Recent Advances in Cyclonucleosides: C-Cyclonucleosides and Spore Photoproducts in Damaged DNA

Abstract
:1. Introduction
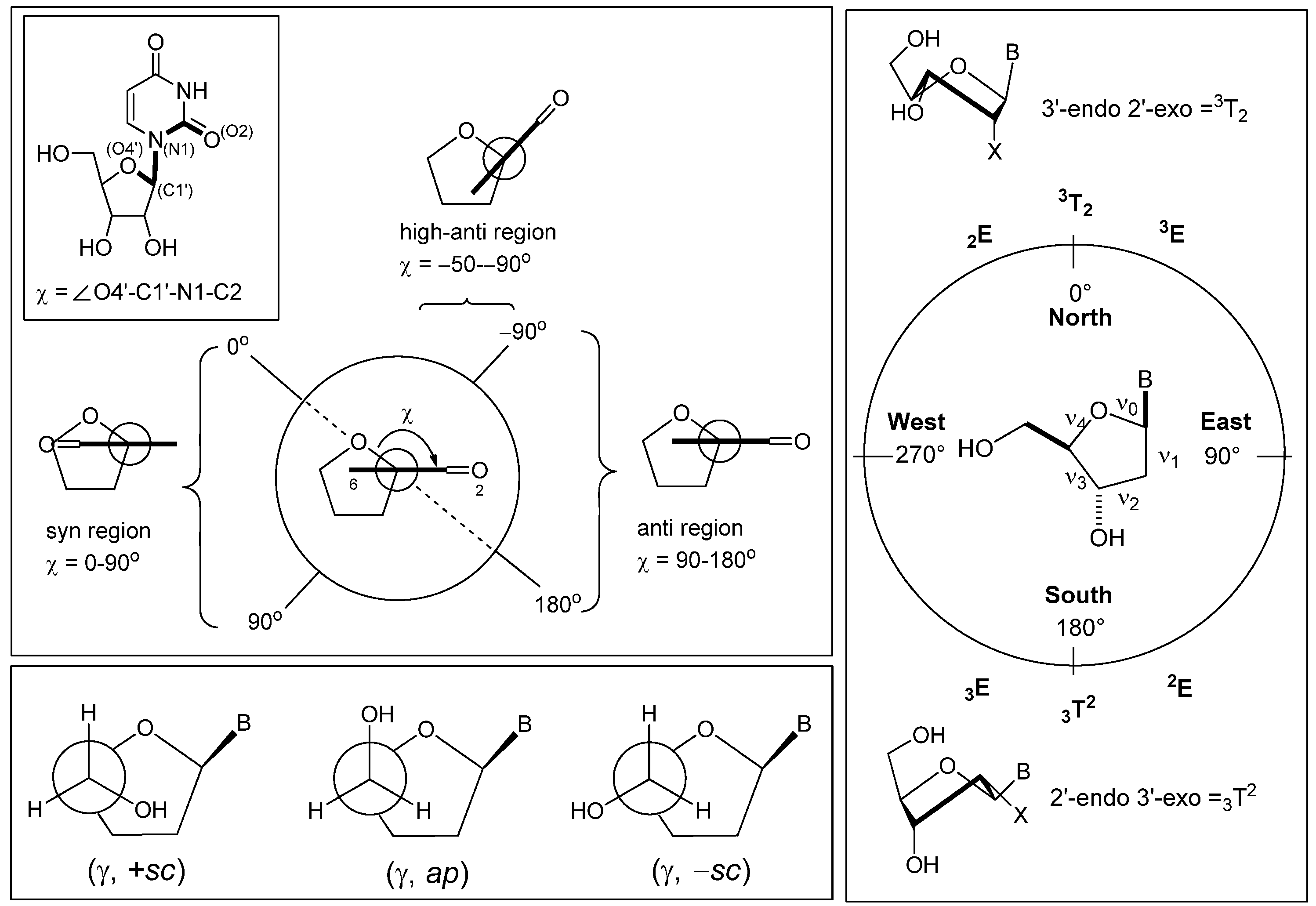
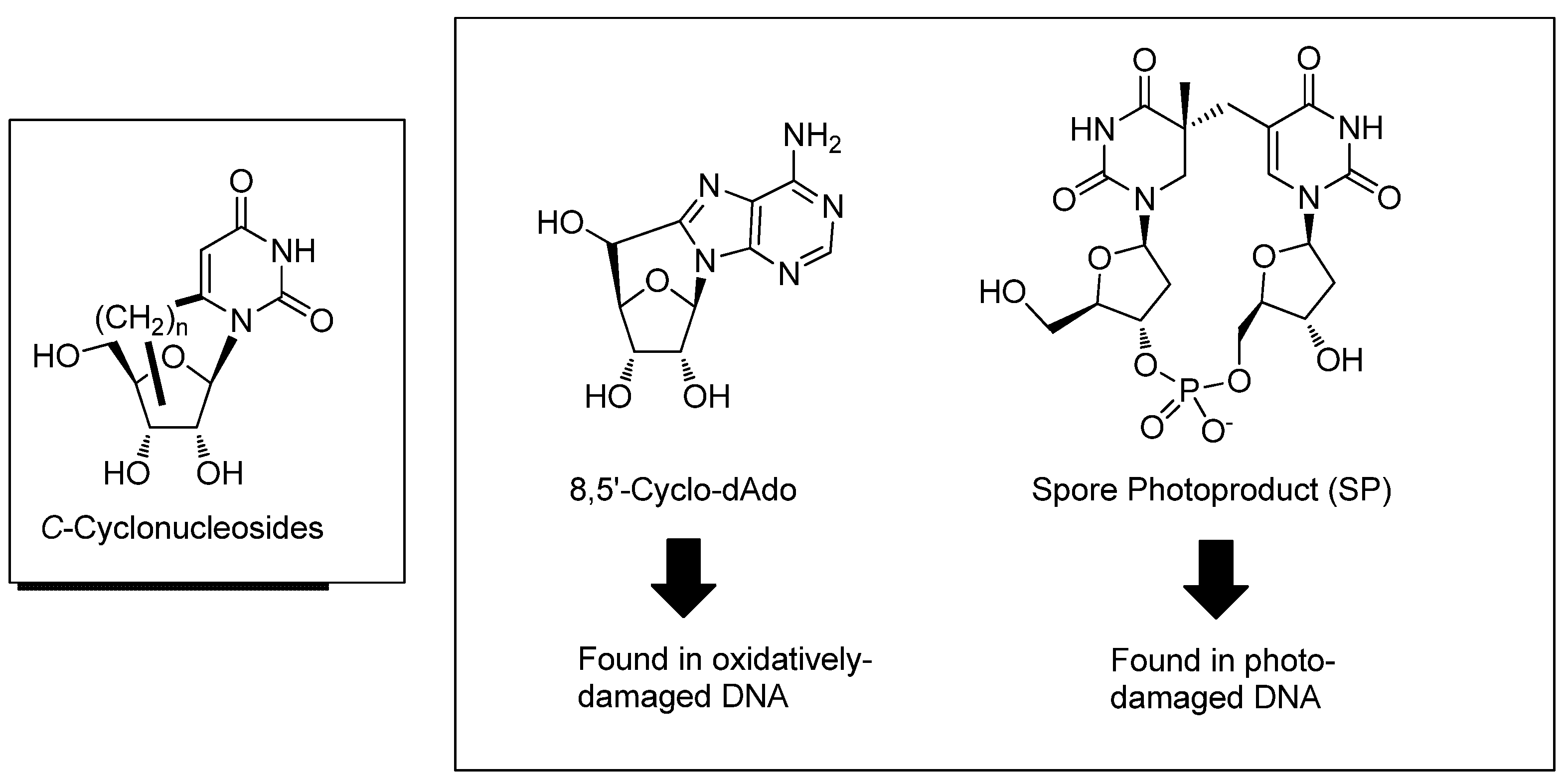
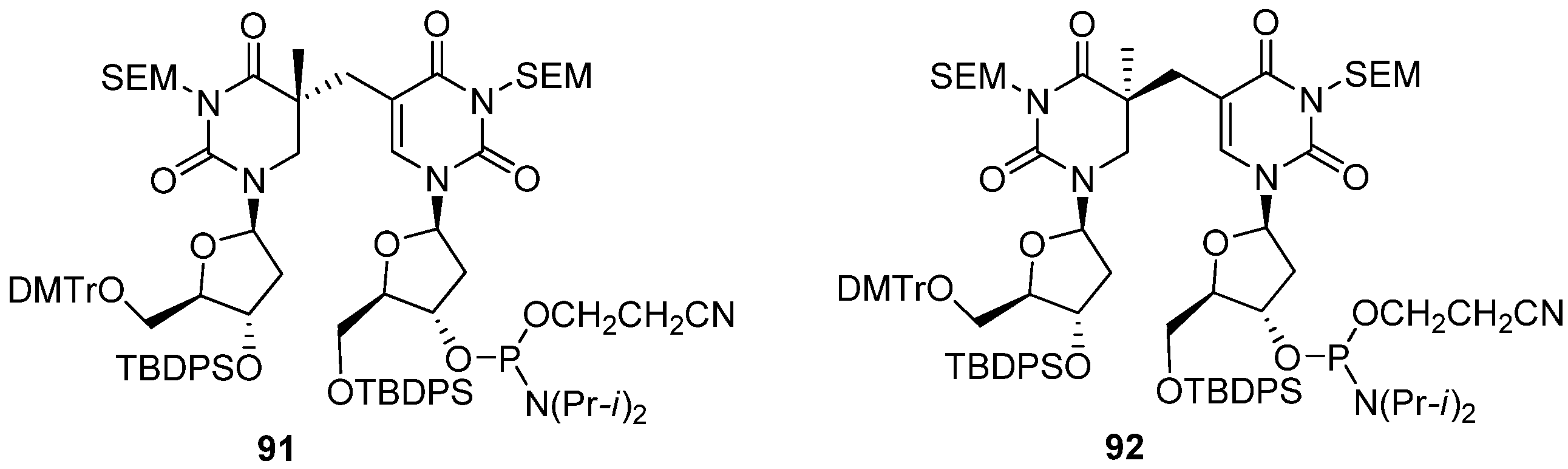
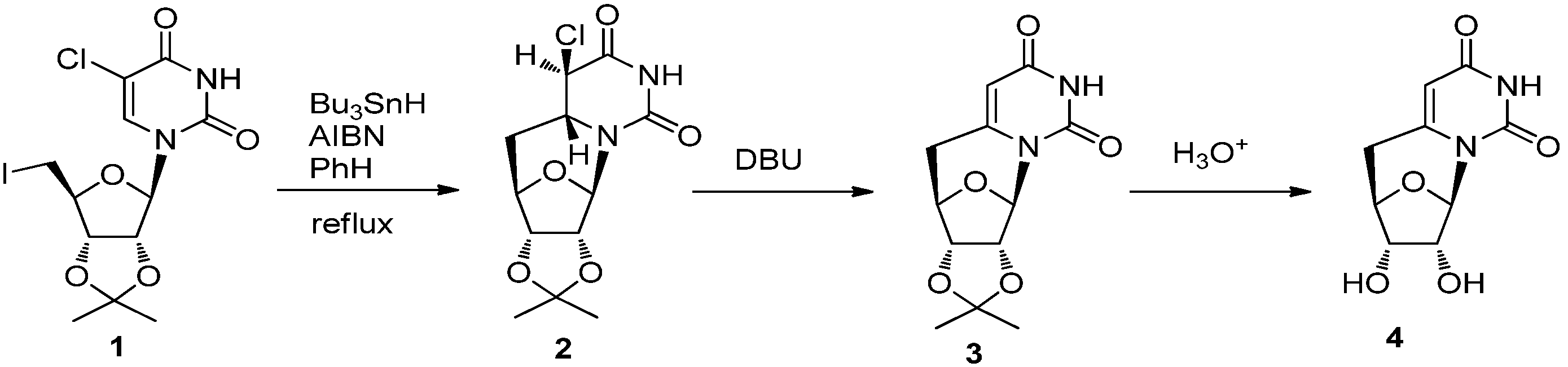
