Crystal Structure and Density Functional Theory Study on Structural Properties and Energies of a Isonicotinohydrazide Compound

Abstract
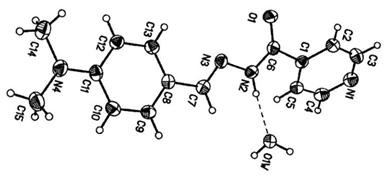
:
1. Introduction
2. Results and Discussion
2.1. Molecular Geometry


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification code | 119 |
| Empirical formula | C15H18N4O2 |
| Formula weight | 286.33 |
| Temperature | 120(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Orthorhombic |
| Space group | P 21 21 21 |
| Unit cell dimensions | a = 7.2272(17) Å |
| b = 11.745(3) Å | |
| c = 17.177(5) Å | |
| Volume | 1458.1(7) Å 3 |
| Z | 4 |
| Density (calculated) | 1.304 Mg/m3 |
| Absorption coefficient | 0.090 mm−1 |
| F(000) | 608 |
| Crystal size | 0.34 × 0.08 × 0.06 mm3 |
| Theta range for data collection | 2.10 to 28.00° |
| Index ranges | −9 ≤ h ≤ 9, −15 ≤ k ≤ 15, −22 ≤ l ≤ 22 |
| Reflections collected | 15039 |
| Independent reflections | 2034 [R(int) = 0.0529] |
| Completeness to theta = 28.00° | 100.0% |
| Absorption correction | None |
| Refinement method | Full-matrix least-squares on F2 |
| Data / restraints / parameters | 2034 / 0 / 192 |
| Goodness-of-fit on F2 | 0.998 |
| Final R indices [for 1694 rfls with I>2sigma(I)] | R1 = 0.0419, wR2 = 0.0848 |
| R indices (all data) | R1 = 0.0562, wR2 = 0.0944 |
| Largest diff. peak and hole | 0.200 and −0.181 e. Å −3 |
| D-H···A | d(D-H) | d(H···A) | d(D···A) | <(DHA) |
|---|---|---|---|---|
| N(2)-H(2B)···O(1W)#1 | 0.86 | 1.898 | 2.730(3) | 162 |
| O(1W)-H(1)···O(1)#2 | 0.85 | 1.962 | 2.717(3) | 147 |
| O(1W)-H(2)···N(1)#3 | 0.85 | 1.977 | 2.818(3) | 170 |

| Bond lengths | X-ray | B3LYP | PBE1PBE |
|---|---|---|---|
| C(1)-C(2) | 1.383(3) | 1.403 | 1.399 |
| C(1)-C(5) | 1.392(3) | 1.402 | 1.397 |
| C(1)-C(6) | 1.504(3) | 1.493 | 1.487 |
| C(3)-N(1) | 1.342(3) | 1.352 | 1.347 |
| C(4)-N(1) | 1.338(3) | 1.350 | 1.345 |
| C(2)-C(3) | 1.382(3) | 1.392 | 1.388 |
| C(4)-C(5) | 1.388(3) | 1.395 | 1.392 |
| C(6)-O(1) | 1.236(3) | 1.255 | 1.250 |
| C(6)-N(2) | 1.342(3) | 1.378 | 1.370 |
| C(7)-N(3) | 1.290(3) | 1.301 | 1.297 |
| C(7)-C(8) | 1.458(3) | 1.467 | 1.462 |
| C(8)-C(9) | 1.398(3) | 1.410 | 1.405 |
| C(8)-C(13) | 1.402(3) | 1.411 | 1.406 |
| C(9)-C(10) | 1.382(3) | 1.386 | 1.383 |
| C(10)-C(11) | 1.410(3) | 1.419 | 1.414 |
| C(11)-N(4) | 1.370(3) | 1.384 | 1.376 |
| C(11)-C(12) | 1.415(3) | 1.418 | 1.413 |
| C(12)-C(13) | 1.385(3) | 1.389 | 1.385 |
| C(14)-N(4) | 1.454(3) | 1.464 | 1.452 |
| C(15)-N(4) | 1.453(3) | 1.464 | 1.452 |
| N(2)-N(3) | 1.398(3) | 1.388 | 1.374 |
| Bond angles | X-ray | B3LYP | PBE1PBE |
|---|---|---|---|
| C(2)-C(1)-C(6) | 118.5(2) | 116.6 | 116.2 |
| C(5)-C(1)-C(6) | 122.7(2) | 125.2 | 125.5 |
| N(1)-C(3)-C(2) | 123.7(2) | 122.9 | 123.0 |
| N(1)-C(4)-C(5) | 123.8(2) | 123.4 | 123.5 |
| O(1)-C(6)-N(2) | 125.0(2) | 117.7 | 116.8 |
| O(1)-C(6)-C(1) | 119.9(2) | 120.7 | 120.6 |
| N(2)-C(6)-C(1) | 115.1(2) | 121.6 | 121.6 |
| N(3)-C(7)-C(8) | 122.2(2) | 131.2 | 130.3 |
| C(13)-C(8)-C(7) | 122.6(2) | 124.5 | 124.0 |
| N(4)-C(11)-C(10) | 121.7(2) | 121.4 | 121.3 |
| N(4)-C(11)-C(12) | 121.2(2) | 121.4 | 121.3 |
| C(4)-N(1)-C(3) | 116.7(2) | 117.5 | 117.5 |
| C(6)-N(2)-N(3) | 119.3(2) | 124.2 | 124.4 |
| C(7)-N(3)-N(2) | 113.7(2) | 119.2 | 119.0 |
| C(11)-N(4)-C(15) | 120.6(2) | 120.3 | 120.1 |
| C(11)-N(4)-C(14) | 121.3(2) | 120.3 | 120.1 |
| C(15)-N(4)-C(14) | 118.1(2) | 119.4 | 119.7 |
2.2. IR Spectrum

