Quantum Mechanics Calculations, Basicity and Crystal Structure: The Route to Transition Metal Complexes of Azahelicenes

Abstract
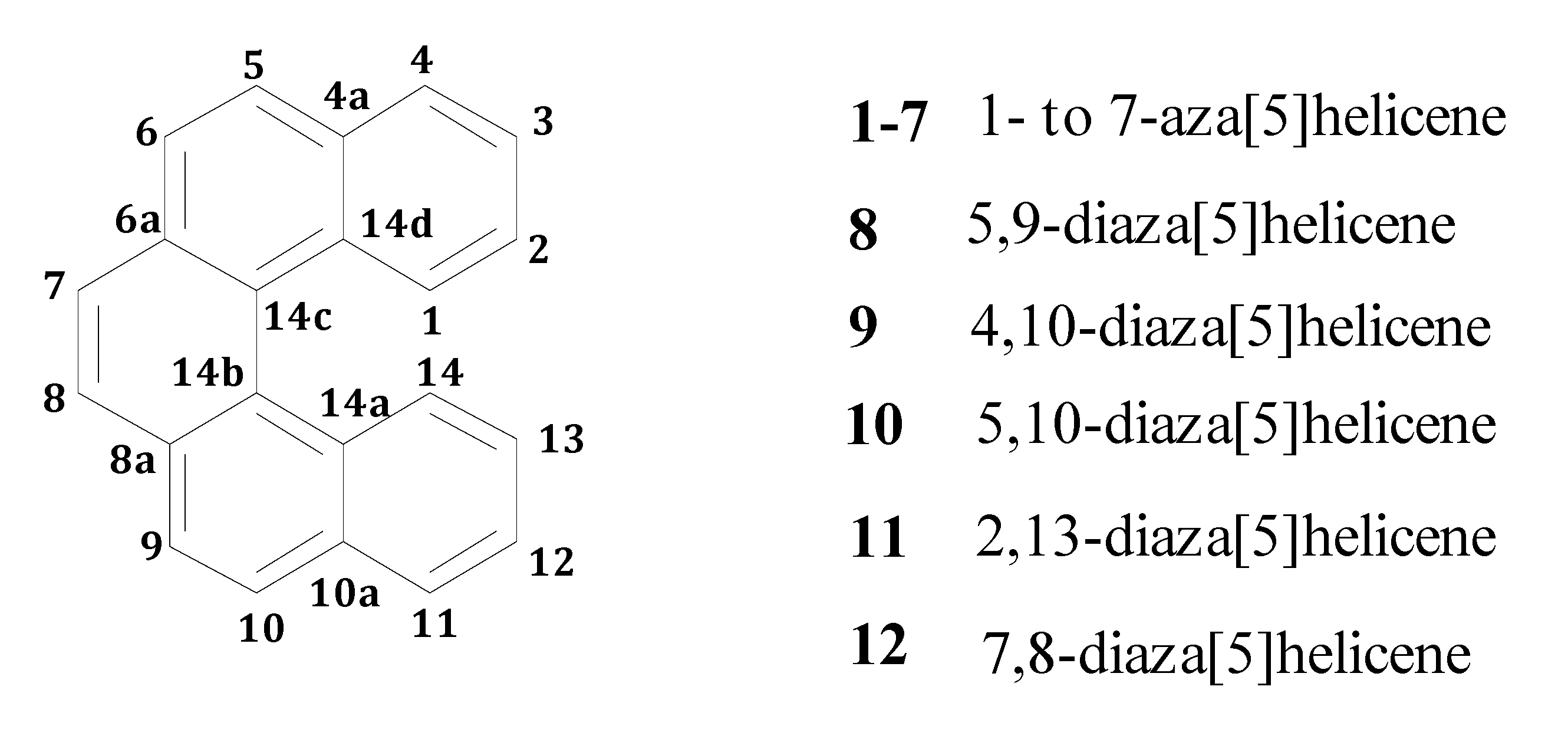
:1. Introduction

2. Results and Discussion
2.1. DFT Calculations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Atomic charges on N atoms | PA (kJ/mol) |
|---|---|---|
| 1 | N1: −0.52 | 1,009.8 |
| 1-H+ | N1: +0.06 | |
| 2 | N2: −0.67 | 1,003.9 |
| 2-H+ | N2: −0.21 | |
| 3 | N3: −0.63 | 999.7 |
| 3-H+ | N3: −0.06 | |
| 4 | N4: −0.69 | 996.1 |
| 4-H+ | N4: −0.28 | |
| 5 | N5: −0.64 | 1,001.3 |
| 5-H+ | N5: −0.26 | |
| 6 | N6: −0.66 | 995.2 |
| 6-H+ | N6: −0.21 | |
| 7 | N7: −0.65 | 1,001.2 |
| 7-H+ | N7: −0.20 | |
| Quinoline | N: −0.61 | 970.9 |
| Quinoline-H+ | N: −0.09 |

| Compound | Atomic charges on electronegative atoms | PA (kJ/mol) * |
|---|---|---|
| 8 | N5: −0.66 N9: −0.66 | |
| 8-H+(H5) | N5: −0.33 N9: −0.66 | 991.8 |
| 8-H+(H9) | N5: −0.58 N9: −0.27 | 976.3 |
| 9 | N4: −0.69 N10: −0.64 | |
| 9-H+(H4) | N4: −0.27 N10: −0.59 | 979.1 |
| 9-H+(H10) | N4: −0.66 N10: −0.26 | 989.0 |
| 10 | N5: −0.64 N10: −0.64 | |
| 10-H+(H5) | N5: −0.30 N10: −0.56 | 979.3 |
| 10-H+(H10) | N5: −0.56 N10: −0.30 | 979.2 |
| 11 | N2: −0.65 N13: −0.65 | |
| 11-H+(H2) | N2: −0.16 N13: −0.65 | 997.2 |