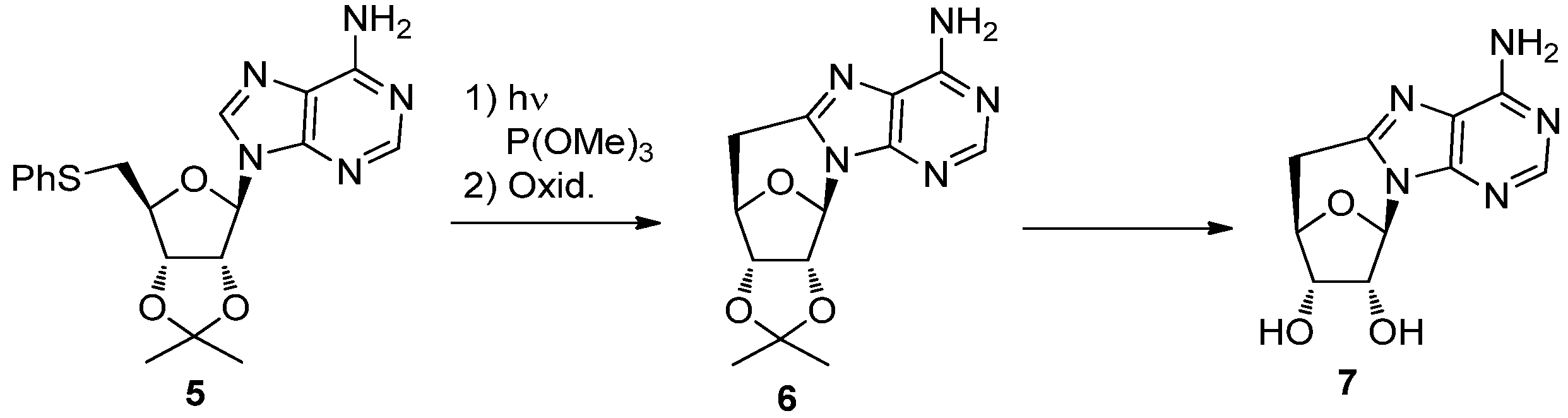
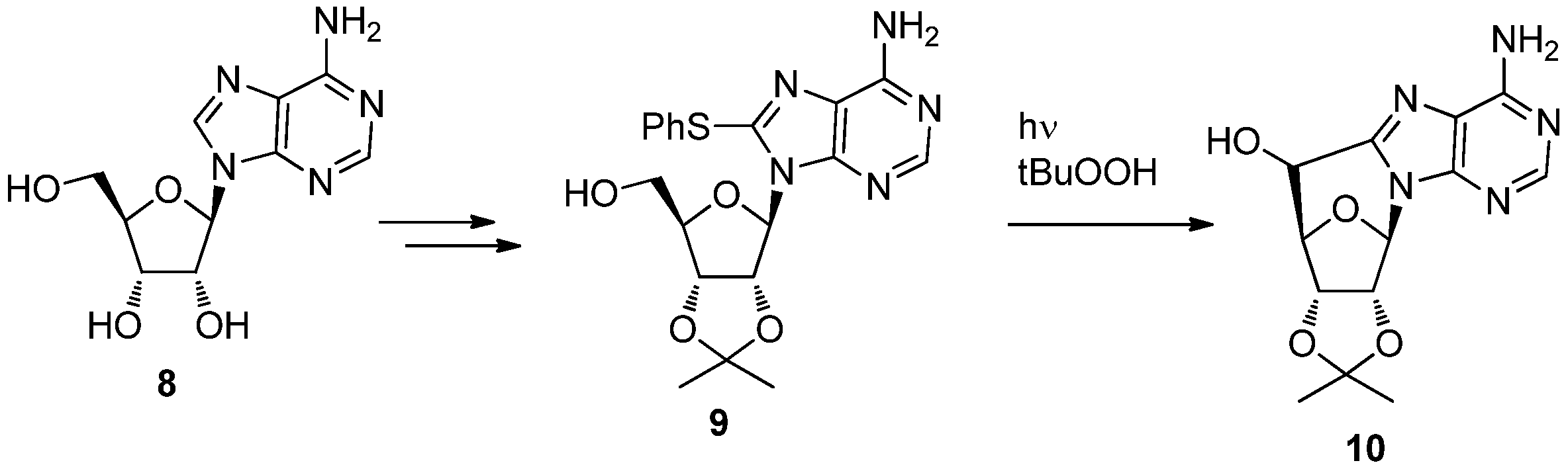
2. Synthesis of C-Cyclonucleosides
2.1. Classical Synthesis of C-Cyclonucleosides
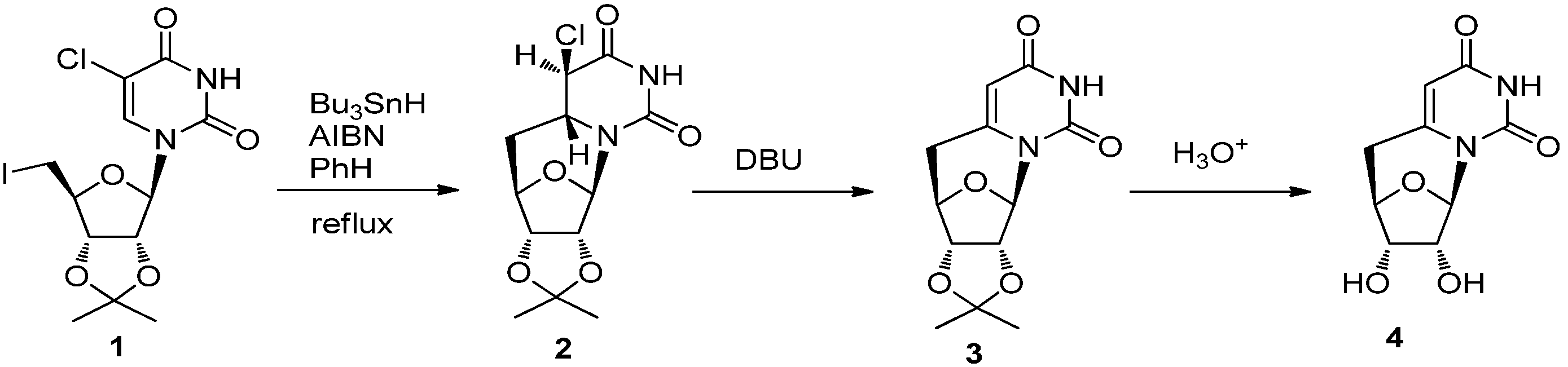


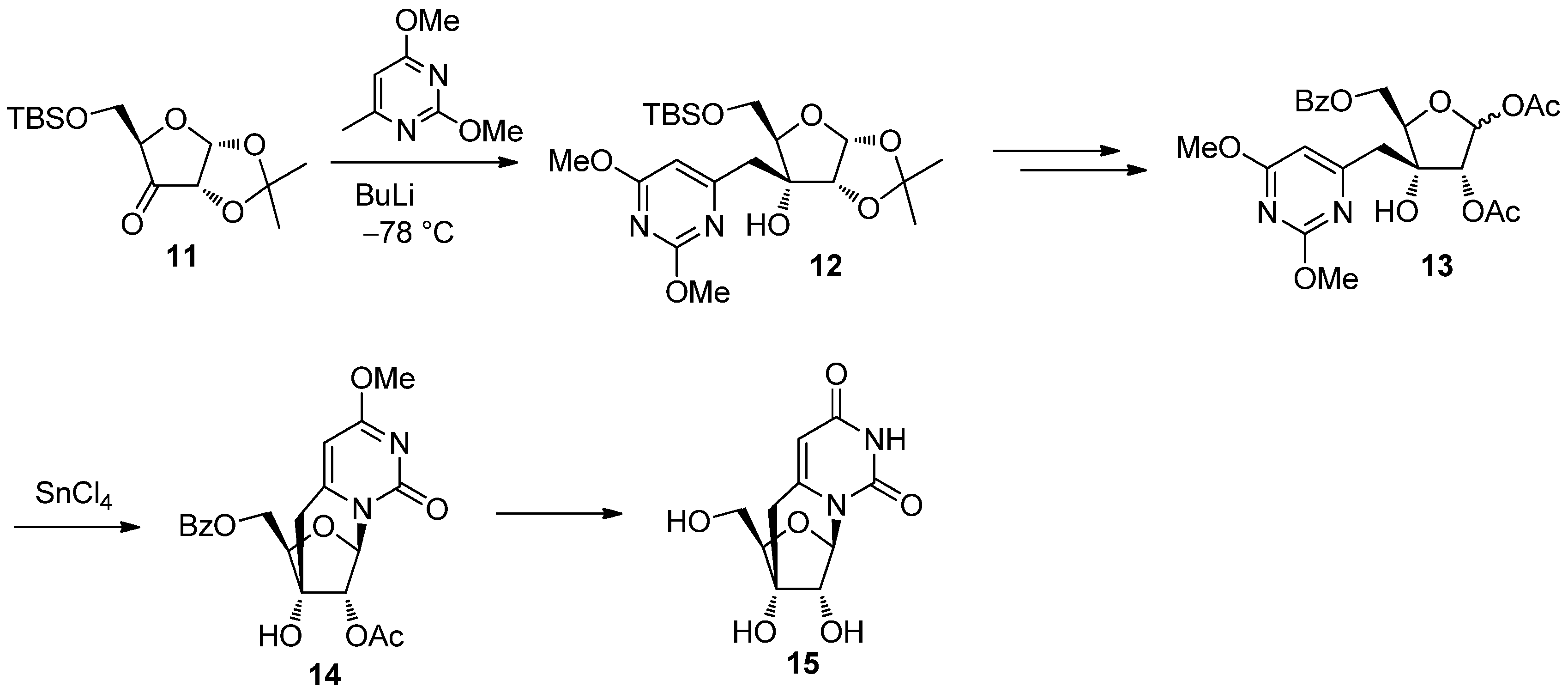
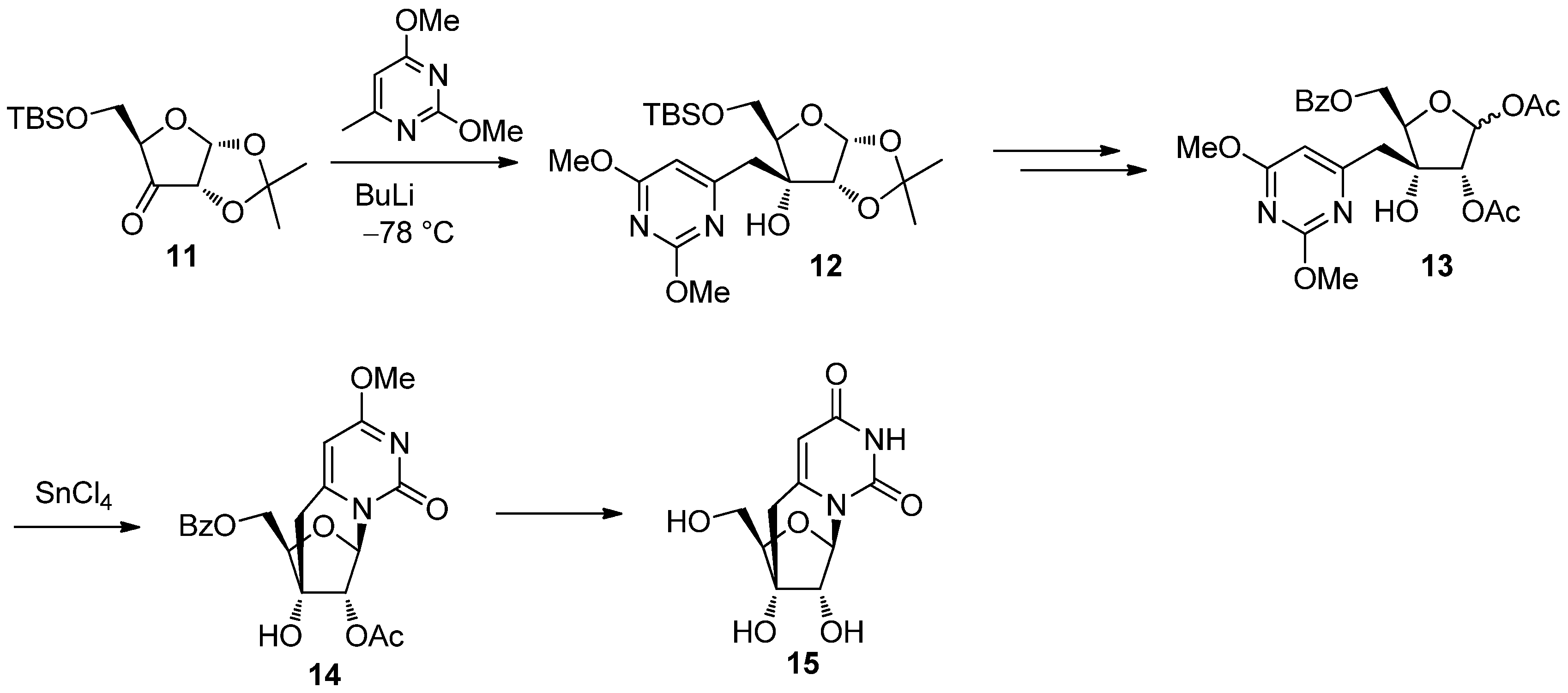