| Freq. | Int. (IR) | B3LYP 6-311G** | Int. (IR) | PBE1PBE 6-311G** | Int. (IR) | Vib. |
|---|---|---|---|---|---|---|
| 3407 | m | 3490 | 30.44 | 3523 | 36.83 | νN–H |
| 3191 | m | 3157 | 18.03 | 3184 | 14.14 | νC–H (aromatic) |
| 2928 | m | 2981 | 12.33 | 3002 | 97.84 | νC–H (CH3 symmetric) |
| 1664 | s | 1633 | 181.51 | 1673 | 216.11 | νC=O |
| 1593 | s | 1617 | 168.64 | 1661 | 200.13 | νC=N |
| 1524 | s | 1577 | 16.21 | 1607 | 16.42 | νC=C (aromatic) |
| 1308 | s | 1340 | 148.77 | 1369 | 112.49 | C–H methyl in plane |
| 1055 | m | 1068 | 55.76 | 1107 | 34.71 | νN–N |
| 974 | w | 989 | 1.56 | 1009 | 1.44 | C–H C–H out of plane |
| 846 | w | 851 | 35.56 | 859 | 31.32 | C–H |
| 813 | m | 836 | 16.82 | 843 | 22.38 | C–H |
| 750 | w | 775 | 50.37 | 785 | 69.11 | C–H |
| 524 | w | 464 | 7.80 | 467 | 7.94 | C–H |
2.3. Orbital Analysis