| 11-H+(H13) | N2: −0.65 N13: −0.17 | 997.2 |
| 12 | N7: −0.36 N8: −0.35 | |
| 12-H+(H7) | N7: +0.15 N8: −0.49 | 1,002.1 |
| 12-H+(H8) | N7: −0.49 N8: +0.15 | 1,002.4 |
| 12a | N7: +0.81 N8: −0.65 O7: −0.50 | |
| 12a -H+ (O7) | N7: +0.56 N8: −0.56 O7: −0.47 | 978.8 |
| 12a -H+ (N8) | N7: +0.56 N8: −0.38 O7: −0.37 | 957.1 |
| 12b | N7: +0.35 N8: +0.34 | |
| O7: −0.42 O8: −0.42 | ||
| 12b -H+ (O7) | N7: +0.12 N8: +0.39 | 973.0 |
| O7: −0.39 O8: −0.41 | ||
| 12b -H+ (O8) | N7: +0.40 N8: +0.11 | 973.1 |
| O7: −0.41 O8: −0.38 | ||
| 13 | N1: +0.05 N9: −0.55 | 1,015.5 |
| 13-H+ | N1: −0.56 N9: +0.06 | 1,015.6 |
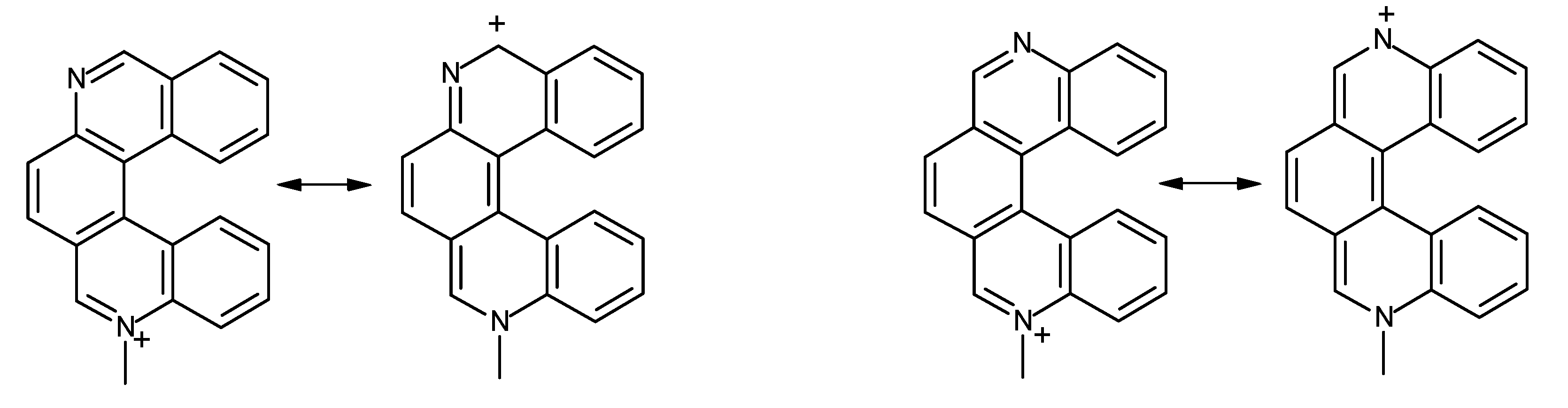
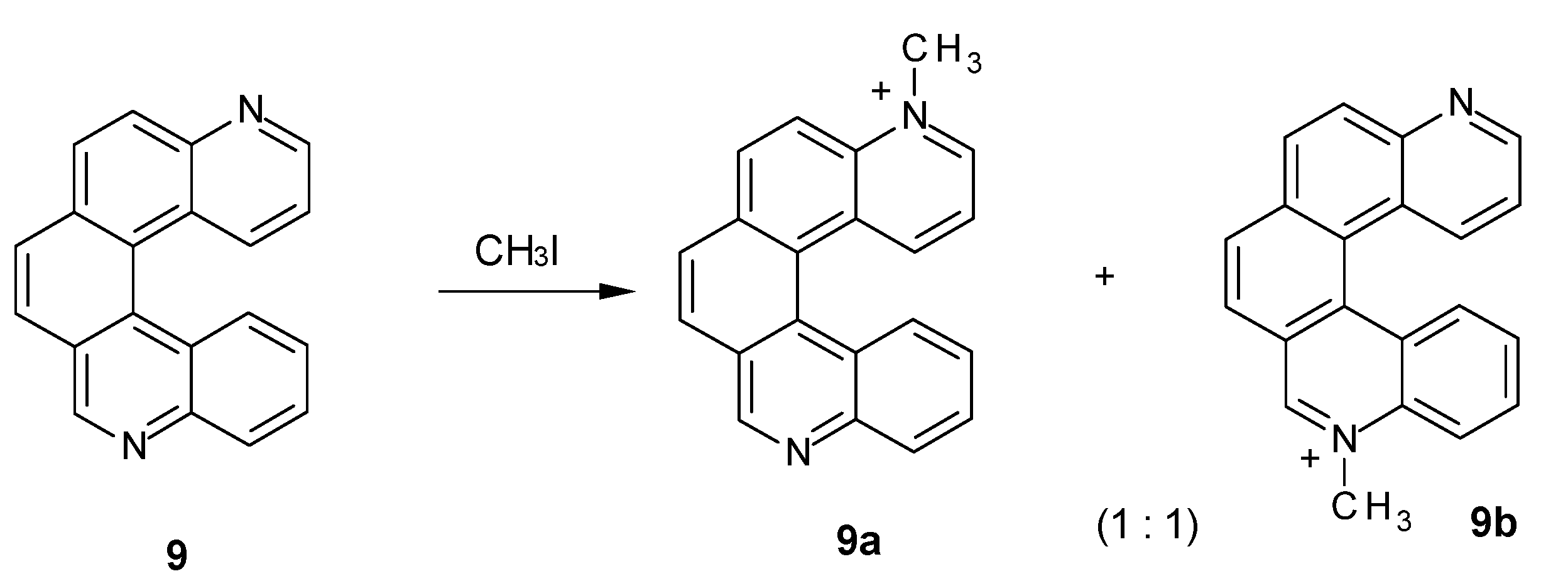
2.2. N-Methylation of Mono-and Diazahelicenes


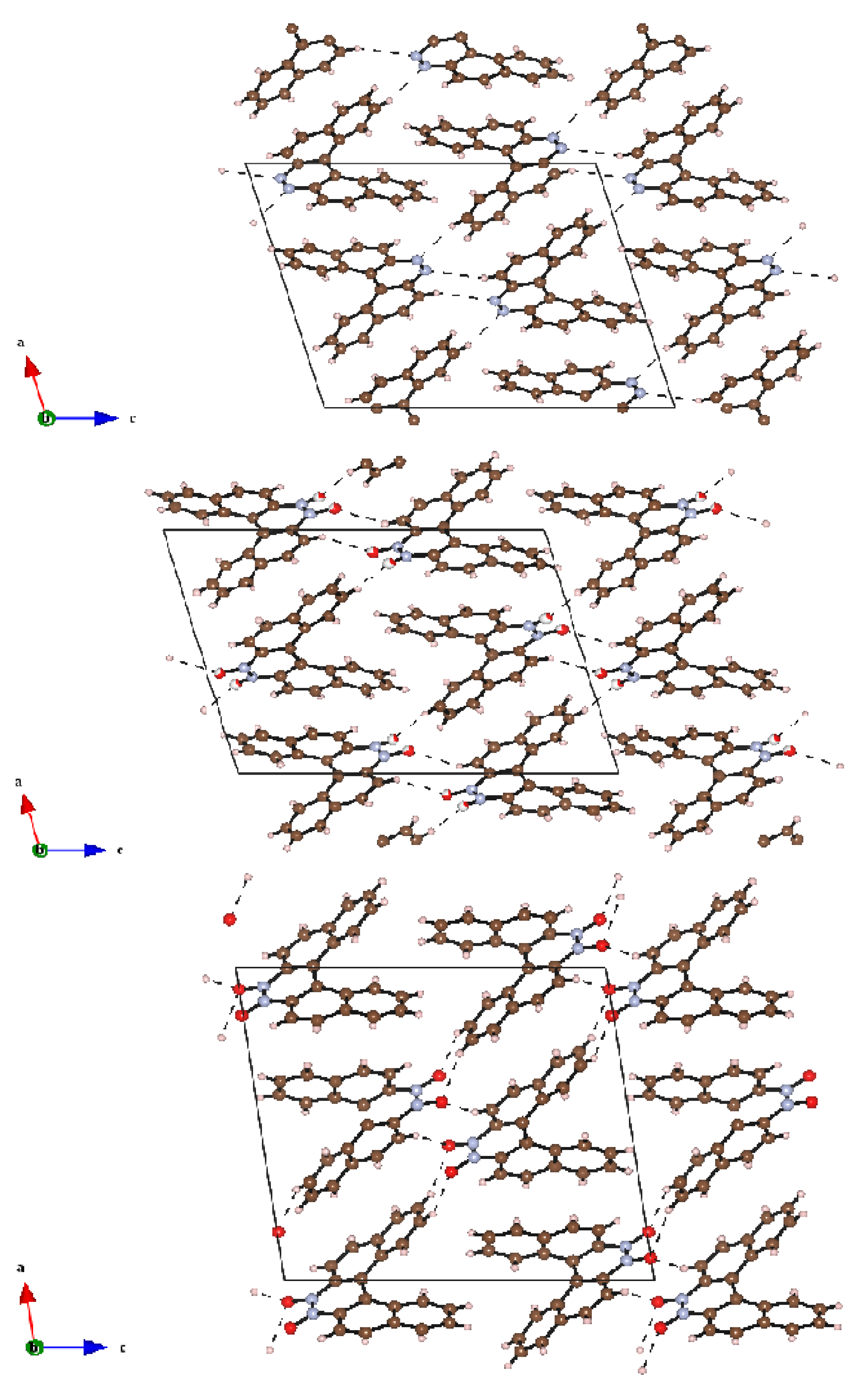
2.3. Crystal Structures and Crystal Packing
| Compound | C-H---X contacts | C----X distances | C-H---X angles |
|---|---|---|---|
| 12 | C(2)-H(2)---N(8) | 3.452(3) Å | 155.3(2)° |
| C(6)-H(6)---N(7) | 3.808(3) Å | 156.0(2)° | |
| 12a | C(2)-H(2)---O(16) | 2.871(5) Å | 137.8(2)° |
| C(6)-H(6)---O(15) | 3.329(3) Å | 143.6(2)° | |
| 12b | C(2)-H(2)--- O(16) | 3.399(3) Å | 147.4(2)° |
| C(3)-H(3)--- O(15) | 3.374(3) Å | 159.5(2)° | |
