Exploiting the Nucleotide Substrate Specificity of Repair DNA Polymerases To Develop Novel Anticancer Agents

Abstract
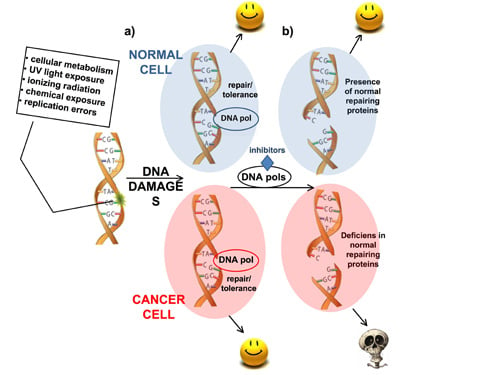
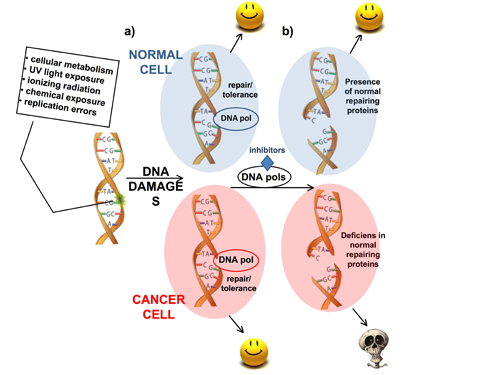
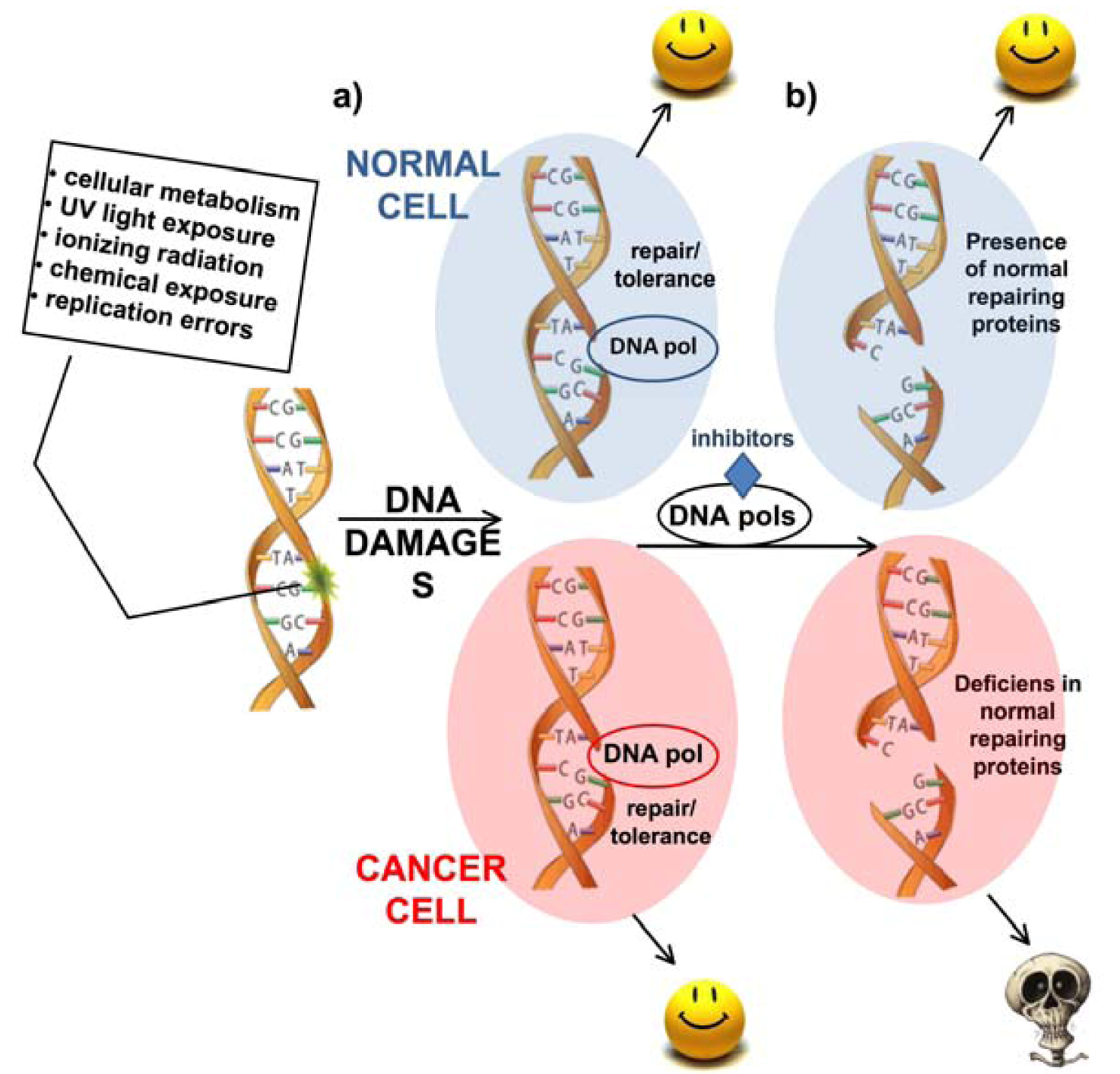
:
1. DNA Damage Response
1.1. Defective DDR in Cancer
1.2. Targeting DDR in Cancer Treatment: The “Synthetic Lethality Model”
2. DNA Polymerases as Anticancer Drug Targets

{kind=link}
{kind=link}
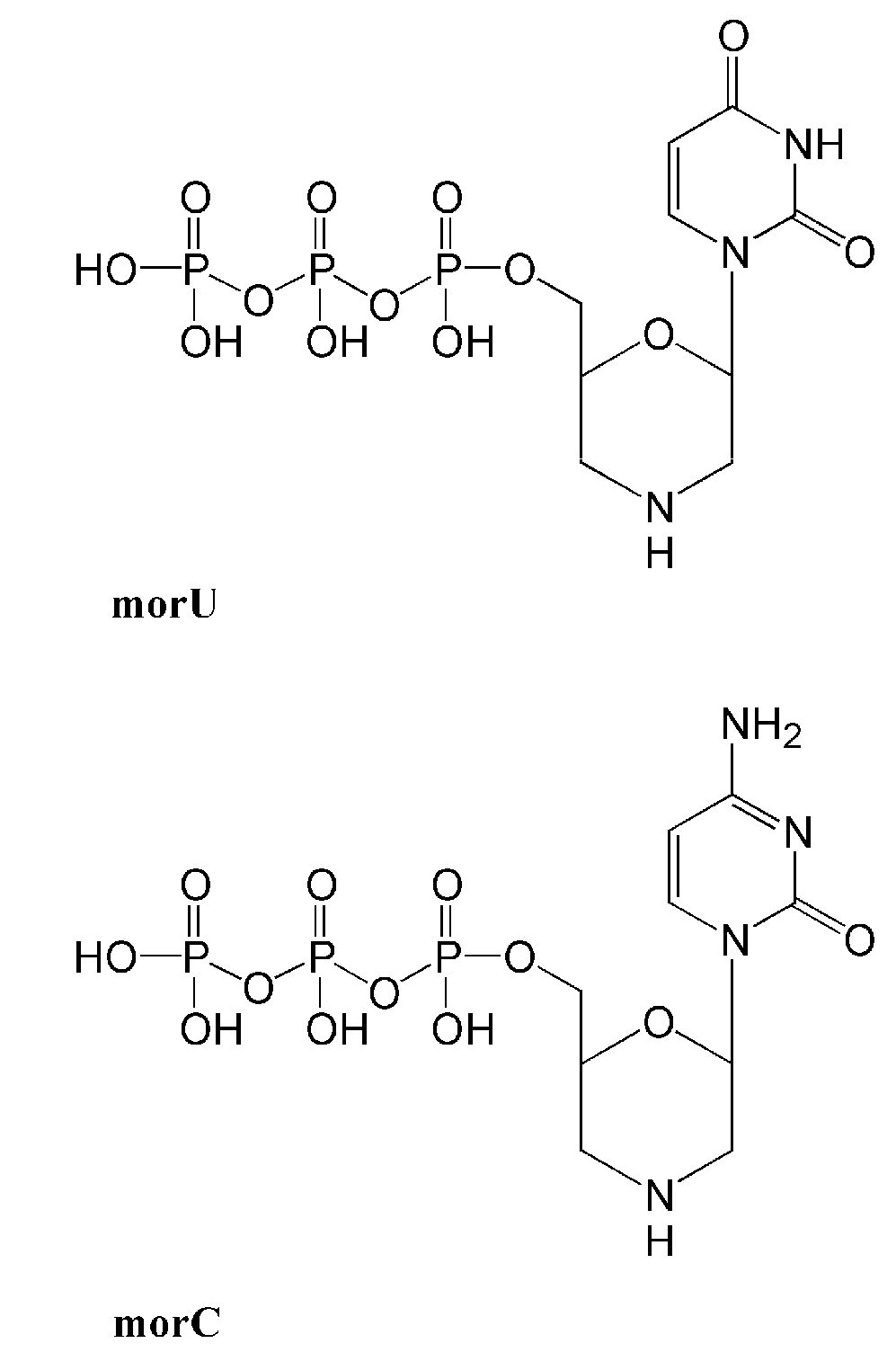{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA repair pathways | DNA lesions recognized and removed | DNA polymerases involved | Related diseases |
|---|---|---|---|
| NUCLEOTIDE EXCISION REPAIR (NER) | Bulky lesions: thymidine dimers, pyrimidine dimers, single-strand breaks | pol α, pol β, pol δ, pol ε, pol κ, pol η | Xeroderma Pigmentosum (XP), Cockayne Syndrome (CS), trichothiodystrophy |
| MISMATCH REPAIR (MMR) | Base-base mismatches | pol β, pol δ | Hereditary nonpolyposis colorectal cancer (HNPCC), sporadic cancer (colorectal, gastric, endometrial, cervical, ovarian, breast, lung, bladder), gliomas, leukemia and lymphomas |
| BASE EXCISION REPAIR (BER) | Non-bulky lesions: base modifications by alkylation and oxidation, single strand breaks (SSBs) | pol β, pol λ, pol δ, pol ε | Solid tumors, chronic myeloid leukemia |
| HOMOLOGOUS RECOMBINATION (HR) | DNA gaps, DNA double strand breaks (DSBs), DNA interstrand crosslinks | pol δ, pol ε | Breast cancer, ovarian cancer, Fanconi anemia |
| NON-HOMOLOGOUS END JOINING (NHEJ) | Double strand breaks (DSBs) | pol μ, pol λ, Terminal transferase (TdT), pol η | Leukemias |
| TRANSLESION SYNTESIS (TLS) | Abasic sites, bulky DNA template adducts, thymidine-thymidine or cyclobutane-pyrimidine dimers, cis-platinum adducts. | TLS polymerases η, ι, κ, ζ, Rev1 | Xeroderma pigmentosum-variant (XPV) |

2.1. DNA Pol λ in Mammalian Cells
2.2. DNA Pol β in Mammalian Cells
3. Nucleoside Analogs as DNA Pol β and λ Inhibitors
3.1. Nucleotide Analogs Cytotoxicity and Metabolism
3.1.1. Substrate Specificity of Human DNA Pols towards Different Nucleotide Analogs

3.1.2. Nucleoside Analogs Interactions with Other Intracellular Targets
3.2. Mono-, Di- and Triphosphate Analogs as Drugs
3.2.1. dNTPs Modified at the γ-Phosphorus

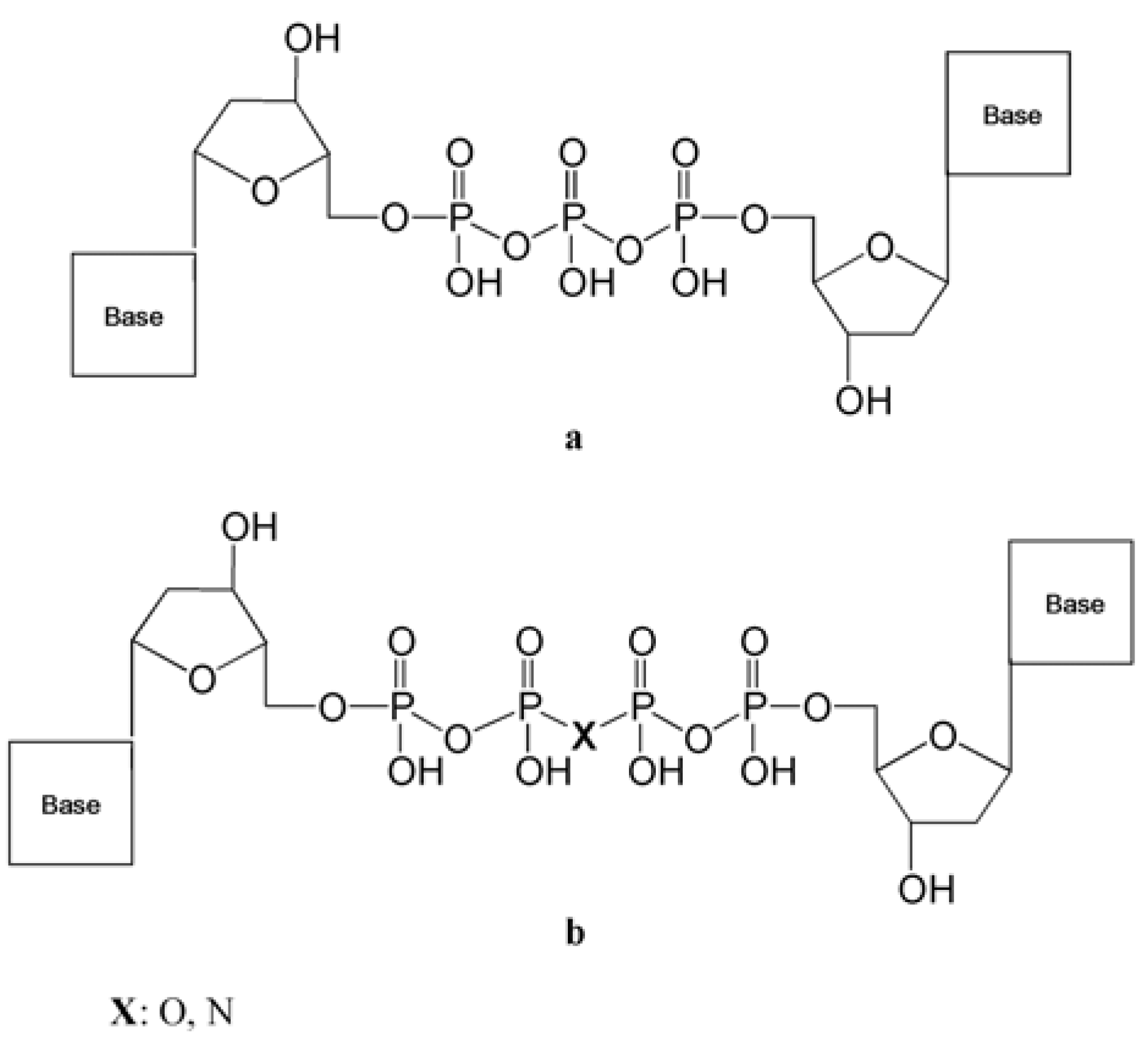
3.2.2. Dinucleoside Tetraphosphates

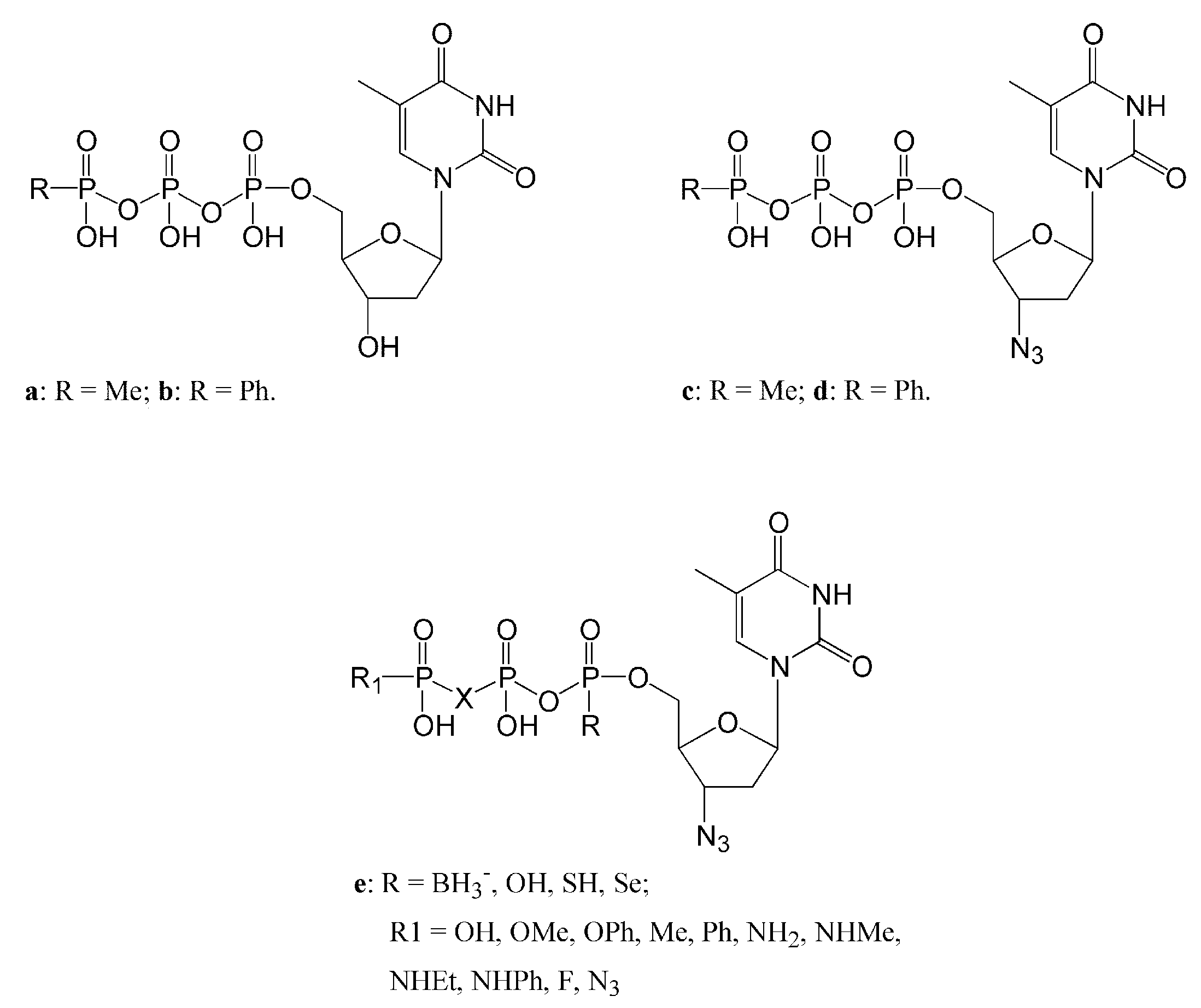
3.2.3. dNTPs Modified at the α-Phosphorus and β,γ Bridging Analogs
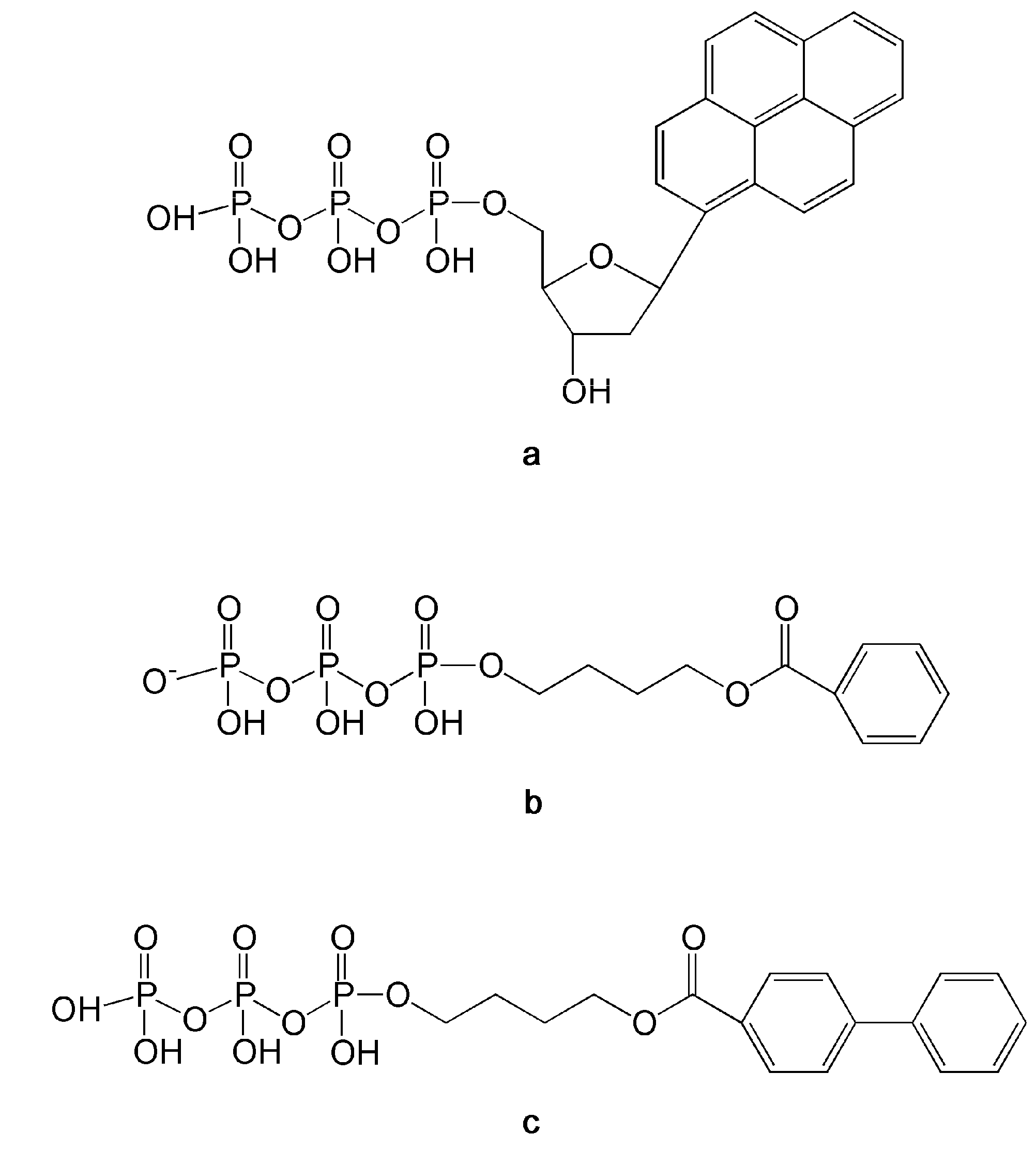
3.3. Nucleotide Analogs Targeting Specific DNA Lesions

3.4. Strategies to Deliver Mono-, Di- and Triphosphate Analogs
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Lord, C.J.; Garrett, M.D.; Ashworth, A. Targeting the double-strand DNA break repair pathway as a therapeutic strategy. Clin. Cancer Res. 2006, 12, 4463–4468. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef]
- de Winter, J.P.; Joenje, H. The genetic and molecular basis of Fanconi anemia. Mutat. Res. 2009, 668, 11–19. [Google Scholar] [CrossRef]
- English, J.S.; Swerdlow, A.J. The risk of malignant melanoma, internal malignancy and mortality in xeroderma pigmentosum patients. Br. J. Dermatol. 1987, 117, 457–461. [Google Scholar] [CrossRef]
- Chompret, A. The Li-Fraumeni syndrome. Biochimie 2002, 84, 75–82. [Google Scholar] [CrossRef]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef]
- Fackenthal, J.D.; Olopade, O.I. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat. Rev. Cancer 2007, 7, 937–948. [Google Scholar] [CrossRef]
- Dobzhansky, T. Genetics of natural populations. Xiii. Recombination and variability in populations of Drosophila pseudoobscura. Genetics 1946, 31, 269–290. [Google Scholar]
- Lucchesi, J.C. Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanogaster. Genetics 1968, 59, 37–44. [Google Scholar]