3. Experimental, Theoretical and Computational Methods
3.1. General
3.2. Preparation of N'-(4-Dimethylaminobenzylidene)isonicotinohydrazide Monohydrate
3.3. X-ray Crystallography
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Supplementary Materials
Supplementary File 1References
- Gilchrist, T.L. Heterocylic Chemistry; John Wiley & Sons: New York, NY, USA, 1988. [Google Scholar]
- Fallas, J.A.; Gonzalez, L.; Corral, I. Density functional theory rationalization of the substituent effects in trifluoromethylpyridinol derivatives. Tetrahedron 2009, 65, 232–239. [Google Scholar] [CrossRef]
- Lizarraga, M.E.; Navarro, R.; Urriolabeitia, E.P. Synthesis and characterization of dinuclear complexes of PdII containing the (–N–C–S)2 skeleton. J. Org. Met. Chem. 1997, 542, 51–60. [Google Scholar]
- Georgopoulou, A.S.; Ulvenlund, S.; Mingos, D.M.P.; Baxter, I.; Williams, D.J. Synthesis of water soluble DTPA complexes with pendant uracil moieties capable of forming complementary hydrogen bonds. J. Chem. Soc. 1999, 4, 547–552. [Google Scholar]
- Liaw, W.; Lee, N.; Chen, C.; Lee, C.; Lee, G.; Peng, S. Dinuclear and mononuclear iron(ii)-thiolate complexes with mixed co/cn− ligands: Synthetic advances for iron sites of [Fe]-only hydrogenases. J. Am. Chem. Soc. 2000, 122, 488–494. [Google Scholar] [CrossRef]
- Trotter, P.J.; White, P.A. Resonance raman determination of the triiodide structure in bis(tetrathiotetracene)triiodide organic conductor compared with the poly(vinyl alcohol)-iodine complex. J. Appl. Spectrosc. 1978, 32, 323–324. [Google Scholar] [CrossRef]
- Rajpure, K.Y.; Bhosale, C.H. Sb2S3 semiconductor-septum rechargeable storage cell. J. Mater. Chem. Phys. 2000, 64, 70–74. [Google Scholar] [CrossRef]
- Licht, S. Electrolyte modified photoelectrochemical solar cells. Solar Energy Mater. Solar Cell. 1995, 38, 305–319. [Google Scholar] [CrossRef]
- Altenburger, J.M.; Lassalle, G.Y.; Matrougui, M.; Galtier, D.; Jetha, J.C.; Bocskei, Z.; Berry, C.N.; Lunven, C.; Lorrain, J.; Herault, J.P.; et al. SSR182289A, a selective and potent orally active thrombin inhibitor. Bioorg. Med. Chem. Lett. 2004, 12, 1713–1730. [Google Scholar] [CrossRef]
- Gallego, M.; García-Vargas, M.; Valcarcel, M. Pyridine-2-carbaldehyde 2-hydroxybenzoylhydrazone as a selective reagent for the extraction and spectrophotometric determination of iron(II). Analyst 1979, 104, 613–619. [Google Scholar] [CrossRef]
- Gallego, M.; Garcı´a-Vargas, M.; Pino, F.; Valcarcel, M. Analytical applications of picolinealdehyde salicyloylhydrazone: Spectrophotometric determination of nickel and zinc. Microchem. J. 1978, 23, 353–359. [Google Scholar] [CrossRef]
- Jayabalakrishnan, C.; Natarajan, K. Synthesis, characterization, and biological activities of ruthenium(II) carbonyl complexes containing bifunctional tridentate schiff bases. Synth. React. Inorg. Met.-Org. Chem. 2001, 31, 983–995. [Google Scholar] [CrossRef]
- Maiti, A.; Ghosh, S. Synthesis and reactivity of some octa coordinated dioxouranium(VI) complexes of diacetyl bis(benzoyl-hydrazone) and benzil bis(benzoyl hydrazone). Indian J. Chem. 1989, 29, 980–983. [Google Scholar]
- Mishra, A.P.; Soni, M. Synthesis, structural, and biological studies of some schiff bases and their metal complexes. Metal-Based Drugs 2008, 2008. 7 pages. [Google Scholar]
- Ainscough, E.W.; Brodie, A.M.; Dobbs, A.J.; Ranford, J.D. Antitumour copper(II) salicylaldehyde benzoylhydrazone (H2sb) complexes: Physicochemical properties and the single-crystal X-ray structures of [{Cu(H2sb)(CCl3CO2)2}2] and [{Cu(Hsb) (ClO4) (C2H5OH)}2] and the related salicylaldehyde acetylhydrazone (H2sa) complex, [Cu(Hsa)Cl(H2O)]·h2O. Inorg. Chim. Acta 1998, 267, 27–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-97, Program for Solution Crystal Structure; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL-97, Program for Solution Crystal Structure and Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; et al. GASSIAN 98 (Revision A. 3); Gaussian Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5653. [Google Scholar] [CrossRef]
- Sample Availability: Available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sahebalzamani, H.; Khaligh, N.; Ghammamy, S.; Salimi, F.; Mehrani, K. Crystal Structure and Density Functional Theory Study on Structural Properties and Energies of a Isonicotinohydrazide Compound. Molecules 2011, 16, 7715-7724. https://doi.org/10.3390/molecules16097715
Sahebalzamani H, Khaligh N, Ghammamy S, Salimi F, Mehrani K. Crystal Structure and Density Functional Theory Study on Structural Properties and Energies of a Isonicotinohydrazide Compound. Molecules. 2011; 16(9):7715-7724. https://doi.org/10.3390/molecules16097715
Chicago/Turabian StyleSahebalzamani, Hajar, Nina Khaligh, Shahriar Ghammamy, Farshid Salimi, and Kheyrollah Mehrani. 2011. "Crystal Structure and Density Functional Theory Study on Structural Properties and Energies of a Isonicotinohydrazide Compound" Molecules 16, no. 9: 7715-7724. https://doi.org/10.3390/molecules16097715