| C(6)-H(6)--- O(15) | 3.329(3) Å | 153.4(2)° |

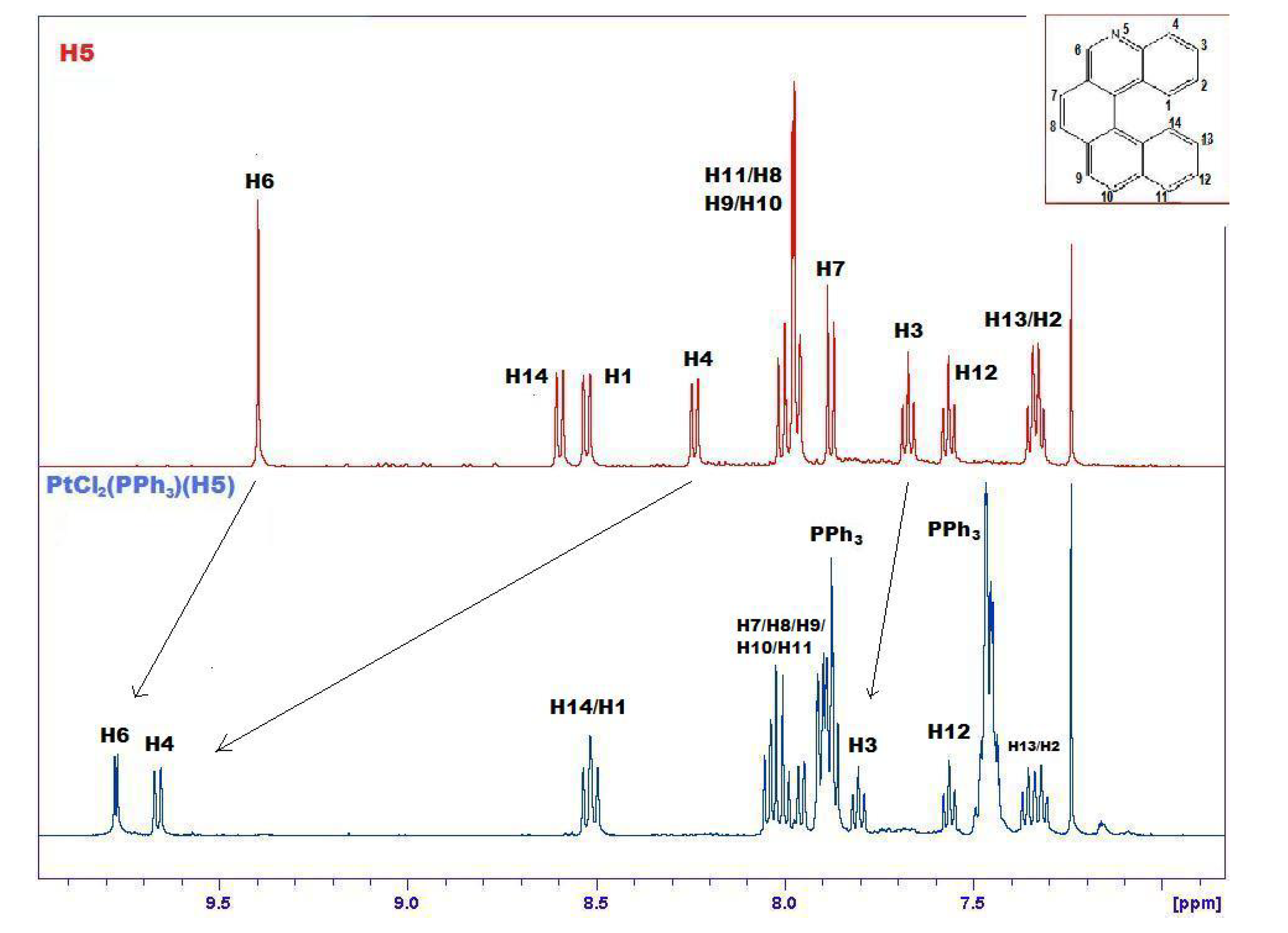
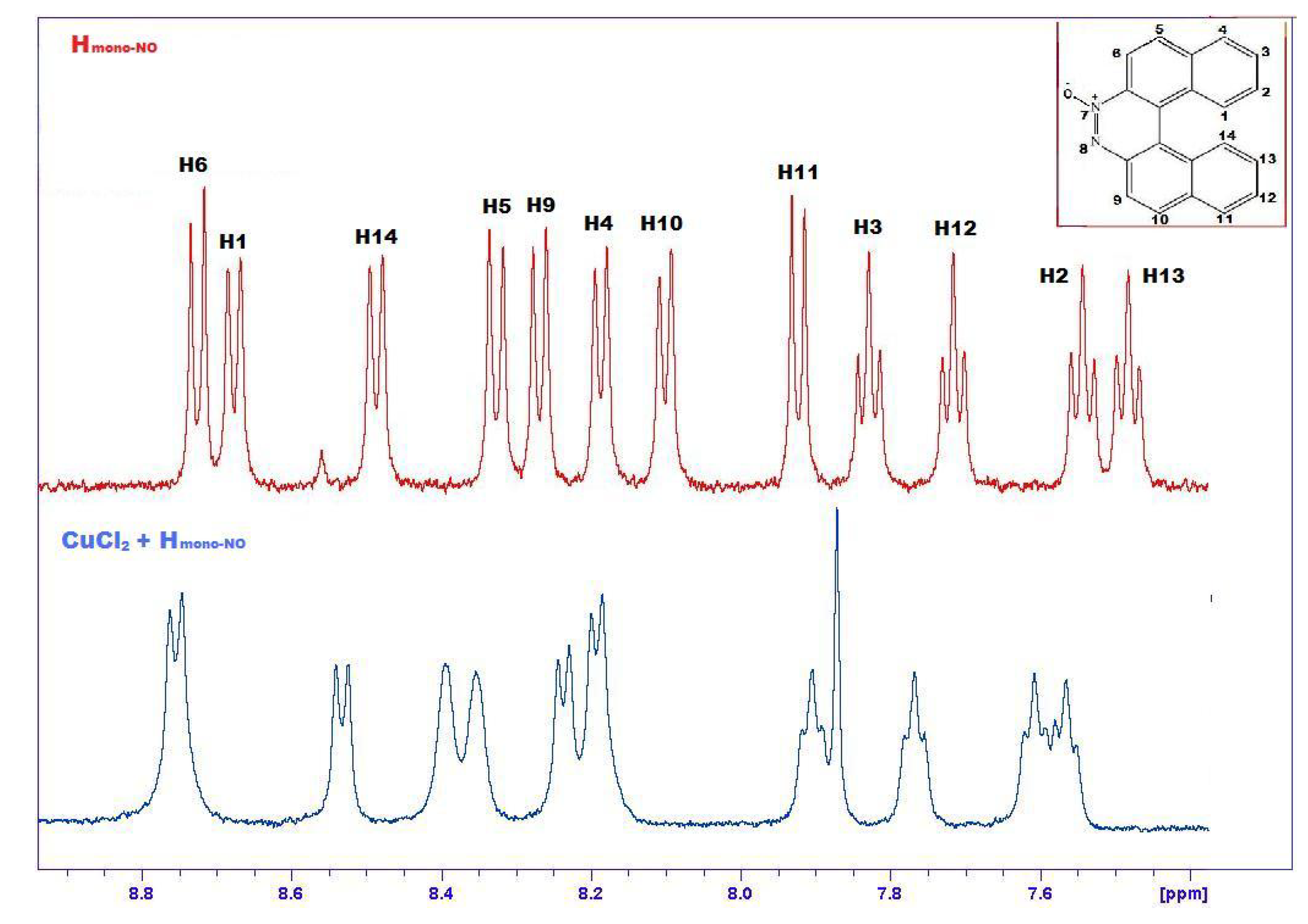
2.4. Transition Metal Complexes





3. Experimental
3.1. General
3.2. Synthesis of Quaternary N-Methylazahelicenium Salts
3.3. Synthesis and Characterization of Metal Complexes
3.3.1. cis-PtCl2(NCEt)(PPh3) [33]
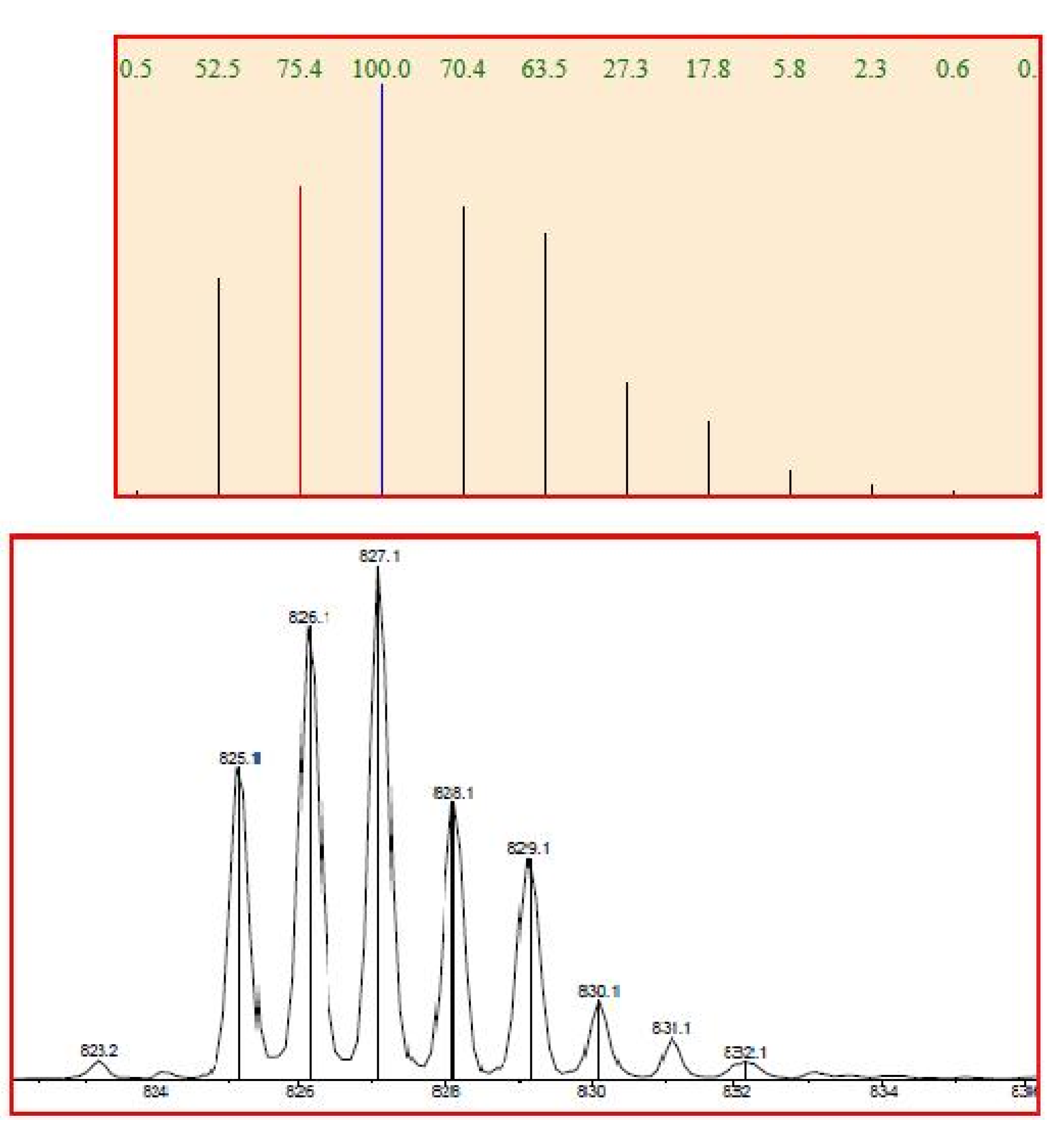
3.3.2. PtCl2(PPh3)(5)
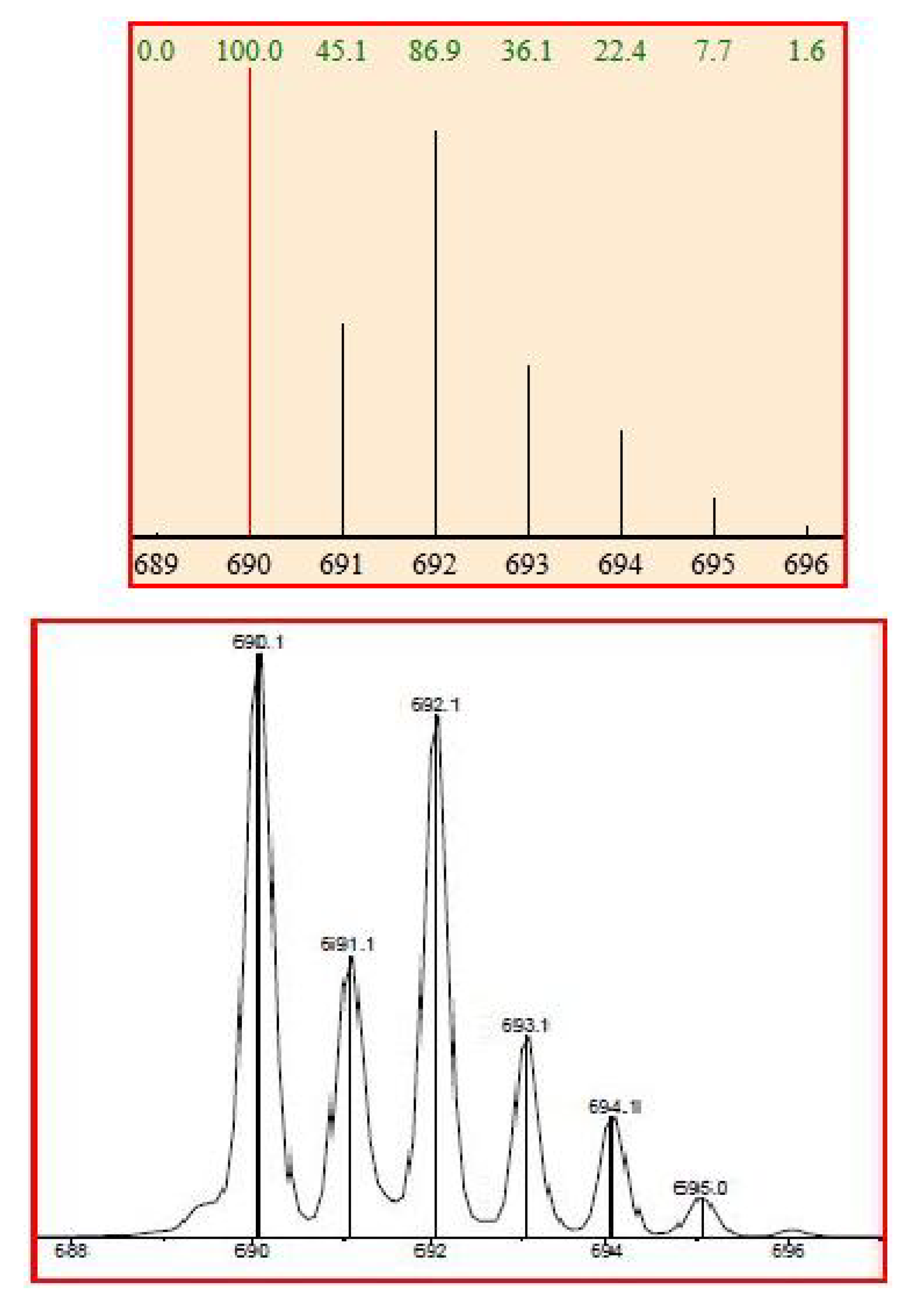
3.3.3. CuCl(12a)2
4. Conclusions
Supplementary Materials
- Samples Availability: Samples of compounds 1–12, 5a, 12a and 12b are available from the authors.
References and Notes
- Dumitrascu, F.; Dumitrescu, D.G.; Aron, I. Azahelicenes and other similar tri and tetracyclic helical molecules. ARKIVOC 2010, i, 1–32. [Google Scholar]
- Shen, Y.; Chen, C.-F. Helicenes: Synthesis and applications. Chem. Rev. 2011. [Google Scholar] [CrossRef]
- Verbiest, T.; Van Elshocht, S.; Kauranen, M.; Hellemans, L.; Snauwaert, J.; Nuckolls, C.; Katz, T.J.; Persoons, A. Strong enhancement of nonlinear optical properties through supramolecular chirality. Science 1998, 282, 913–915. [Google Scholar] [CrossRef]