- Hartwell, L.H.; Szankasi, P.; Roberts, C.J.; Murray, A.W.; Friend, S.H. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997, 278, 1064–1068. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Virag, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef]
- Southan, G.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibitors. Curr. Med. Chem. 2003, 10, 321–340. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar]
- Hubscher, U.; Maga, G.; Spadari, S. Eukaryotic DNA polymerases. Annu. Rev. Biochem. 2002, 71, 133–163. [Google Scholar] [CrossRef]
- Colussi, C.; Parlanti, E.; Degan, P.; Aquilina, G.; Barnes, D.; Macpherson, P.; Karran, P.; Crescenzi, M.; Dogliotti, E.; Bignami, M. The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr. Biol. 2002, 12, 912–918. [Google Scholar] [CrossRef]
- Macpherson, P.; Barone, F.; Maga, G.; Mazzei, F.; Karran, P.; Bignami, M. 8-oxoguanine incorporation into DNA repeats in vitro and mismatch recognition by MutSalpha. Nucleic Acids Res. 2005, 33, 5094–5105. [Google Scholar] [CrossRef]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef]
- Morrison, A.; Johnson, A.L.; Johnston, L.H.; Sugino, A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993, 12, 1467–1473. [Google Scholar]
- Martin, S.A.; McCabe, N.; Mullarkey, M.; Cummins, R.; Burgess, D.J.; Nakabeppu, Y.; Oka, S.; Kay, E.; Lord, C.J.; Ashworth, A. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell 2010, 17, 235–248. [Google Scholar] [CrossRef]
- Albertella, M.R.; Lau, A.; O’Connor, M.J. The overexpression of specialized DNA polymerases in cancer. DNA Repair (Amst.) 2005, 4, 583–593. [Google Scholar] [CrossRef]
- Frechet, M.; Canitrot, Y.; Bieth, A.; Dogliotti, E.; Cazaux, C.; Hoffmann, J.S. Deregulated DNA polymerase beta strengthens ionizing radiation-induced nucleotidic and chromosomal instabilities. Oncogene 2002, 21, 2320–2327. [Google Scholar] [CrossRef]
- Frechet, M.; Canitrot, Y.; Cazaux, C.; Hoffmann, J.S. DNA polymerase beta imbalance increases apoptosis and mutagenesis induced by oxidative stress. FEBS Lett. 2001, 505, 229–232. [Google Scholar] [CrossRef]
- Yang, J.; Chen, Z.; Liu, Y.; Hickey, R.J.; Malkas, L.H. Altered DNA polymerase iota expression in breast cancer cells leads to a reduction in DNA replication fidelity and a higher rate of mutagenesis. Cancer Res. 2004, 64, 5597–5607. [Google Scholar] [CrossRef]
- Maga, G.; Villani, G.; Crespan, E.; Wimmer, U.; Ferrari, E.; Bertocci, B.; Hubscher, U. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature 2007, 447, 606–608. [Google Scholar]
- Zhang, Y.; Yuan, F.; Wu, X.; Taylor, J.S.; Wang, Z. Response of human DNA polymerase iota to DNA lesions. Nucleic Acids Res. 2001, 29, 928–935. [Google Scholar] [CrossRef]
- Ramadan, K.; Shevelev, I.; Hubscher, U. The DNA-polymerase-X family: Controllers of DNA quality? Nat. Rev. Mol. Cell Biol. 2004, 5, 1038–1043. [Google Scholar] [CrossRef]
- Wimmer, U.; Ferrari, E.; Hunziker, P.; Hubscher, U. Control of DNA polymerase [lambda] stability by phosphorylation and ubiquitination during the cell cycle. EMBO Rep. 2008, 9, 1027–1033. [Google Scholar] [CrossRef]
- Fan, W.; Wu, X. DNA polymerase lambda can elongate on DNA substrates mimicking non-homologous end joining and interact with XRCC4-ligase IV complex. Biochem. Biophys. Res. Commun. 2004, 323, 1328–1333. [Google Scholar] [CrossRef]
- Garcia-Diaz, M.; Bebenek, K.; Kunkel, T.A.; Blanco, L. Identification of an intrinsic 5'-deoxyribose-5-phosphate lyase activity in human DNA polymerase lambda: A possible role in base excision repair. J. Biol. Chem. 2001, 276, 34659–34663. [Google Scholar]
- Maga, G.; Hubscher, U. Repair and translesion DNA polymerases as anticancer drug targets. Anticancer Agents Med. Chem. 2008, 8, 431–447. [Google Scholar]