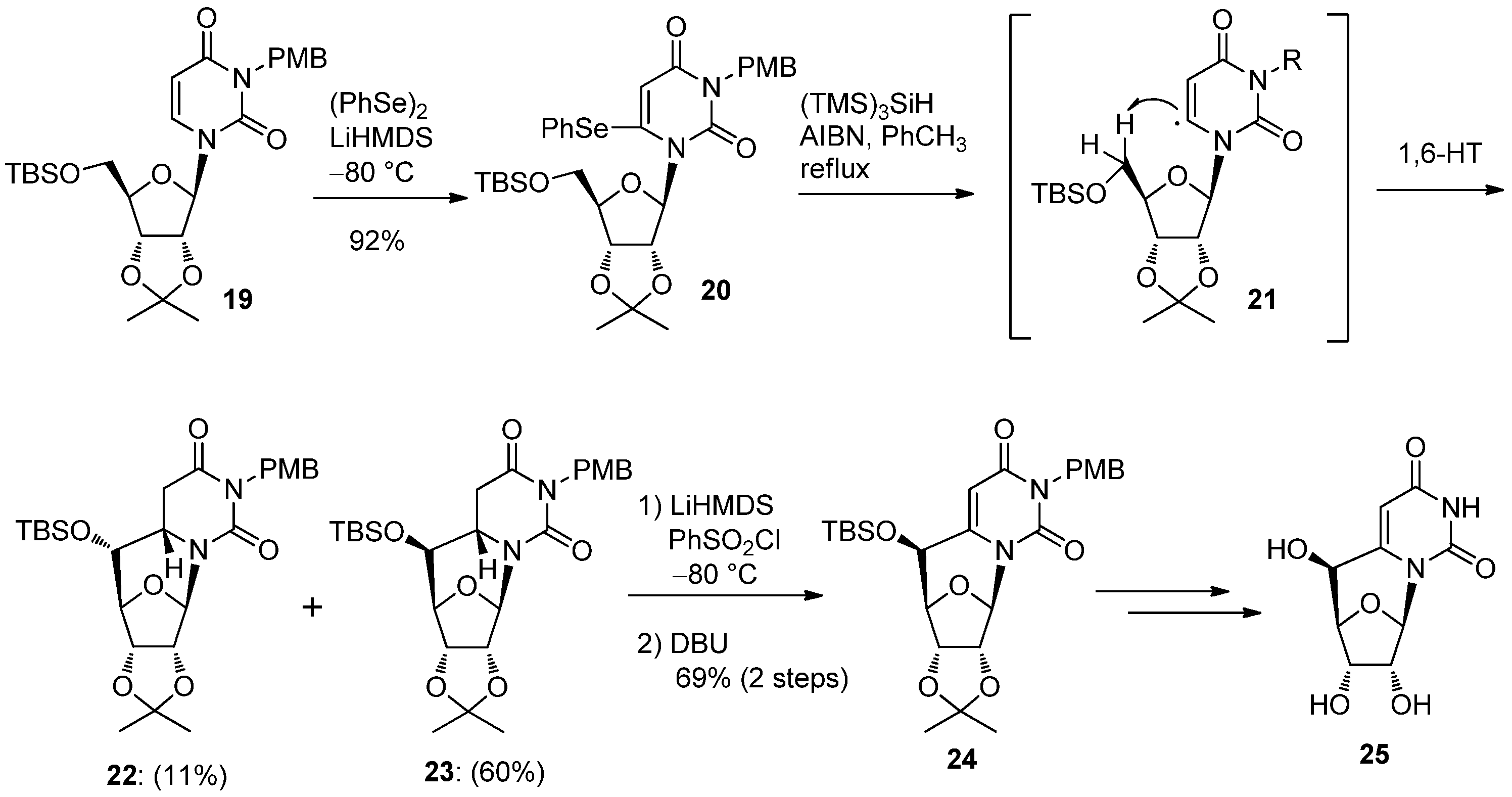
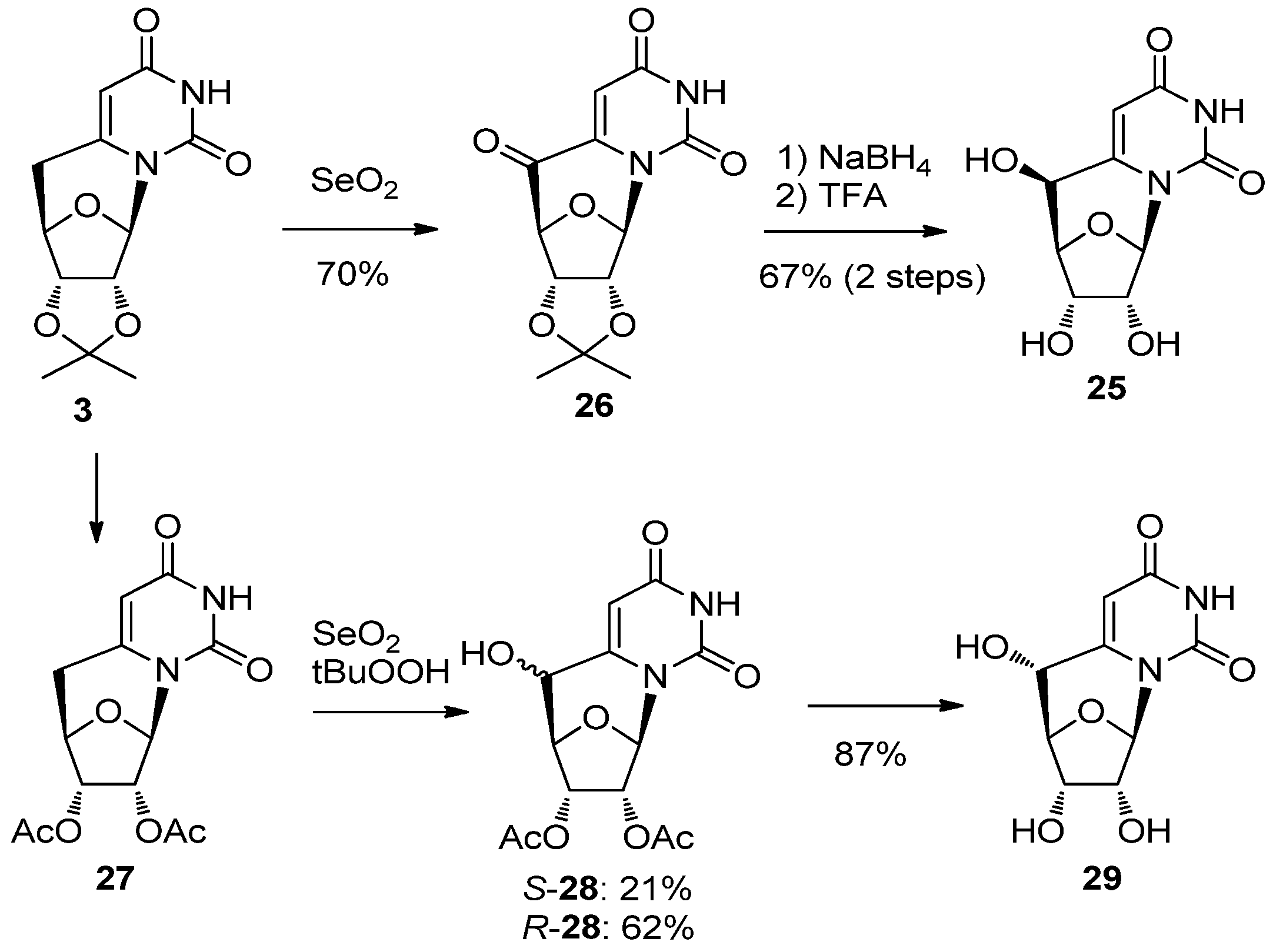
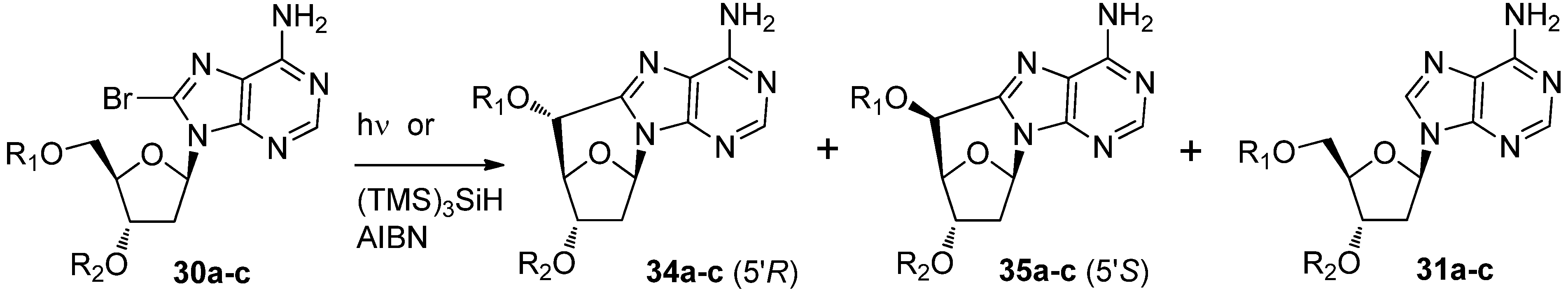
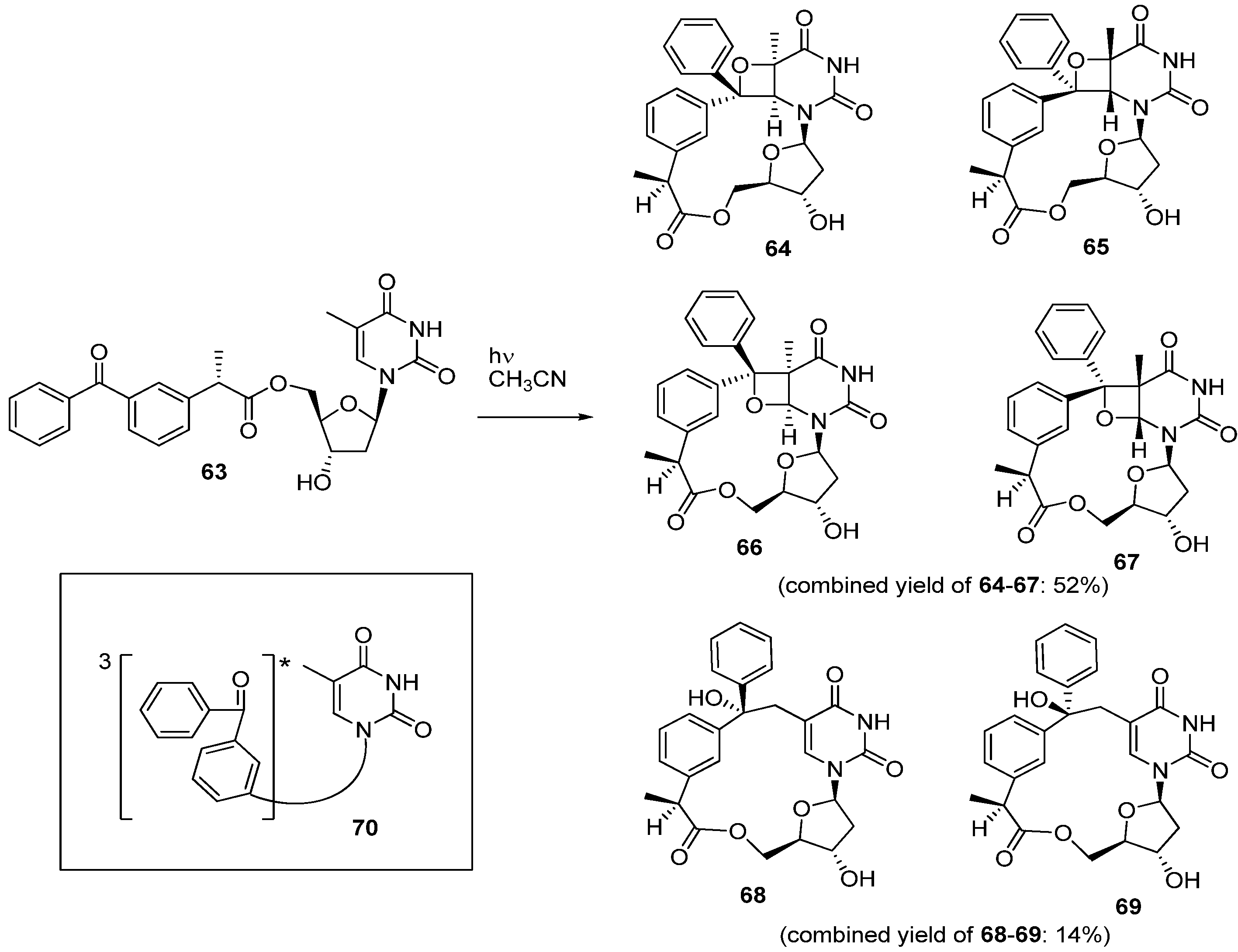
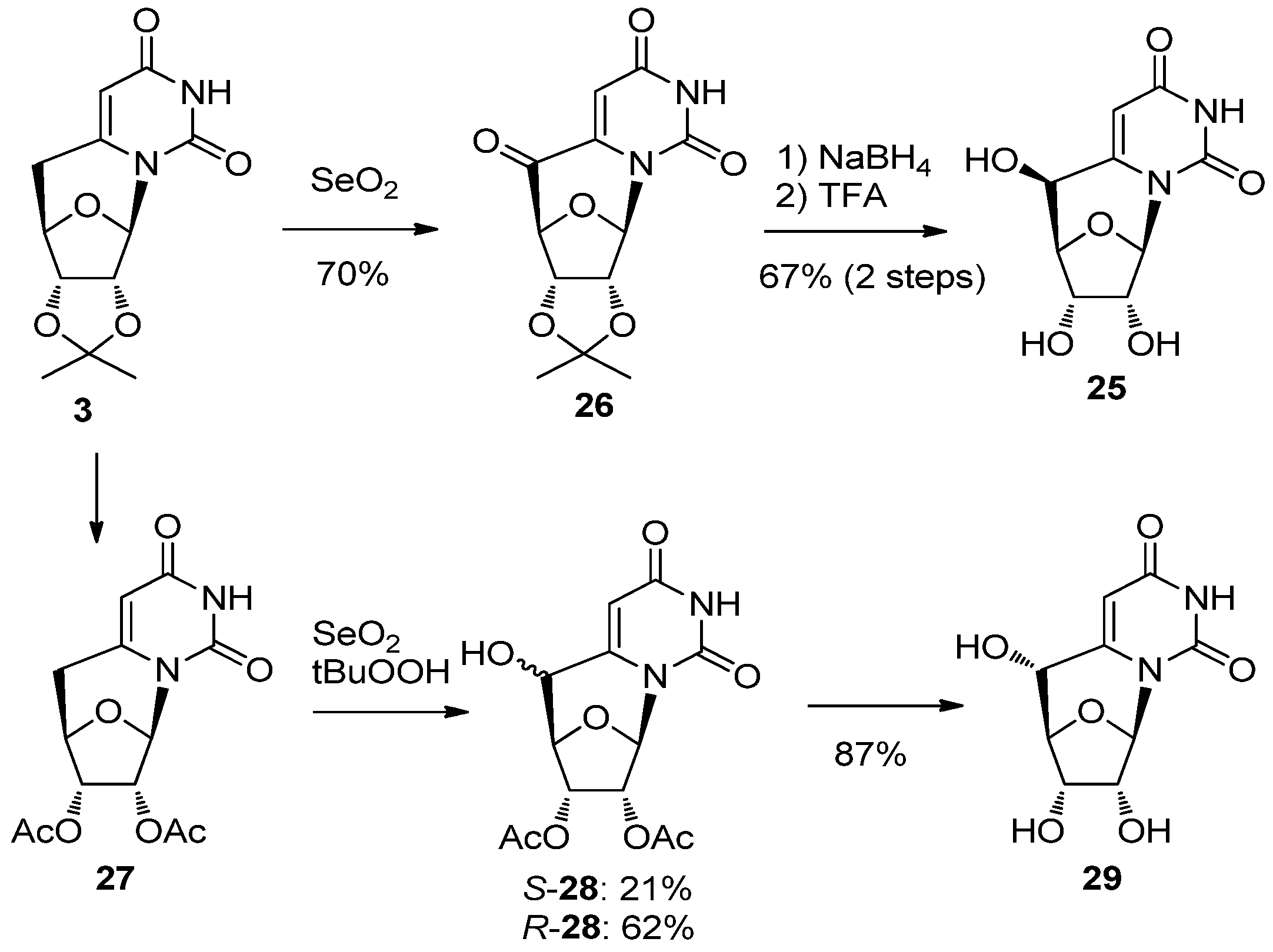
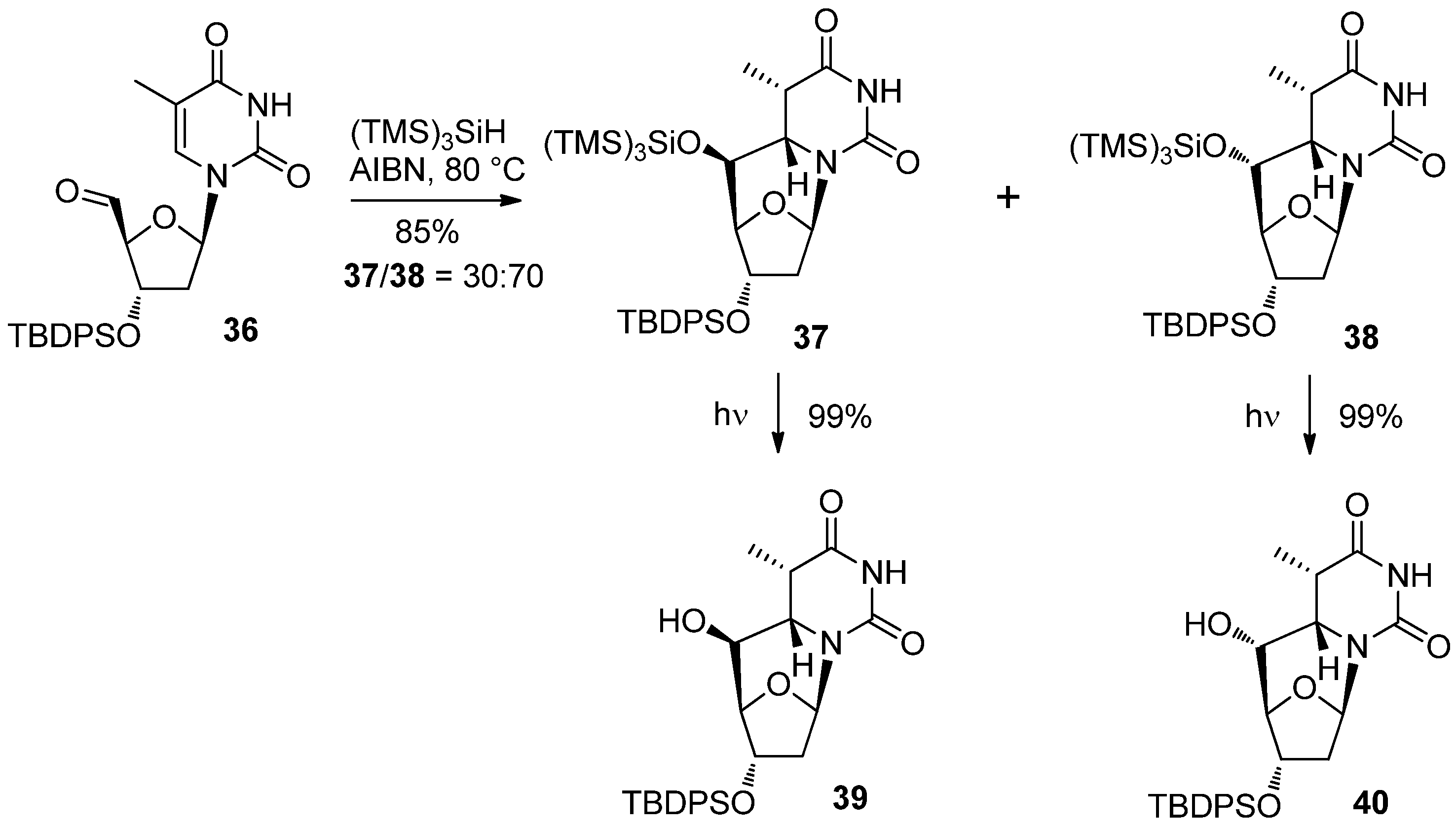