- Fontana, F.; Caronna, T.; Donato, N.; Latino, M.; Bonavita, A.; Rizzo, G.; Neri, G. A novel organic/MWCNTs semiconductor composite for resistive gas sensors. In Sensors and Microsystems; Neri, G., Donato, N., d’Amico, A., Di Natale, C., Eds.; Springer: Dordrecht, The Netherlands, 2011; pp. 61–66. [Google Scholar]
- Beljonne, D.; Shuai, Z.; Brédas, J.L.; Kauranen, M.; Verbiest, T.; Persoons, A. Electro-optic response of chiral helicenes in isotropic media. J. Chem. Phys. 1998, 108, 1301–1304. [Google Scholar]
- Botek, E.; Champagne, B.; Turki, M.; Andre, J.M. Theoretical design of substituted tetrathia[7]helicenes with large second-order nonlinear optical responses. J. Chem. Phys. 2004, 120, 2042–2048. [Google Scholar] [CrossRef]
- Passeri, R.; Aloisi, G.G.; Latterini, L.; Elisei, F.; Caronna, T.; .Fontana, F.; Natali Sora, I. Photophysical properties of N-alkylated aza-helicene derivatives as DNA intercalators: Counterion effects. Photochem. Photobiol. Sci. 2009, 8, 1574–1582. [Google Scholar]
- Latterini, L.; Galletti, E.; Passeri, R.; Barbafina, A.; Urbanelli, L.; Emiliani, C.; Elisei, F.; Fontana, F.; Mele, A.; Caronna, T. Fluorescence properties of aza-helicenium derivatives for cell imaging. J. Photochem. Photobiol. A Chem. 2011, 222, 307–313. [Google Scholar] [CrossRef]