- Bebenek, K.; Garcia-Diaz, M.; Blanco, L.; Kunkel, T.A. The frameshift infidelity of human DNA polymerase lambda. Implications for function. J. Biol. Chem. 2003, 278, 34685–34690. [Google Scholar]
- Maga, G.; Ramadan, K.; Locatelli, G.A.; Shevelev, I.; Spadari, S.; Hubscher, U. DNA elongation by the human DNA polymerase lambda polymerase and terminal transferase activities are differentially coordinated by proliferating cell nuclear antigen and replication protein A. J. Biol. Chem. 2005, 280, 1971–1981. [Google Scholar]
- Ramadan, K.; Maga, G.; Shevelev, I.V.; Villani, G.; Blanco, L.; Hubscher, U. Human DNA polymerase lambda possesses terminal deoxyribonucleotidyl transferase activity and can elongate RNA primers: Implications for novel functions. J. Mol. Biol. 2003, 328, 63–72. [Google Scholar] [CrossRef]
- Amoroso, A.; Concia, L.; Maggio, C.; Raynaud, C.; Bergounioux, C.; Crespan, E.; Cella, R.; Maga, G. Oxidative DNA damage bypass in Arabidopsis thaliana requires DNA polymerase lambda and proliferating cell nuclear antigen 2. Plant Cell 2011, 23, 806–822. [Google Scholar] [CrossRef]
- Amoroso, A.; Crespan, E.; Wimmer, U.; Hubscher, U.; Maga, G. DNA polymerases and oxidative damage: Friends or foes? Curr. Mol. Pharmacol. 2008, 1, 162–170. [Google Scholar]
- Maga, G.; Crespan, E.; Wimmer, U.; van Loon, B.; Amoroso, A.; Mondello, C.; Belgiovine, C.; Ferrari, E.; Locatelli, G.; Villani, G.; et al. Replication protein A and proliferating cell nuclear antigen coordinate DNA polymerase selection in 8-oxo-guanine repair. Proc. Natl. Acad. Sci. USA 2008, 105, 20689–20694. [Google Scholar]
- Markkanen, E.; van Loon, B.; Ferrari, E.; Hubscher, U. Ubiquitylation of DNA polymerase lambda. FEBS Lett. 2011, 585, 2826–2830. [Google Scholar] [CrossRef]
- van Loon, B.; Hubscher, U. An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase lambda. Proc. Natl. Acad. Sci. USA 2009, 106, 18201–18206. [Google Scholar] [CrossRef] [Green Version]
- Idriss, H.T.; Al-Assar, O.; Wilson, S.H. DNA polymerase beta. Int. J. Biochem. Cell Biol. 2002, 34, 321–324. [Google Scholar] [CrossRef]
- Vaisman, A.; Chaney, S.G. The efficiency and fidelity of translesion synthesis past cisplatin and oxaliplatin GpG adducts by human DNA polymerase beta. J. Biol. Chem. 2000, 275, 13017–13025. [Google Scholar] [CrossRef]
- Efrati, E.; Tocco, G.; Eritja, R.; Wilson, S.H.; Goodman, M.F. Abasic translesion synthesis by DNA polymerase beta violates the “A-rule”. Novel types of nucleotide incorporation by human DNA polymerase beta at an abasic lesion in different sequence contexts. J. Biol. Chem. 1997, 272, 2559–2569. [Google Scholar]
- Louat, T.; Servant, L.; Rols, M.P.; Bieth, A.; Teissie, J.; Hoffmann, J.S.; Cazaux, C. Antitumor activity of 2',3'-dideoxycytidine nucleotide analog against tumors up-regulating DNA polymerase beta. Mol. Pharmacol. 2001, 60, 553–558. [Google Scholar]
- Bergoglio, V.; Frechet, M.; Philippe, M.; Bieth, A.; Mercier, P.; Morello, D.; Lacroix-Tricki, M.; Delsol, G.; Hoffmann, J.S.; Cazaux, C. Evidence of finely tuned expression of DNA polymerase beta in vivo using transgenic mice. FEBS Lett. 2004, 566, 147–150. [Google Scholar] [CrossRef]
- Cabelof, D.C.; Guo, Z.; Raffoul, J.J.; Sobol, R.W.; Wilson, S.H.; Richardson, A.; Heydari, A.R. Base excision repair deficiency caused by polymerase beta haploinsufficiency: Accelerated DNA damage and increased mutational response to carcinogens. Cancer Res. 2003, 63, 5799–5807. [Google Scholar]
- Parsons, J.L.; Tait, P.S.; Finch, D.; Dianova, I.I.; Edelmann, M.J.; Khoronenkova, S.V.; Kessler, B.M.; Sharma, R.A.; McKenna, W.G.; Dianov, G.L. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009, 28, 3207–3215. [Google Scholar] [CrossRef]