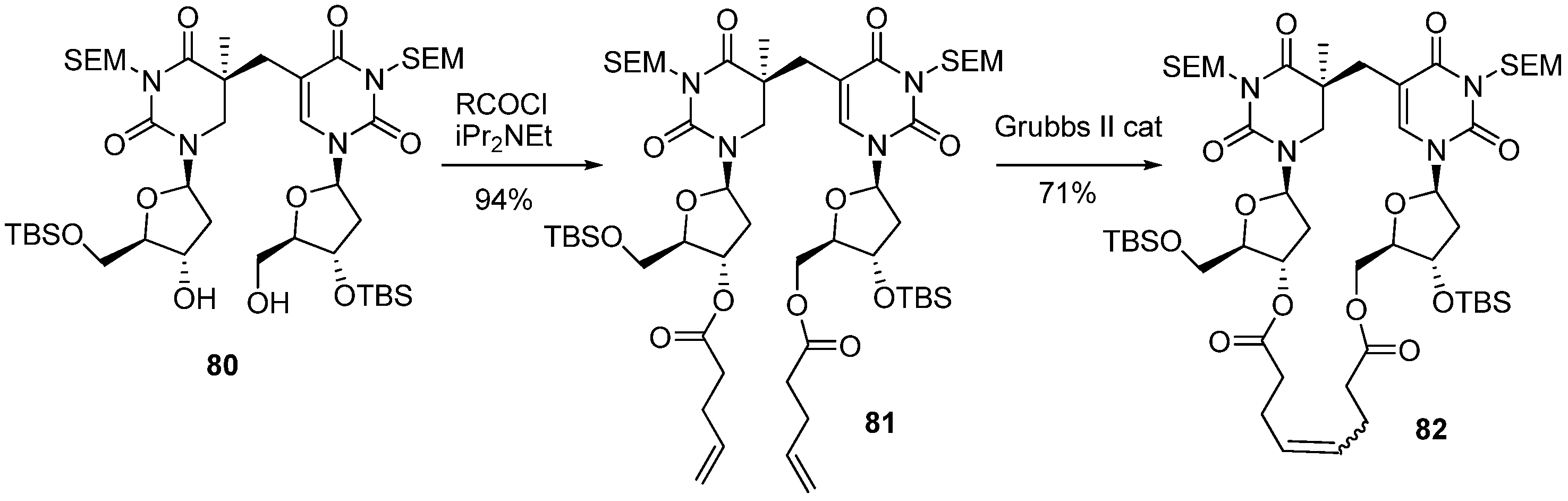
2.2. Recent Advances in the Synthesis of C-Cyclonucleosides
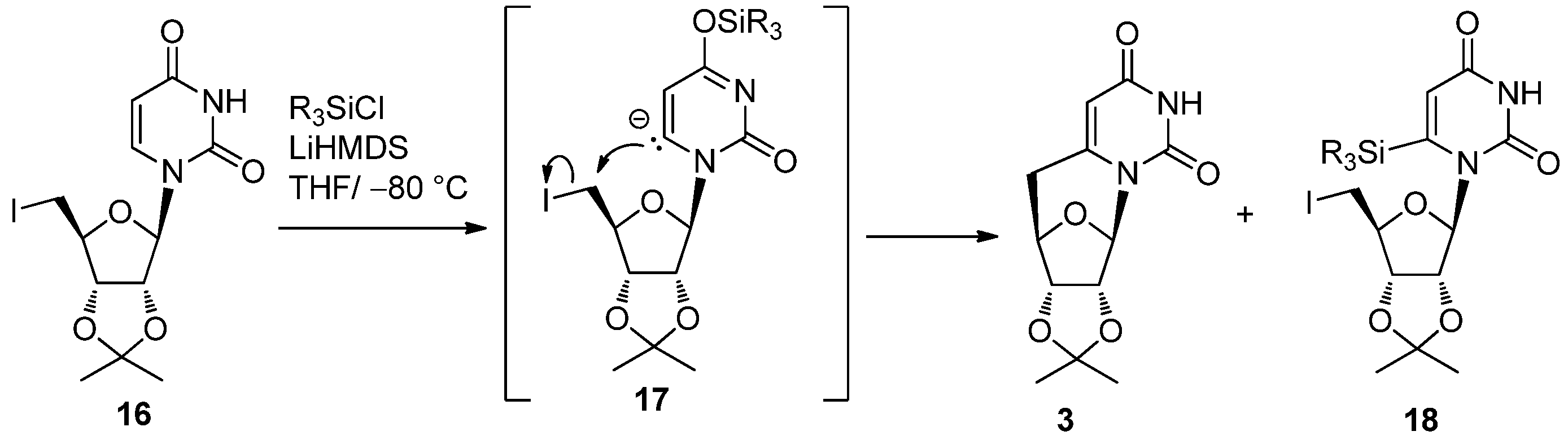

{kind=link}
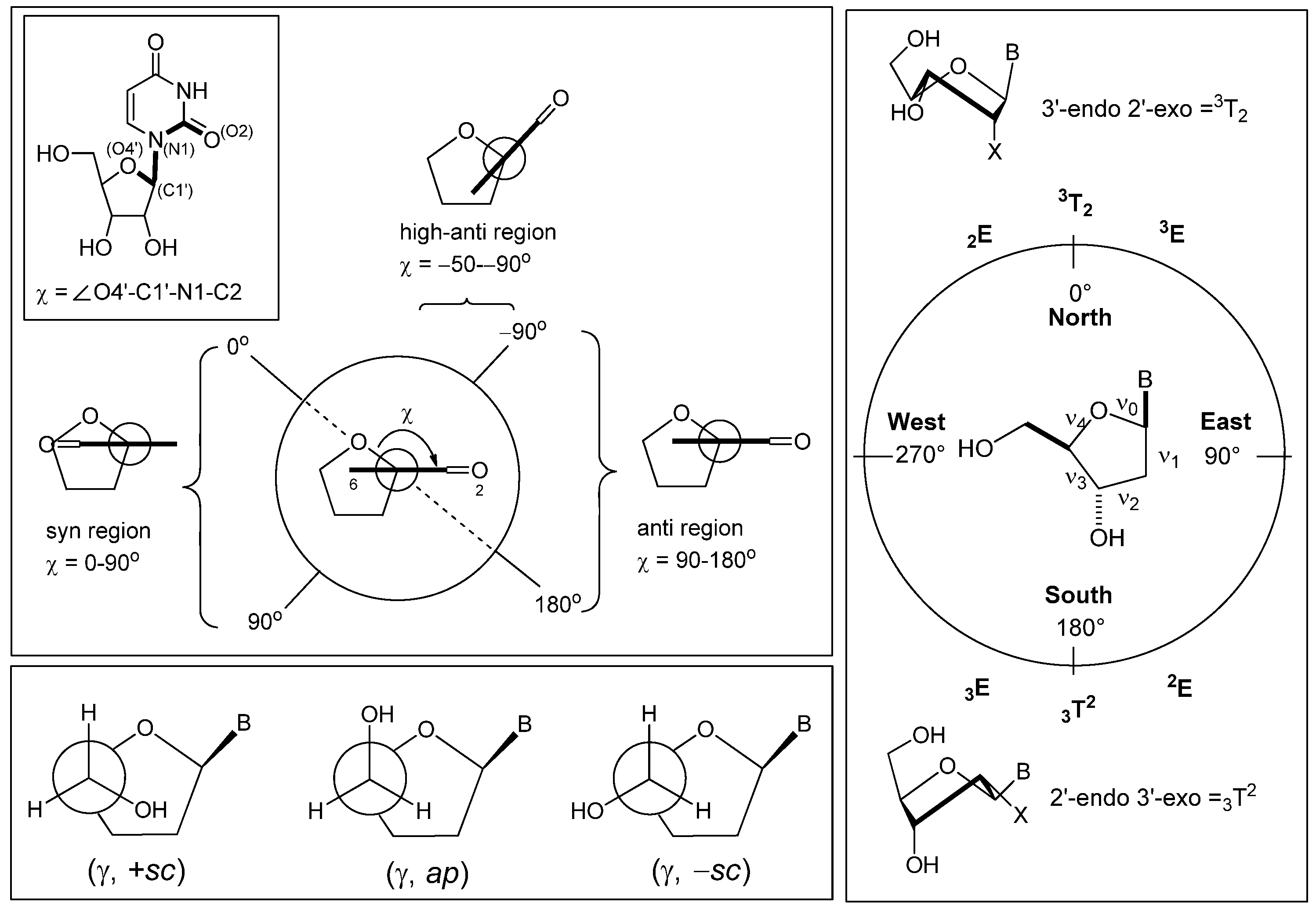
{kind=link}
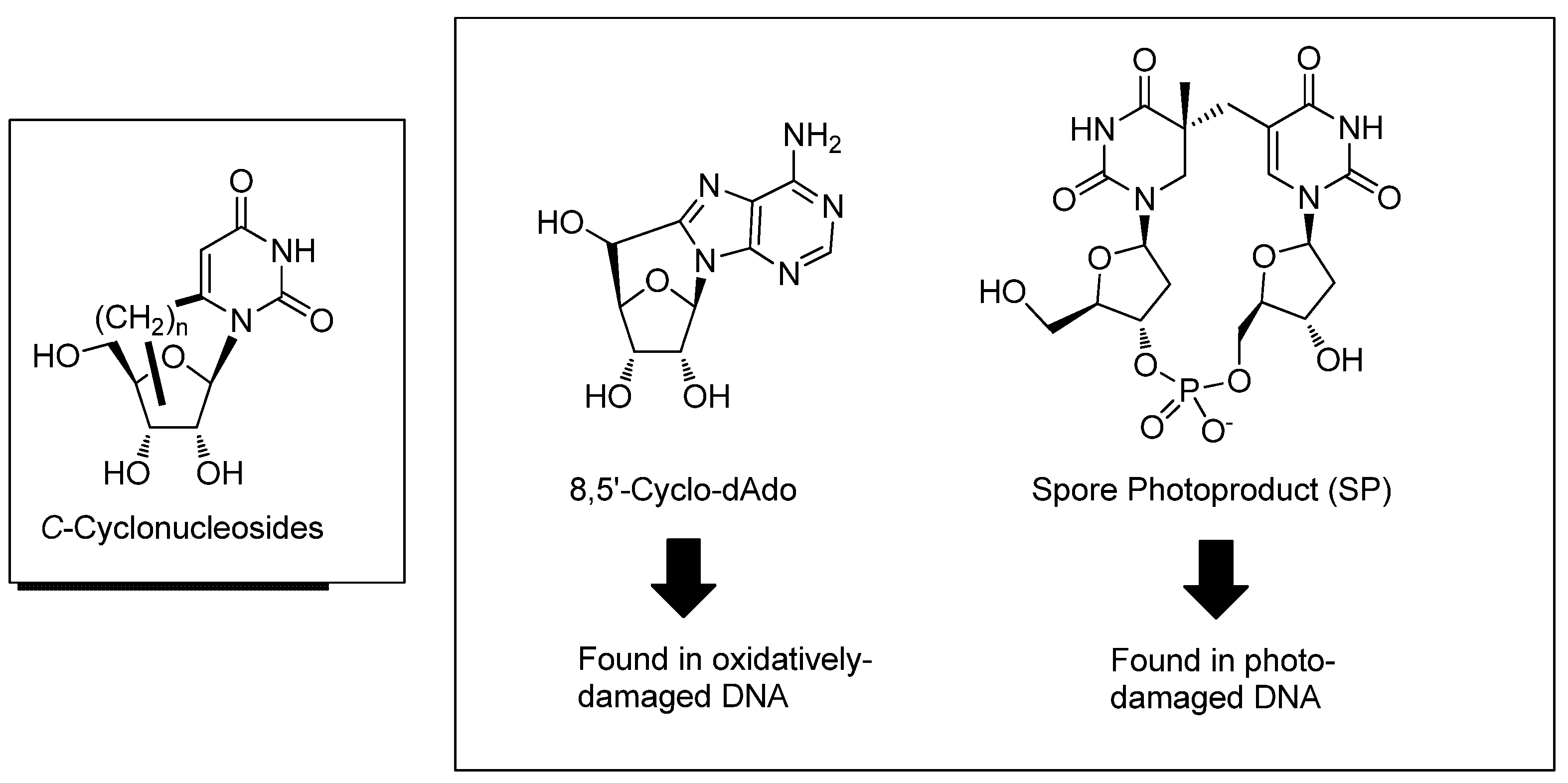
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
