Synthesis of Quinoxaline 1,4-di-N-Oxide Analogues and Crystal Structure of 2-Carbomethoxy-3-hydroxyquinoxaline-di-N-oxide

Abstract
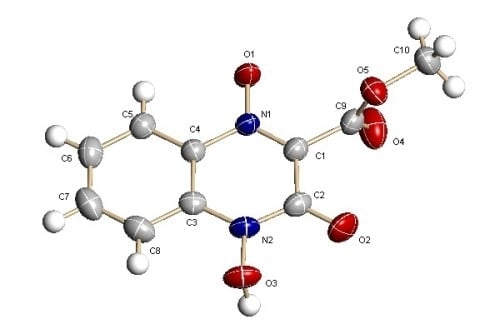
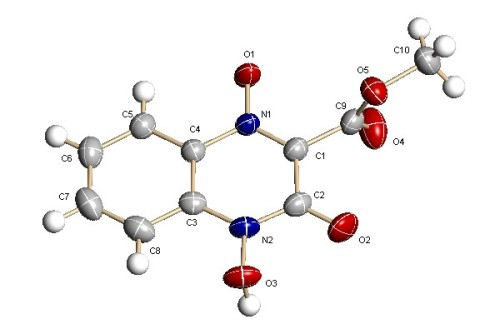
:
1. Introduction
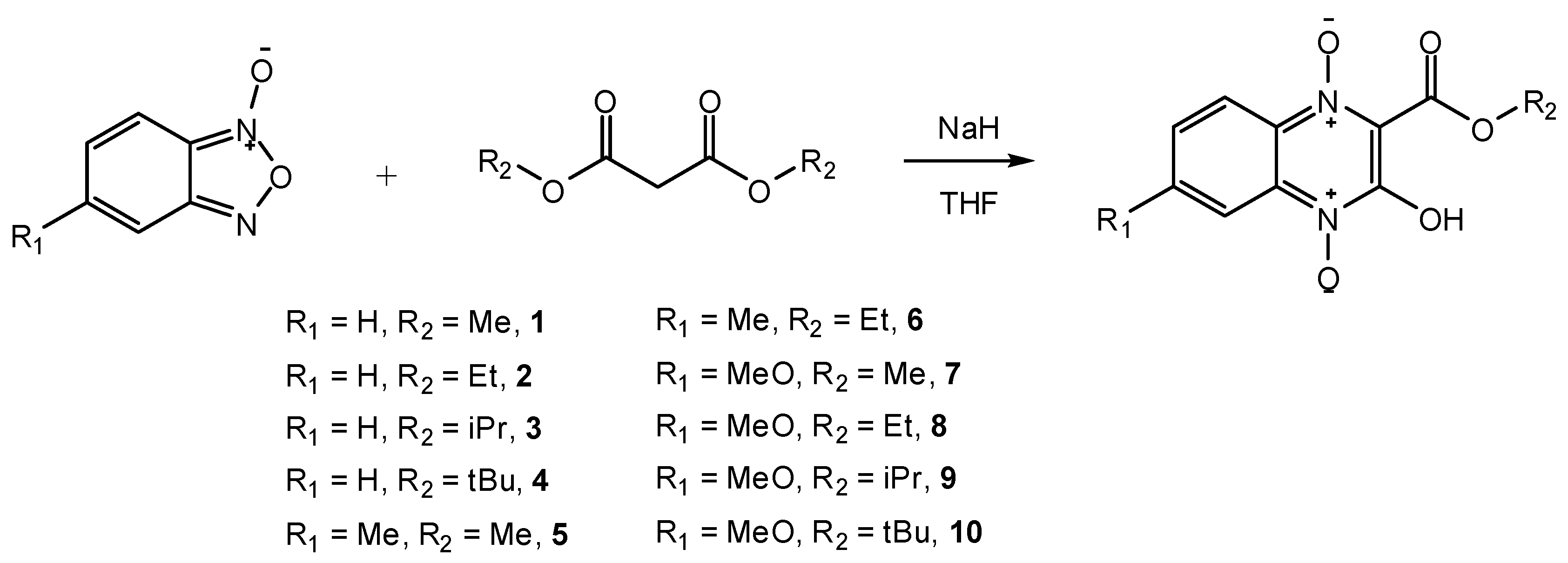
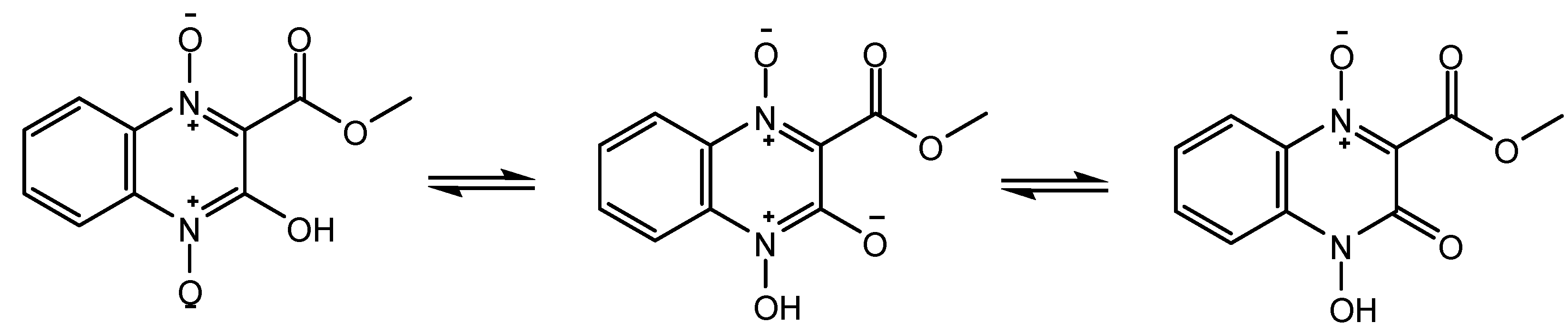
2. Results and Discussion
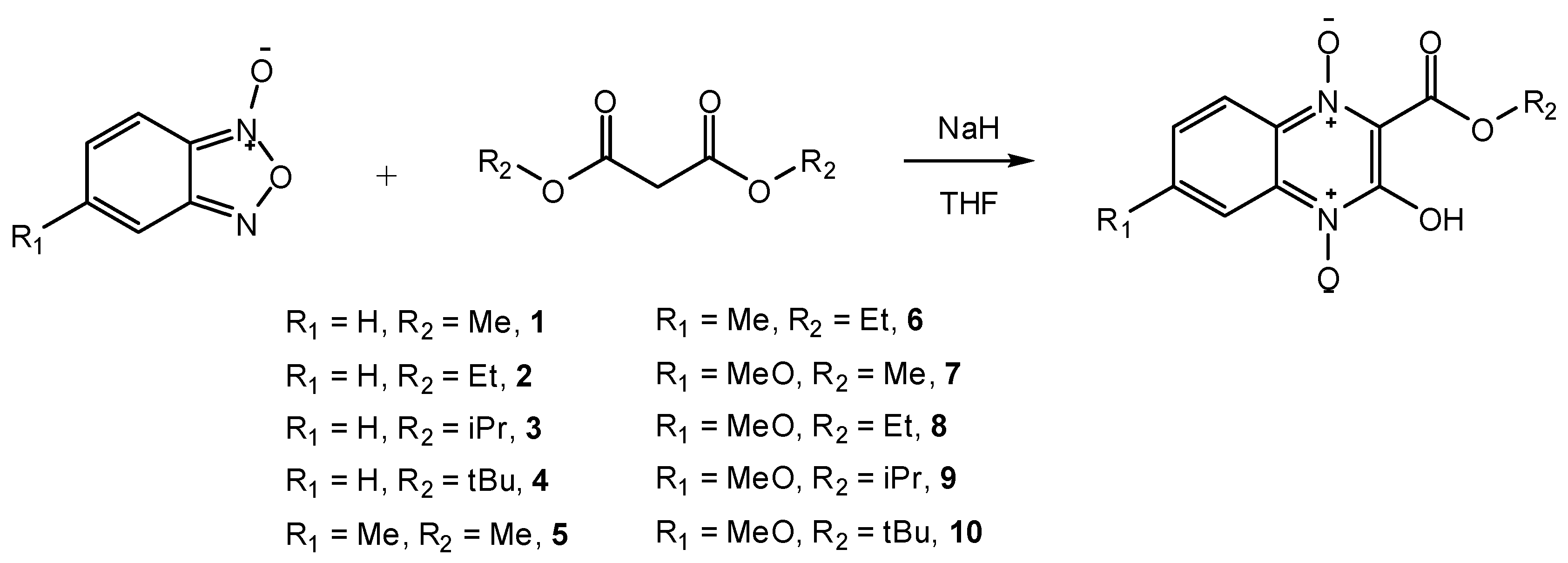


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Bond length |
|---|---|
| O(1)–N(1) | 1.2903 (13) |
| O(2)–C(2) | 1.2115 (17) |
| O(3)–N(2) | 1.3844 (14) |

3. Experimental
3.1. General
3.2. General Synthesis Procedure
3.3. Crystal structure determination
4. Supplementary Material
5. Conclusions
Acknowledgments
References and Notes
- Johnsbon, J.D.; Saybrock, O. Novel Antibacterial Agents. U.S. Patent 3,371,090, 27 February 1968. [Google Scholar]
- Dirlam, J.P.; Presslitz, J.E.; Williams, B.J. Synthesis and antibacterial activity of some 3-[(alkylthio)methyl]quinoxaline 1-oxide derivatives. J. Med. Chem. 1983, 26, 1122–1128. [Google Scholar] [CrossRef]
- Andrés, J.; Belén, Z.; Ignacio, A. Synthesis of New Quinoxaline-2-carboxylate 1,4-Dioxide Derivatives as Anti-Mycobacterium tuberculosis Agents. J. Med. Chem. 2005, 48, 2019–2024. [Google Scholar] [CrossRef]
- Issidorides, C.H.; Haddadin, M.J. Quinoxaline derivatives. U.S. Patent 4,343,942, 10 August 1982. [Google Scholar]
- Haddadin, M.J.; Issidorides, C.H. Enamines with isobenzofuroxan: A novel synthesis of quinoxaline-di-N-oxide. Tetrahedron Lett. 1965, 36, 3253–3256. [Google Scholar]