- Yang, J.; Parsons, J.; Nicolay, N.H.; Caporali, S.; Harrington, C.F.; Singh, R.; Finch, D.; D’Atri, S.; Farmer, P.B.; Johnston, P.G.; et al. Cells deficient in the base excision repair protein, DNA polymerase beta, are hypersensitive to oxaliplatin chemotherapy. Oncogene 2010, 29, 463–468. [Google Scholar]
- Hubscher, U.; Spadari, S. DNA replication and chemotherapy. Physiol. Rev. 1994, 74, 259–304. [Google Scholar] [CrossRef]
- Balzarini, J.; van Aerschot, A.; Pauwels, R.; Baba, M.; Schols, D.; Herdewijn, P.; de Clercq, E. 5-Halogeno-3'-fluoro-2',3'-dideoxyuridines as inhibitors of human immunodeficiency virus (HIV): Potent and selective anti-HIV activity of 3'-fluoro-2',3'-dideoxy-5-chlorouridine. Mol. Pharmacol. 1989, 35, 571–577. [Google Scholar]
- Cheng, Y.C.; Dutschman, G.E.; Bastow, K.F.; Sarngadharan, M.G.; Ting, R.Y. Human immunodeficiency virus reverse transcriptase. General properties and its interactions with nucleoside triphosphate analogs. J. Biol. Chem. 1987, 262, 2187–2189. [Google Scholar]
- Brown, J.A.; Pack, L.R.; Fowler, J.D.; Suo, Z. Pre-steady-state kinetic analysis of the incorporation of anti-HIV nucleotide analogs catalyzed by human X- and Y-family DNA polymerases. Antimicrob. Agents Chemother. 2011, 55, 276–283. [Google Scholar]
- Feng, J.Y.; Johnson, A.A.; Johnson, K.A.; Anderson, K.S. Insights into the molecular mechanism of mitochondrial toxicity by AIDS drugs. J. Biol. Chem. 2001, 276, 23832–23837. [Google Scholar]
- Bouayadi, K.; Hoffmann, J.S.; Fons, P.; Tiraby, M.; Reynes, J.P.; Cazaux, C. Overexpression of DNA polymerase beta sensitizes mammalian cells to 2',3'-deoxycytidine and 3'-azido-3'-deoxythymidine. Cancer Res. 1997, 57, 110–116. [Google Scholar]
- Skuta, G.; Fischer, G.M.; Janaky, T.; Kele, Z.; Szabo, P.; Tozser, J.; Sumegi, B. Molecular mechanism of the short-term cardiotoxicity caused by 2',3'-dideoxycytidine (ddC): Modulation of reactive oxygen species levels and ADP-ribosylation reactions. Biochem. Pharmacol. 1999, 58, 1915–1925. [Google Scholar]
- Cherrington, J.M.; Allen, S.J.; McKee, B.H.; Chen, M.S. Kinetic analysis of the interaction between the diphosphate of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, ddCTP, AZTTP, and FIAUTP with human DNA polymerases beta and gamma. Biochem. Pharmacol. 1994, 48, 1986–1988. [Google Scholar]
- Hart, G.J.; Orr, D.C.; Penn, C.R.; Figueiredo, H.T.; Gray, N.M.; Boehme, R.E.; Cameron, J.M. Effects of (-)-2'-deoxy-3'-thiacytidine (3TC) 5'-triphosphate on human immunodeficiency virus reverse transcriptase and mammalian DNA polymerases alpha, beta, and gamma. Antimicrob. Agents Chemother. 1992, 36, 1688–1694. [Google Scholar] [CrossRef]
- Faderl, S.; Gandhi, V.; Kantarjian, H.M. Potential role of novel nucleoside analogs in the treatment of acute myeloid leukemia. Curr. Opin. Hematol. 2008, 15, 101–107. [Google Scholar] [CrossRef]
- Burnett, A.K.; Milligan, D.; Prentice, A.G.; Goldstone, A.H.; McMullin, M.F.; Hills, R.K.; Wheatley, K. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer 2007, 109, 1114–1124. [Google Scholar] [CrossRef]
- Crino, L.; Mosconi, A.M.; Scagliotti, G.; Selvaggi, G.; Novello, S.; Rinaldi, M.; Della, G.M.; Gridelli, C.; Rossi, A.; Calandri, C.; et al. Gemcitabine as second-line treatment for advanced non-small-cell lung cancer: A phase II trial. J. Clin. Oncol. 1999, 17, 2081–2085. [Google Scholar]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar]
- Tempero, M.; Plunkett, W.; van Haperen, V.R.; Hainsworth, J.; Hochster, H.; Lenzi, R.; Abbruzzese, J. Randomized phase II comparison of dose-intense gemcitabine: Thirty-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J. Clin. Oncol. 2003, 21, 3402–3408. [Google Scholar] [CrossRef]
- Hensley, M.L.; Correa, D.D.; Thaler, H.; Wilton, A.; Venkatraman, E.; Sabbatini, P.; Chi, D.S.; Dupont, J.; Spriggs, D.; Aghajanian, C. Phase I/II study of weekly paclitaxel plus carboplatin and gemcitabine as first-line treatment of advanced-stage ovarian cancer: Pathologic complete response and longitudinal assessment of impact on cognitive functioning. Gynecol. Oncol. 2006, 102, 270–277. [Google Scholar] [CrossRef]