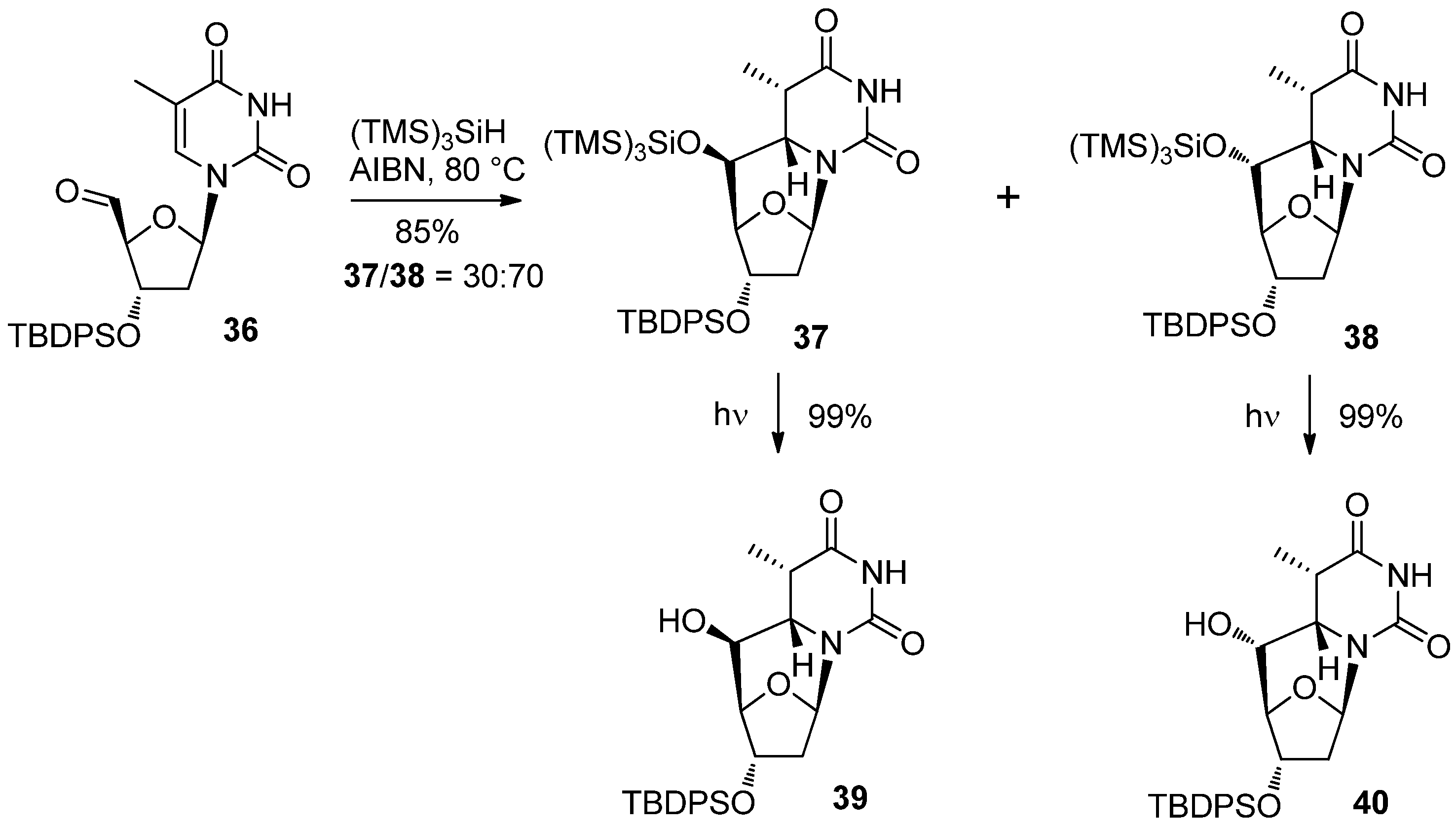{kind=link}
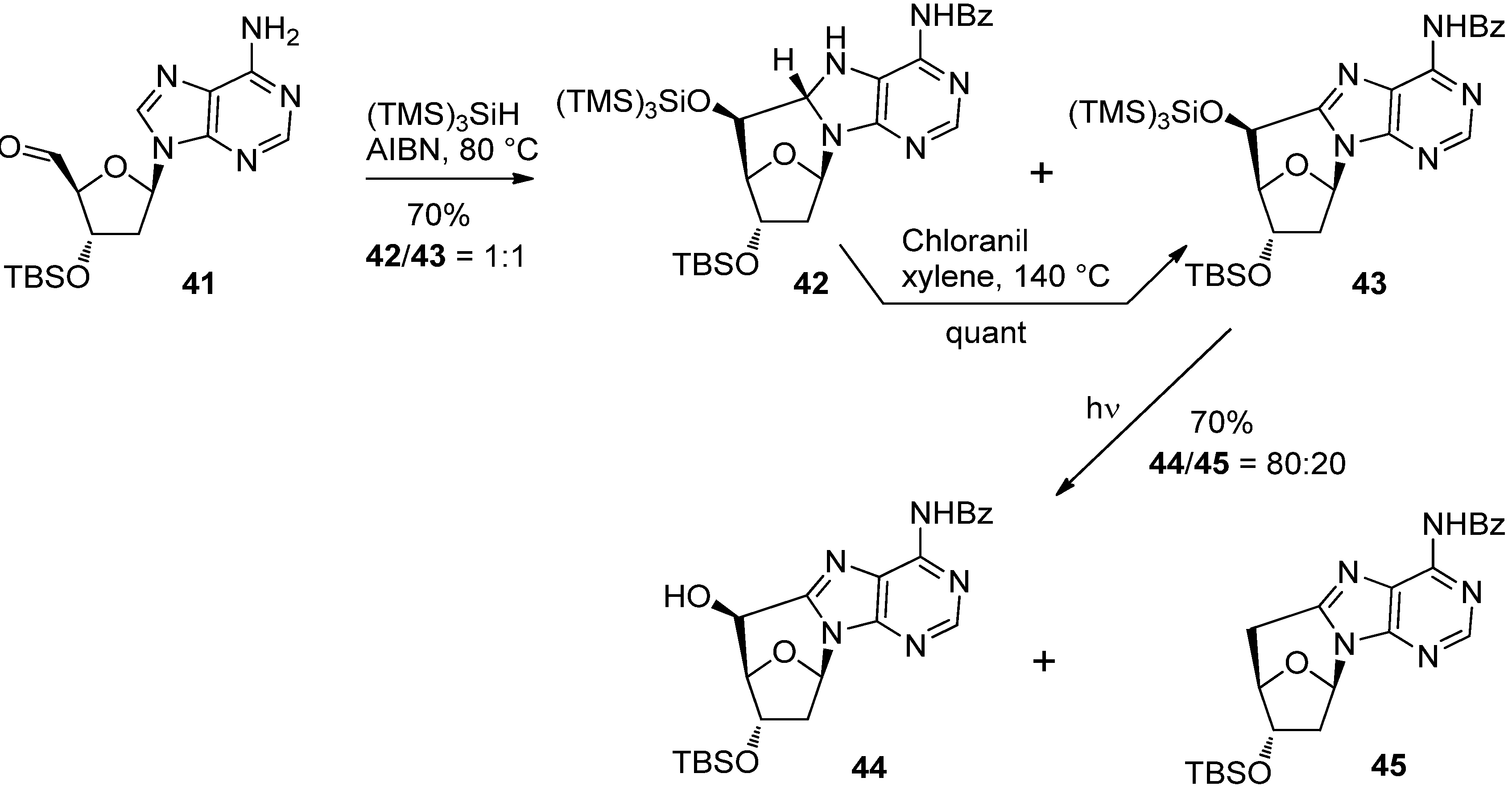{kind=link}
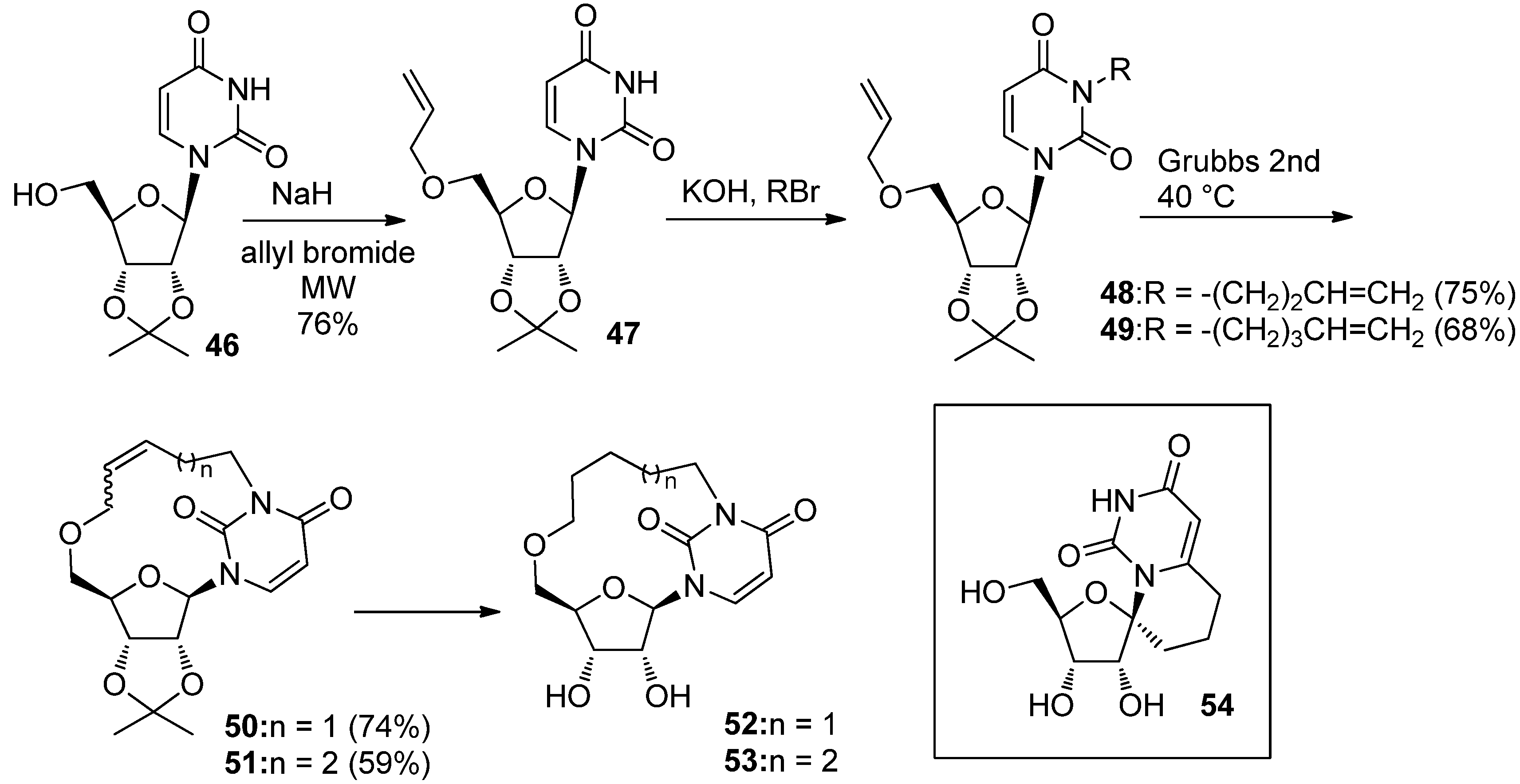{kind=link}
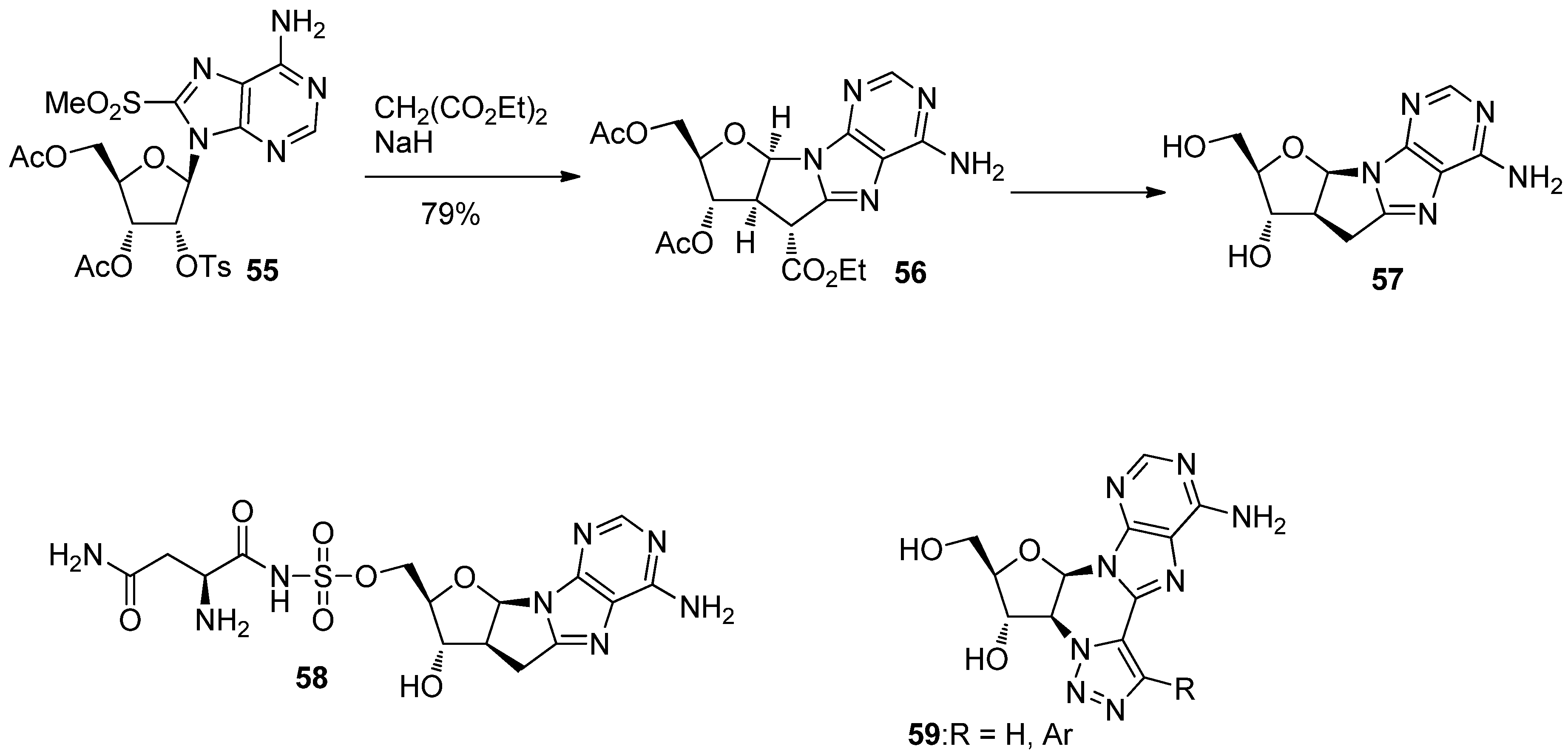{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R3SiCl (equiv.) | Yield (%) | |
