Chim. 2007, 10, 666–676. [Google Scholar]
- Agostinho, M.; Braunstein, P.; Welter, R. Phosphinito- and phosphonito-oxazoline Pd(ii) complexes as CO/ethylene insertion intermediates: Synthesis and structural characterization. Dalton Trans. 2007, 759–770. [Google Scholar]
- Agostinho, M.; Braunstein, P. Structurally characterized intermediates in the stepwise insertion of CO-ethylene or CO-methyl acrylate into the metal-carbon bond of Pd(II) complexes stabilized by (phosphinomethyl)oxazoline ligands. Chem. Commun. 2007, 58–60. [Google Scholar] [CrossRef]
- Buono, G.; Toselli, N.; Martin, D. Chiral Diazaphospholidine Ligands in Phosphorus Ligands in Asymmetric Catalysis; Börner, A., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp. 529–546. [Google Scholar]
- Breeden, S.; Cole-Hamilton, D.J.; Foster, D.F.; Schwarz, G.J.; Will, M. Rhodium-mediated asymmetric hydroformylation with a novel bis(diazaphospholidine) ligand. Angew. Chem. Int. Ed. 2000, 39, 4106–4108. [Google Scholar] [CrossRef]
- Myagmarsuren, G.; Tkack, V.S.; Shmidt, F.K.; Mohamad, M.; Suslov, D.S. Selective dimerization of styrene ot 1,3-diphenyl-1-butene with bis(beta-diketonato)palladium/boron trifloride etherate catalyst system. J. Mol. Cat. A Chem. 2005, 235, 154–160. [Google Scholar] [CrossRef]
- Peng, J.; Li, J.; Qiu, H.; Jiang, J.; Jiang, K.; Mao, J.; Lai, G. Dimerization of styrene to 1,3-diphenyl-1-butene catalyzed by palladium-Lewis acid in ionic liquid. J. Mol. Cat. A Chem. 2006, 255, 16–18. [Google Scholar] [CrossRef]
- Lee, D.W.; Yi, C.S. Chain-Selective and Regioselective Ethylene and Styrene Dimerization Reactions Catalyzed by a Well-Defined Cationic Ruthenium Hydride Complex: New Insights on the Styrene Dimerization Mechanism. Organometallics 2010, 29, 3413–3417. [Google Scholar] [CrossRef]
- Cabrero-Antonino, J.R.; Leyva-Perez, A.; Corma, A. Iron-Catalysed Regio- and Stereoselective Head-to-Tail Dimerisation of Styrenes. Adv. Synth. Catal. 2010, 352, 1571–1576. [Google Scholar] [CrossRef]
- Vogt, D. Cobalt-Catalyzed Asymmetric Hydrovinylation. Angew. Chem. Int. Ed. 2010, 49, 7166–7168. [Google Scholar]
- RajanBabu, T.V. Asymmetric Hydrovinylation Reaction. Chem. Rev. 2003, 103, 2845–2860. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Herald, D.L.; Lloyd-Williams, P.; Kantoci, D.; Burkett, D.D.; Barkoczy, J.; Hogan, F.; Wardlaw, T.R. The Dolastatins. 17. Synthesis of Dolaproine and Related Diasteroisomers. J. Org. Chem. 1994, 59, 6287–6295. [Google Scholar]
- Xu, X.-Y.; Tang, Z.; Wang, Y.-Z.; Luo, S.-W.; Cun, L.-F.; Gong, L.-Z. Asymmetric Organocatalytic Direct Aldol Reactions of Ketones with alpha-Keto acids and Their Application to the Synthesis of 2-Hydroxy-gamma-butyrolactones. J. Org. Chem. 2007, 72, 9905–9913. [Google Scholar] [CrossRef]
- Luo, S.; Xu, H.; Li, J.; Zhang, L.; Mi, X.; Zheng, X.; Cheng, J.-P. Facile evolution of asymmetric organocatalysts in water assisted by surfactant Brønsted acids. Tetrahedron 2007, 63, 11307–11314. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Dalpozzo, R.; Giuliani, A.; Marcantoni, E.; Mercozzi, T.; Sambri, L.; Torregiani, E. An Efficient Procedure for the Preparation of (E)-alpha-Alkylidene Cycloalkanones Mediated by CeCl3*7H2O-NaI System. Novel Methodology for the Synthesis of (S)-(-)-Pulegone. J. Org. Chem. 2002, 67, 9111–9114. [Google Scholar] [CrossRef]
- Torisawa, Y.; Hashimoto, A.; Okouchi, M.; Limori, T.; Nagasawa, M.; Hino, T.; Nakagawa, M. Manzamine C Congeners with Modified Azacyclic Rings: Synthesis and Biological Evaluation. Bioorg. Med. Chem. Lett. 1996, 6, 2565–2570. [Google Scholar] [CrossRef]