- Carmichael, J.; Walling, J. Phase II activity of gemcitabine in advanced breast cancer. Semin. Oncol. 1996, 23, 77–81. [Google Scholar]
- Morandi, P. Biological agents and gemcitabine in the treatment of breast cancer. Ann. Oncol. 2006, 17, v177–v180. [Google Scholar] [CrossRef]
- Nabhan, C.; Krett, N.; Gandhi, V.; Rosen, S. Gemcitabine in hematologic malignancies. Curr. Opin. Oncol. 2001, 13, 514–521. [Google Scholar] [CrossRef]
- Iwasaki, H.; Huang, P.; Keating, M.J.; Plunkett, W. Differential incorporation of ara-C, gemcitabine, and fludarabine into replicating and repairing DNA in proliferating human leukemia cells. Blood 1997, 90, 270–278. [Google Scholar]
- Crul, M.; van Waardenburg, R.C.A.M.; Bocxe, S.; van Eijndhoven, M.A.J.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.M. DNA repair mechanisms involved in gemcitabine cytotoxicity and in the interaction between gemcitabine and cisplatin. Biochem. Pharmacol. 2003, 65, 275–282. [Google Scholar]
- Prakasha Gowda, A.S.; Polizzi, J.M.; Eckert, K.A.; Spratt, T.E. Incorporation of Gemcitabine and Cytarabine into DNA by DNA Polymerase β and Ligase III/XRCC1. Biochemistry 2010, 49, 4833–4840. [Google Scholar]
- Cavanaugh, N.A.; Beard, W.A.; Wilson, S.H. DNA polymerase beta ribonucleotide discrimination: Insertion, misinsertion, extension, and coding. J. Biol. Chem. 2010, 285, 24457–24465. [Google Scholar]
- Garcia-Diaz, M.; Murray, M.S.; Kunkel, T.A.; Chou, K.M. Interaction between DNA polymerase lambda and anticancer nucleoside analogs. J. Biol. Chem. 2010, 285, 16874–16879. [Google Scholar] [CrossRef]
- Lebedeva, N.A.; Seredina, T.A.; Silnikov, V.N.; Abramova, T.V.; Levina, A.S.; Khodyreva, S.N.; Rechkunova, N.I.; Lavrik, O.I. Analysis of interactions of DNA polymerase beta and reverse transcriptases of human immunodeficiency and mouse leukemia viruses with dNTP analogs containing a modified sugar residue. Biochemistry (Mosc.) 2005, 70, 1–7. [Google Scholar]
- Blanca, G.; Shevelev, I.; Ramadan, K.; Villani, G.; Spadari, S.; Hubscher, U.; Maga, G. Human DNA polymerase lambda diverged in evolution from DNA polymerase beta toward specific Mn(++) dependence: A kinetic and thermodynamic study. Biochemistry 2003, 42, 7467–7476. [Google Scholar]
- Berdis, A.J. DNA polymerases as therapeutic targets. Biochemistry 2008, 47, 8253–8260. [Google Scholar] [CrossRef]
- Devadoss, B.; Berdis, A.J. Non-Natural nucleotide analogs as probes of DNA polymerase activity. Curr. Chem. Biol. 2007, 1, 241–264. [Google Scholar]
- Robak, T.; Korycka, A.; Lech-Maranda, E.; Robak, P. Current status of older and new purine nucleoside analogues in the treatment of lymphoproliferative diseases. Molecules 2009, 14, 1183–1226. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Sugawara, F. New cancer chemotherapy agents: Inhibitors of DNA polymerase. Curr. Drug Ther. 2008, 3, 44–53. [Google Scholar] [CrossRef]
- Lim, S.E.; Ponamarev, M.V.; Longley, M.J.; Copeland, W.C. Structural determinants in human DNA polymerase gamma account for mitochondrial toxicity from nucleoside analogs. J. Mol. Biol. 2003, 329, 45–57. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kennedy, W.D.; Yin, Y.W. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 2009, 139, 312–324. [Google Scholar] [CrossRef]
- Pelletier, H.; Sawaya, M.R.; Kumar, A.; Wilson, S.H.; Kraut, J. Structures of ternary complexes of rat DNA polymerase beta, a DNA template-primer, and ddCTP. Science 1994, 264, 1891–1903. [Google Scholar]