- Issidorides, C.H.; Haddadin, M.J. Benzofurazan oxide: II Reactions with enolate anions. J. Org. Chem. 1966, 31, 4067–4068. [Google Scholar] [CrossRef]
- Lin, S. Syntheses and spectroscopic investigations on eleven 2,3-disubstituted quinoxaline-1,4-dioxide antibiotics. Appl. Chem. 1988, 5, 17–23. [Google Scholar]
- Lin, S.; Wang, H. An improved method for synthesis of substituted quinoxaline-1,4-dioxides. Youji Huaxue. 1986, 298–303. [Google Scholar]
- Zhang, D.L.; Liang, J.P.; Liu, Y.; Wang, X.H.; Hua, L.Y. Synthesis and antibacterial activity of quinoxalines 1,4-di-N-oxide. Chinese J. Pharm. 2008, 39, 410–412. [Google Scholar]
- Ley, K.; Eholzer, U.; Nast, R.; Seng, F. Preparation of Quinoxaline-di-N-oxides. U.S. Patent 3,660,398, 1972. [Google Scholar]
- Wang, J.L.; Liu, F.C. Synthesis of biscarbonyl side chain substituted quinoxaline-1,4-dioxide. Hecheng Huaxue 1996, 4, 171–173. [Google Scholar]
- Simone, C.A.; Silva, R.P.; Goulart, M.; Silva, R,; Lobato, A.P.; Pinto, M.C.; Pinto, A.V. Synthesis and crystal structure of the N4-oxide of an hydroxybenzo[f]quinoxalin-6(5H)-one obtained by oxidative cleavage of beta-lapachone-quinoxaline. J. Chem. Crystallography 2006, 36, 551–556. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-97. Acta Cryst. 1990, A46, 467–475. [Google Scholar]
- DIRDIF94: Beurskens, P.T.; Admiraal, G.; Beurskens, G.; Bosman, W.P.; Garcia-Granda, S.; Gould, R.O.; Smits, J.M.; Smykalla, C. The DIRDIF-94 Program System; University of Nijmegen: Nijmegen, The Netherlands, 1994; Technical Report of the Crystallography Laboratory. [Google Scholar]