- Bazzini, C.; Brovelli, S.; Caronna, T.; Gambarotti, C.; Giannone, M.; Macchi, P.; Meinardi, F.; Mele, A.; Panzeri, W.; Recupero, F.; et al. Synthesis and characterization of some aza[5]helicenes. Eur. J. Org. Chem. 2005, 1247–1257. [Google Scholar]
- Abbate, S.; Bazzini, C.; Caronna, T.; Fontana, F.; Gambarotti, C.; Gangemi, F.; Longhi, G.; Mele, A.; Natali Sora, I.; Panzeri, W. Monoaza[5]helicenes. Part 2: Synthesis, characterisation and theoretical calculations. Tetrahedron 2006, 62, 139–148. [Google Scholar]
- Caronna, T.; Fontana, F.; Longhi, G.; Mele, A.; Natali Sora, I.; Viganò, L. 2,13-diaza[5]helicene: synthesis, theoretical calculations and spectroscopic properties. ARKIVOC 2009, 7, 145–155. [Google Scholar]
- Caronna, T.; Fontana, F.; Mele, A.; Natali Sora, I.; Panzeri, W.; Viganò, L. A simple approach to the synthesis of 7,8-diaza[5]helicene. Synthesis 2008, 413–416. [Google Scholar]
- Bazzini, C.; Caronna, T.; Fontana, F.; Macchi, P.; Mele, A.; Natali Sora, I.; Panzeri, W; Sironi, A. Synthesis, crystal structure and crystal packing of diaza[5]helicenes. New J. Chem. 2008, 32, 1710–1717. [Google Scholar]
- Abbate, S.; Caronna, T.; Longo, A.; Ruggirello, A.; Turco Liveri, V. Study of confined 5-aza[5]helicene in ytterbium (III) bis (2-ethylhexyl) sulfosuccinate. Reversed micelles. J. Phys. Chem. B 2007, 111, 4089–4097. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, J.S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S. ; Schmidt M.W. Advances in electronic structure theory: GAMESS a decade later. In Theory and Applications of Computational Chemistry, the First Forty Years; Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. [Google Scholar]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford Scientific: Oxford, UK, 1989. [Google Scholar]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory; Wiley VCH: New York, NY, USA, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry; Wiley and Sons: Chichester, UK, 2007. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chablowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650. [Google Scholar] [CrossRef]
- Gavezzotti, A. The calculation of molecular volumes and the use of volume analysis in the investigation of structured media and of solid-state organic reactivity. J. Am. Chem. Soc. 1983, 105, 5220–5225. [Google Scholar] [CrossRef]
- Gal, J.F.; Maria, P.C.; Raczyńska, E.D. Thermochemical aspects of proton transfer in the gas phase. J. Mass Spectrom. 2001, 36, 699–716. [Google Scholar] [CrossRef]
- Alder, R.W. Design of C2-chiral diamines that are computationally predicted to be a million-fold more basic than the original proton sponges. J. Am. Chem. Soc. 2005, 127, 7924–931. [Google Scholar] [CrossRef]
- Kovacevic, K.; Maksic, Z.B. High basicity of phosphorus-proton affinity of tris-(tetramethylguanidinyl)phosphine and tris- (hexamethyltriaminophosphazenyl)phosphine by DFT calculations. Chem. Commun. 2005, 1524–1526. [Google Scholar]
- Roithová, J.; Schröder, D.; Mıšek, J.; Stará, I.G.; Stary, I. Chiral superbases: The proton affinities of 1- and 2-aza[6]helicene in the gas phase. J. Mass Spectrom. 2007, 42, 1233–1237. [Google Scholar] [CrossRef]
- Rochon, F.D.; Morneau, A. 195Pt and 1H-NMR studies of platinum (II) complexes with ethylenediamine derivatives. Magn. Reson. Chem. 1991, 29, 120–126. [Google Scholar]
- Banci, L.; Bertini, I.; Luchinat, C. Nuclear and Electron Relaxation. The Magnetic Nucleus-Unpaired Electron Coupling in Solution; VCH: Weinheim, Germany, 1991. [Google Scholar]
- Banci, L.; Bertini, I.; Luchinat, C. Solution NMR of Paramagnetic Molecules: Applications to Metallobiomolecules; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Kukushkin, V.Y.; Oskarsson, Å.; Elding, L.I.; Jonasdottir, S. Tetrakis(propanenitrile) platinum(II) trifluoromethanesulfonate as a suitable intermediate in synthetic Pt(II) chemistry. Inorg. Synth. 1997, 31, 279–284. [Google Scholar]