- Joyce, C.M.; Benkovic, S.J. DNA polymerase fidelity: Kinetics, structure, and checkpoints. Biochemistry 2004, 43, 14317–14324. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Prasad, R.; Wilson, S.H.; Kraut, J.; Pelletier, H. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: Evidence for an induced fit mechanism. Biochemistry 1997, 36, 11205–11215. [Google Scholar] [CrossRef]
- Hübscher, U.; Spadari, S.; Villani, G.; Maga, G. DNA Polymerases: Discovery, Characterization and Functions in Cellular DNA Transactions; World Scientific: Singapore, 2010; pp. 11–152. [Google Scholar]
- Belt, J.A.; Marina, N.M.; Phelps, D.A.; Crawford, C.R. Nucleoside transport in normal and neoplastic cells. Adv. Enzyme Regul. 1993, 33, 235–252. [Google Scholar] [CrossRef]
- Griffith, D.A.; Jarvis, S.M. Nucleoside and nucleobase transport systems of mammalian cells. Biochim. Biophys. Acta (BBA) 1996, 1286, 153–181. [Google Scholar] [CrossRef]
- Lamba, J.K. Genetic factors influencing cytarabine therapy. Pharmacogenomics 2009, 10, 1657–1674. [Google Scholar] [CrossRef]
- Capizzi, R.L.; Yang, J.L.; Cheng, E.; Bjornsson, T.; Sahasrabudhe, D.; Tan, R.S.; Cheng, Y.C. Alteration of the pharmacokinetics of high-dose ara-C by its metabolite, high ara-U in patients with acute leukemia. J. Clin. Oncol. 1983, 1, 763–771. [Google Scholar]
- Mackey, J.R.; Mani, R.S.; Selner, M.; Mowles, D.; Young, J.D.; Belt, J.A.; Crawford, C.R.; Cass, C.E. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998, 58, 4349–4357. [Google Scholar]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17, v7–v12. [Google Scholar] [CrossRef]
- Bergman, A.M.; Pinedo, H.M.; Peters, G.J. Determinants of resistance to 2',2'-difluorodeoxycytidine (gemcitabine). Drug Resist. Updat. 2002, 5, 19–33. [Google Scholar] [CrossRef]
- Hunsucker, S.A.; Mitchell, B.S.; Spychala, J. The 5'-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol. Ther. 2005, 107, 1–30. [Google Scholar] [CrossRef]
- Dumontet, C.; Fabianowska-Majewska, K.; Mantincic, D.; Callet Bauchu, E.; Tigaud, I.; Gandhi, V.; Lepoivre, M.; Peters, G.J.; Rolland, M.O.; Wyczechowska, D.; et al. Common resistance mechanisms to deoxynucleoside analogues in variants of the human erythroleukaemic line K562. Br. J. Haematol. 1999, 106, 78–85. [Google Scholar] [CrossRef]
- Hunsucker, S.A.; Spychala, J.; Mitchell, B.S. Human cytosolic 5'-nucleotidase I: Characterization and role in nucleoside analog resistance. J. Biol. Chem. 2001, 276, 10498–10504. [Google Scholar]
- Schröder, J.K.; Kirch, C.; Seeber, S.; Schütte, J. Structural and functional analysis of the cytidine deaminase gene in patients with acute myeloid leukaemia. Br. J. Haematol. 1998, 103, 1096–1103. [Google Scholar] [CrossRef]
- Ueno, H.; Kiyosawa, K.; Kaniwa, N. Pharmacogenomics of gemcitabine: Can genetic studies lead to tailor-made therapy? Br. J. Cancer 2007, 97, 145–151. [Google Scholar] [CrossRef]
- Huang, P.; Chubb, S.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Action of 2',2'-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991, 51, 6110–6117. [Google Scholar]
- Gandhi, V.; Legha, J.; Chen, F.; Hertel, L.W.; Plunkett, W. Excision of 2',2'-difluorodeoxycytidine (gemcitabine) monophosphate residues from DNA. Cancer Res. 1996, 56, 4453–4459. [Google Scholar]
- Schy, W.E.; Hertel, L.W.; Kroin, J.S.; Bloom, L.B.; Goodman, M.F.; Richardson, F.C. Effect of a template-located 2',2'-difluorodeoxycytidine on the kinetics and fidelity of base insertion by Klenow (3'→5'exonuclease-) fragment. Cancer Res. 1993, 53, 4582–4587. [Google Scholar]
- Ruiz van Haperen, V.W.; Veerman, G.; Vermorken, J.B.; Peters, G.J. 2',2'-Difluoro-deoxycytidine (gemcitabine) incorporation into RNA and DNA of tumour cell lines. Biochem. Pharmacol. 1993, 46, 762–766. [Google Scholar]
- Baker, C.H.; Banzon, J.; Bollinger, J.M.; Stubbe, J.; Samano, V.; Robins, M.J.; Lippert, B.; Jarvi, E.; Resvick, R. 2'-Deoxy-2'-methylenecytidine and 2'-deoxy-2',2'-difluorocytidine 5'-diphosphates: Potent mechanism-based inhibitors of ribonucleotide reductase. J. Med. Chem. 1991, 34, 1879–1884. [Google Scholar] [CrossRef]