- Legrand, O.; Brumel, J.M.; Constantieux, T.; Buono, G. Totally Stereoselective P-O to P-C Migration Rearrangement: Application to the Synthesis of New Chiral o-Hydroxyaryl Phosphine Oxides. Chem. Eur. J. 1998, 4, 1061–1067. [Google Scholar] [CrossRef]
- del Campo, O.; Carbayo, A.; Cuevas, J.V.; García-Herbosa, G.; Muñoz, A. Isomeric Preference in Complexes of Palladium(II) with Chelating P,N-Donor Ligands. Eur. J. Inorg. Chem. 2009, 2254–2260. [Google Scholar]
- Morita, D.K.; Stille, J.K.; Norton, J.R. Methyl/Phenyl Exchange between Palladium and a Phosphine Ligand. Consequences for Catalytic Coupling Reactions. J. Am. Chem. Soc. 1995, 117, 8576–8581. [Google Scholar]
- D'Amora, A.; Fanfoni, L.; Cozzula, D.; Guidolin, N.; Zangrando, E.; Felluga, F.; Gladiali, S.; Benedetti, F.; Milani, B. Addressing the Poly- to Oligo-ketone selectivity in styrene Carbonylation Catalyzed by Palladium/bpy Complexes. Effect of the 6-Alkyl Substitution. Organometallics 2010, 29, 4472–4485. [Google Scholar]
- Durand, J.; Scarel, A.; Milani, B.; Seraglia, R.; Gladiali, S.; Carfagna, C.; Binotti, B. Palladium-Promoted Carbon Monoxide/Ethylene/Styrene Terpolymerization Reaction: Throwing Light on the Different Reactivity of the Two Alkenes. Helv. Chim. Acta 2006, 89, 1752–1771. [Google Scholar]
- Drent, E.; Budzelaar, P.H.M. Palladium catalyzed alternating copolymerization of alkenes and carbon monoxide. Chem. Rev. 1996, 96, 663–681. [Google Scholar] [CrossRef]
- Milani, B.; Anzilutti, A.; Vicentini, L.; Sessanta o Santi, A.; Zangrando, E.; Geremia, S.; Mestroni, G. Bis-chelated palladium(II) complexes with nitrogen-donor chelating ligands are efficient catalyst precursors for the CO/styrene copolymerization reaction. Organometallics 1997, 16, 5064–5075. [Google Scholar] [CrossRef]
- Scarel, A.; Durand, J.; Franchi, D.; Zangrando, E.; Mestroni, G.; Carfagna, C.; Mosca, L.; Seraglia, R.; Consiglio, G.; Milani, B. Mono- and dinuclear bioxazoline-palladium complexes for the stereocontrolled synthesis of CO/styrene polyketones. Chem. Eur. J. 2005, 11, 6014–6023. [Google Scholar] [CrossRef]
- Axet, M.R.; Amoroso, F.; Bottari, G.; D'Amora, A.; Zangrando, E.; Faraone, F.; Drommi, D.; Saporita, M.; Carfagna, C.; Natanti, P.; Seraglia, R.; Milani, B. Application of chiral amine-imine ligands in palladium-catalyzed polyketones synthesis: Effect of ligand backbone on the polymer stereochemistry. Organometallics 2009, 28, 4464–4474. [Google Scholar] [CrossRef]
- Sample Availability: No sample is available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fanfoni, L.; Meduri, A.; Zangrando, E.; Castillon, S.; Felluga, F.; Milani, B. New Chiral P-N Ligands for the Regio- and Stereoselective Pd-Catalyzed Dimerization of Styrene. Molecules 2011, 16, 1804-1824. https://doi.org/10.3390/molecules16021804
Fanfoni L, Meduri A, Zangrando E, Castillon S, Felluga F, Milani B. New Chiral P-N Ligands for the Regio- and Stereoselective Pd-Catalyzed Dimerization of Styrene. Molecules. 2011; 16(2):1804-1824. https://doi.org/10.3390/molecules16021804
Chicago/Turabian StyleFanfoni, Lidia, Angelo Meduri, Ennio Zangrando, Sergio Castillon, Fulvia Felluga, and Barbara Milani. 2011. "New Chiral P-N Ligands for the Regio- and Stereoselective Pd-Catalyzed Dimerization of Styrene" Molecules 16, no. 2: 1804-1824. https://doi.org/10.3390/molecules16021804