- Mendola, D. Reattività verso la dietilammina di complessi di platino (II) con leganti insaturi. Tesi di Laurea in Chimica. Università di Pisa: Pisa, Italy, 2009/2010. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Caronna, T.; Castiglione, F.; Famulari, A.; Fontana, F.; Malpezzi, L.; Mele, A.; Mendola, D.; Sora, I.N. Quantum Mechanics Calculations, Basicity and Crystal Structure: The Route to Transition Metal Complexes of Azahelicenes. Molecules 2012, 17, 463-479. https://doi.org/10.3390/molecules17010463
Caronna T, Castiglione F, Famulari A, Fontana F, Malpezzi L, Mele A, Mendola D, Sora IN. Quantum Mechanics Calculations, Basicity and Crystal Structure: The Route to Transition Metal Complexes of Azahelicenes. Molecules. 2012; 17(1):463-479. https://doi.org/10.3390/molecules17010463
Chicago/Turabian StyleCaronna, Tullio, Franca Castiglione, Antonino Famulari, Francesca Fontana, Luciana Malpezzi, Andrea Mele, Daniele Mendola, and Isabella Natali Sora. 2012. "Quantum Mechanics Calculations, Basicity and Crystal Structure: The Route to Transition Metal Complexes of Azahelicenes" Molecules 17, no. 1: 463-479. https://doi.org/10.3390/molecules17010463