- Heinemann, V.; Xu, Y.Z.; Chubb, S.; Sen, A.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2',2'-difluorodeoxycytidine. Mol. Pharmacol. 1990, 38, 567–572. [Google Scholar]
- Momparler, R.L.; Fischer, G.A. Mammalian deoxynucleoside kinase. I. Deoxycytidine kinase: Purification, properties, and kinetic studies with cytosine arabinoside. J. Biol. Chem. 1968, 243, 4298–4304. [Google Scholar]
- Avramis, V.I.; Nandy, P.; Kwock, R.; Solorzano, M.M.; Mukherjee, S.K.; Danenberg, P.; Cohen, L.J. Increased p21/WAF-1 and p53 protein levels following sequential three drug combination regimen of fludarabine, cytarabine and docetaxel induces apoptosis in human leukemia cells. Anticancer Res. 1998, 18, 2327–2338. [Google Scholar]
- Gandhi, V.; Estey, E.; Du, M.; Keating, M.J.; Plunkett, W. Minimum dose of fludarabine for the maximal modulation of 1-beta-D-arabinofuranosylcytosine triphosphate in human leukemia blasts during therapy. Clin. Cancer Res. 1997, 3, 1539–1545. [Google Scholar]
- Gandhi, V.; Estey, E.; Du, M.; Nowak, B.; Keating, M.J.; Plunkett, W. Modulation of the cellular metabolism of cytarabine and fludarabine by granulocyte-colony-stimulating factor during therapy of acute myelogenous leukemia. Clin. Cancer Res. 1995, 1, 169–178. [Google Scholar]
- Gandhi, V.; Kemena, A.; Keating, M.J.; Plunkett, W. Fludarabine infusion potentiates arabinosylcytosine metabolism in lymphocytes of patients with chronic lymphocytic leukemia. Cancer Res. 1992, 52, 897–903. [Google Scholar]
- Sigmond, J.; Kamphuis, J.A.; Laan, A.C.; Hoebe, E.K.; Bergman, A.M.; Peters, G.J. The synergistic interaction of gemcitabine and cytosine arabinoside with the ribonucleotide reductase inhibitor triapine is schedule dependent. Biochem. Pharmacol. 2007, 73, 1548–1557. [Google Scholar]
- Gandhi, V.V.; Samuels, D.C. A review comparing deoxyribonucleoside triphosphate (dNTP) concentrations in the mitochondrial and cytoplasmic compartments of normal and transformed cells. Nucleosides Nucleotides Nucleic Acids 2011, 30, 317–339. [Google Scholar] [CrossRef]
- de Clercq, E. Antiviral drugs: Current state of the art. J. Clin. Virol. 2001, 22, 73–89. [Google Scholar] [CrossRef]
- de Clercq, E. The clinical potential of the acyclic (and cyclic) nucleoside phosphonates. The magic of the phosphonate bond. Biochem. Pharmacol. 2011, 82, 99–109. [Google Scholar]
- Peterson, L.W.; McKenna, C.E. Prodrug approaches to improving the oral absorption of antiviral nucleotide analogues. Expert Opin. Drug Deliv. 2009, 6, 405–420. [Google Scholar] [CrossRef]
- McKenna, C.E.; Kashemirov, B.A.; Peterson, L.W.; Goodman, M.F. Modifications to the dNTP triphosphate moiety: From mechanistic probes for DNA polymerases to antiviral and anti-cancer drug design. Biochim. Biophys. Acta (BBA) 2010, 1804, 1223–1230. [Google Scholar]
- Arzumanov, A.A.; Semizarov, D.G.; Victorova, L.S.; Dyatkina, N.B.; Krayevsky, A.A. Gamma-phosphate-substituted 2'-deoxynucleoside 5'-triphosphates as substrates for DNA polymerases. J. Biol. Chem. 1996, 271, 24389–24394. [Google Scholar]
- Meyer, P.R.; Smith, A.J.; Matsuura, S.E.; Scott, W.A. Chain-terminating dinucleoside tetraphosphates are substrates for DNA polymerization by human immunodeficiency virus type 1 reverse transcriptase with increased activity against thymidine analogue-resistant mutants. Antimicrob. Agents Chemother. 2006, 50, 3607–3614. [Google Scholar] [CrossRef]
- Victorova, L.; Sosunov, V.; Skoblov, A.; Shipytsin, A.; Krayevsky, A. New substrates of DNA polymerases. FEBS Lett. 1999, 453, 6–10. [Google Scholar] [CrossRef]