- Cromer, D.T.; Waber, J.T. International Tables for X-ray Crystallography; Kynoch Press: Birmingham, UK, 1974; Volume 4, Table 2.2 A. [Google Scholar]
- Ibers, J.A.; Hamiton, W.C. Dispersion corrections and crystal structure refinements. Acta Cryst. 1964, 17, 781. [Google Scholar] [CrossRef]
- Creagh, D.C.; McAuley, W.J. International Tables for Crystallography; Wilson, A.J., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 1992; Volume C, pp. 219–222, Table 4.2.6.3.8. [Google Scholar]
- Creagh, D.C. and Hubbell, J.H. International Tables for Crystallography; Wilson, A.J., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 1992; Volume C, pp. 200–206, Table 4.2.4.3. [Google Scholar]
- TeXsan: Crystal Structure Analysis Package; Molecular Structure Corporation: The Woodlands, TX, USA, 1985.
- Sample Availability: Contact the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, Y.; Wu, F.; Yao, Z.; Zhang, M.; Jiang, S. Synthesis of Quinoxaline 1,4-di-N-Oxide Analogues and Crystal Structure of 2-Carbomethoxy-3-hydroxyquinoxaline-di-N-oxide. Molecules 2011, 16, 6894-6901. https://doi.org/10.3390/molecules16086894
Xu Y, Wu F, Yao Z, Zhang M, Jiang S. Synthesis of Quinoxaline 1,4-di-N-Oxide Analogues and Crystal Structure of 2-Carbomethoxy-3-hydroxyquinoxaline-di-N-oxide. Molecules. 2011; 16(8):6894-6901. https://doi.org/10.3390/molecules16086894
Chicago/Turabian StyleXu, Yingjun, Fanhong Wu, Zhiyi Yao, Minmin Zhang, and Sheng Jiang. 2011. "Synthesis of Quinoxaline 1,4-di-N-Oxide Analogues and Crystal Structure of 2-Carbomethoxy-3-hydroxyquinoxaline-di-N-oxide" Molecules 16, no. 8: 6894-6901. https://doi.org/10.3390/molecules16086894