- Wang, G.; Boyle, N.; Chen, F.; Rajappan, V.; Fagan, P.; Brooks, J.L.; Hurd, T.; Leeds, J.M.; Rajwanshi, V.K.; Jin, Y.; et al. Synthesis of AZT 5'-triphosphate mimics and their inhibitory effects on HIV-1 reverse transcriptase. J. Med. Chem. 2004, 47, 6902–6913. [Google Scholar] [CrossRef]
- Kinsella, T.J. Coordination of DNA mismatch repair and base excision repair processing of chemotherapy and radiation damage for targeting resistant cancers. Clin. Cancer Res. 2009, 15, 1853–1859. [Google Scholar] [CrossRef]
- Matray, T.J.; Kool, E.T. A specific partner for abasic damage in DNA. Nature 1999, 399, 704–708. [Google Scholar]
- Crespan, E.; Alexandrova, L.; Khandazhinskaya, A.; Jasko, M.; Kukhanova, M.; Villani, G.; Hubscher, U.; Spadari, S.; Maga, G. Expanding the repertoire of DNA polymerase substrates: Template-instructed incorporation of non-nucleoside triphosphate analogues by DNA polymerases beta and lambda. Nucleic Acids Res. 2007, 35, 45–57. [Google Scholar]
- Crespan, E.; Zanoli, S.; Khandazhinskaya, A.; Shevelev, I.; Jasko, M.; Alexandrova, L.; Kukhanova, M.; Blanca, G.; Villani, G.; Hubscher, U.; et al. Incorporation of non-nucleoside triphosphate analogues opposite to an abasic site by human DNA polymerases beta and lambda. Nucleic Acids Res. 2005, 33, 4117–4127. [Google Scholar] [CrossRef]
- Meier, C.; Lomp, A.; Meerbach, A.; Wutzler, P. CycloSal-BVDUMP pronucleotides: How to convert an antiviral-inactive nucleoside analogue into a bioactive compound against EBV. J. Med. Chem. 2002, 45, 5157–5172. [Google Scholar] [CrossRef]
- Tan, X.; Chu, C.K.; Boudinot, F.D. Development and optimization of anti-HIV nucleoside analogs and prodrugs: A review of their cellular pharmacology, structure-activity relationships and pharmacokinetics. Adv. Drug Deliv. Rev. 1999, 39, 117–151. [Google Scholar] [CrossRef]
- Wagner, C.R.; Iyer, V.V.; McIntee, E.J. Pronucleotides: Toward the in vivo delivery of antiviral and anticancer nucleotides. Med. Res. Rev. 2000, 20, 417–451. [Google Scholar] [CrossRef]
- Bonnaffé, D.; Dupraz, B.; Ughetto-Monfrin, J.; Namane, A.; Huynh Dinh, T. Synthesis of acyl pyrophosphates. Application to the synthesis of nucleotide lipophilic prodrugs. Tetrahedron Lett. 1995, 36, 531–534. [Google Scholar] [CrossRef]
- Kreimeyer, A.; Ughetto-Monfrin, J.; Namane, A.; Huynh-Dinh, T. Synthesis of acylphosphates of purine ribonucleosides. Tetrahedron Lett. 1996, 37, 8739–8742. [Google Scholar] [CrossRef]
- Jessen, H.J.; Schulz, T.; Balzarini, J.; Meier, C. Bioreversible protection of nucleoside diphosphates. Angew. Chem. Int. Ed. Engl. 2008, 47, 8719–8722. [Google Scholar] [CrossRef]
- Schulz, T.; Jessen, H.J.; Balzarini, J.; Meier, C. Bioreversible protection of nucleosidediphosphates-synthesis and properties. Antiviral Res. 2009, 82, A63. [Google Scholar]
- Vinogradov, S.V.; Kohli, E.; Zeman, A.D. Comparison of nanogel drug carriers and their formulations with nucleoside 5'-triphosphates. Pharm. Res. 2006, 23, 920–930. [Google Scholar] [CrossRef]
- Raemdonck, K.; Demeester, J.; de Smedt, S. Advanced nanogel engineering for drug delivery. Soft Matter 2009, 5, 707–715. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Crespan, E.; Garbelli, A.; Amoroso, A.; Maga, G. Exploiting the Nucleotide Substrate Specificity of Repair DNA Polymerases To Develop Novel Anticancer Agents. Molecules 2011, 16, 7994-8019. https://doi.org/10.3390/molecules16097994
Crespan E, Garbelli A, Amoroso A, Maga G. Exploiting the Nucleotide Substrate Specificity of Repair DNA Polymerases To Develop Novel Anticancer Agents. Molecules. 2011; 16(9):7994-8019. https://doi.org/10.3390/molecules16097994
Chicago/Turabian StyleCrespan, Emmanuele, Anna Garbelli, Alessandra Amoroso, and Giovanni Maga. 2011. "Exploiting the Nucleotide Substrate Specificity of Repair DNA Polymerases To Develop Novel Anticancer Agents" Molecules 16, no. 9: 7994-8019. https://doi.org/10.3390/molecules16097994