|---|---|---|
| 3 | 18 | |
| None | 0 | 0 |
| TMSCl (3.0) | 56 | 42 |
| Me(Ph)2SiCl (1.5) | 83 | trace |
| Ph2SiCl (3.0) | 88 | trace |



| Comp. | Substituents | Conditions | Solvent | Relative yields (%) | 34/35 ratio | ||
|---|---|---|---|---|---|---|---|
| 34 | 35 | 31 | |||||
| 31a | R1 = R2 = H | (TMS)3SiH | CH3CN | 38 | 47 | 15 | 45/55 |
| hν | H2O | 31 | 7 | ND | 82/18 | ||
| 31b | R1 = H, R2 = TBS | (TMS)3SiH | CH3CN | 32 | 38 | 30 | 45/55 |
| hν | CH3CN/H2O | 60 | 25 | 15 | 70/30 | ||
| 31c | R1 = R2 = TBS | (TMS)3SiH | CH3CN | 8 | 70 | 22 | 10/90 |



3. Formation of C-Cyclonucleosides in Damaged DNA
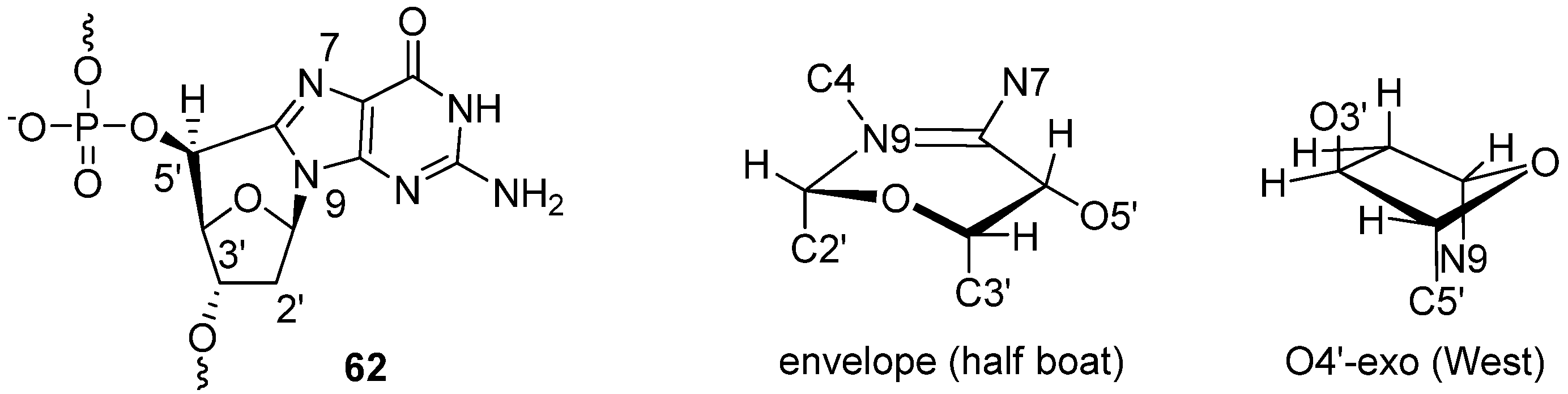
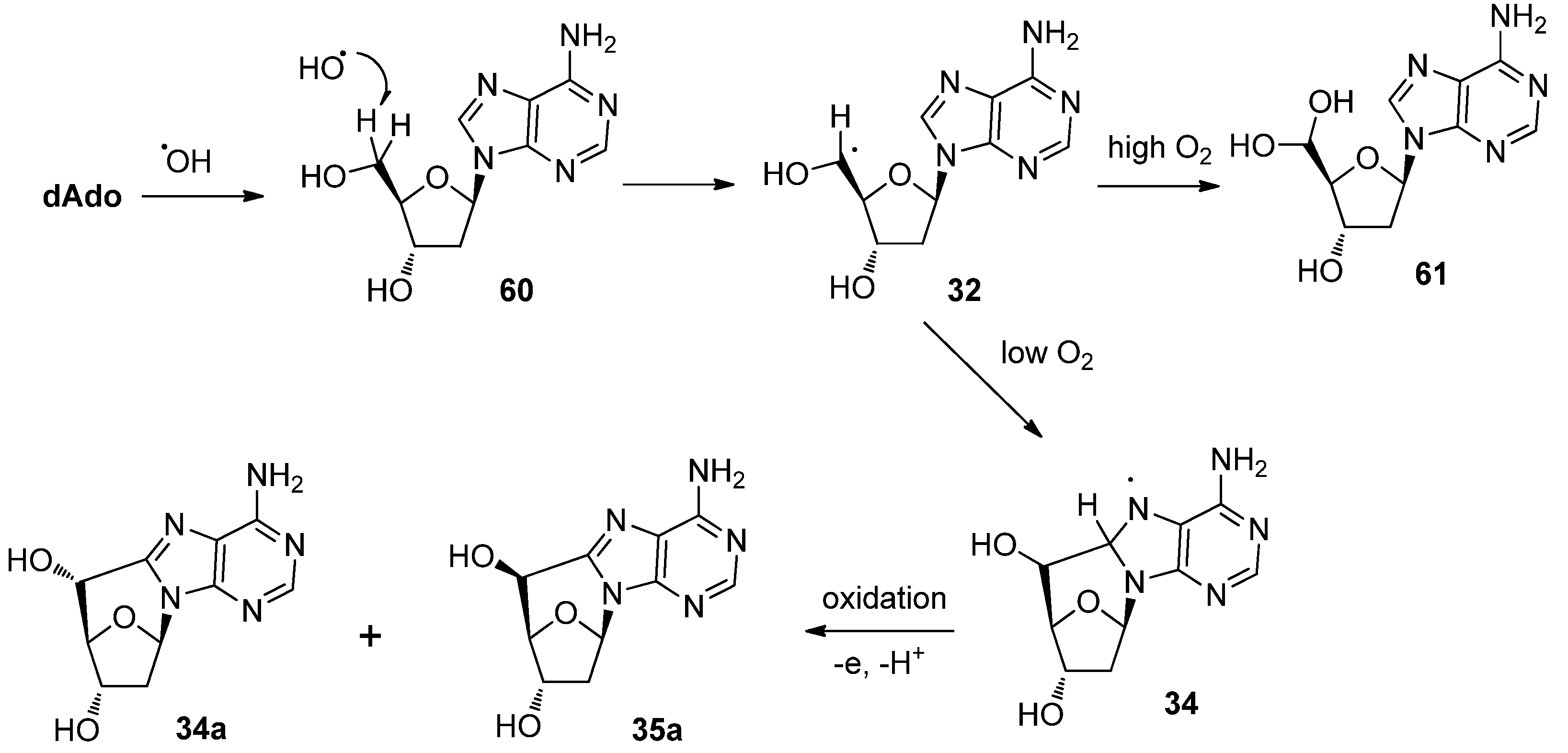
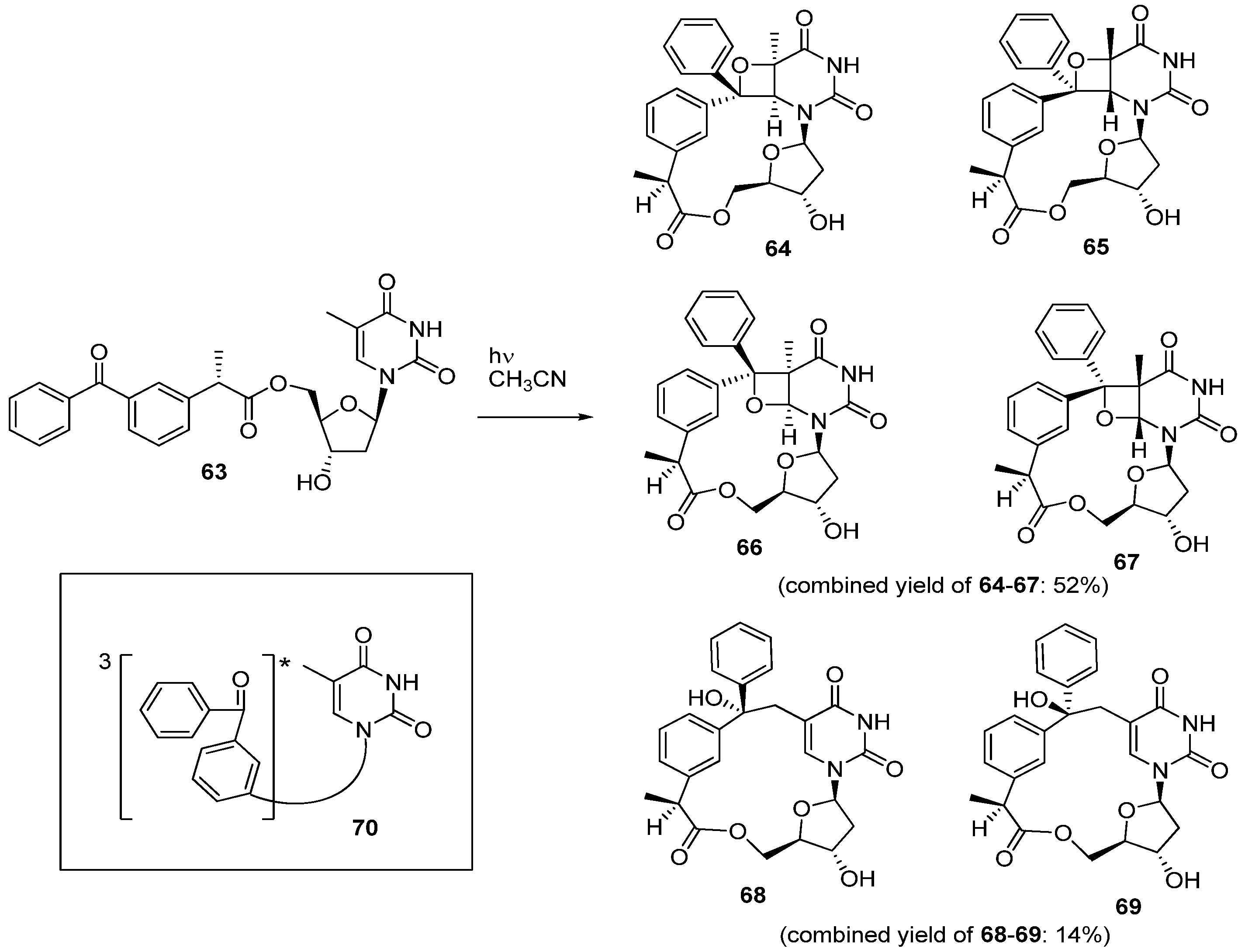
3.1. Purine C-Cyclonucleosides Found in Oxidatively-damaged Nucleic Acids


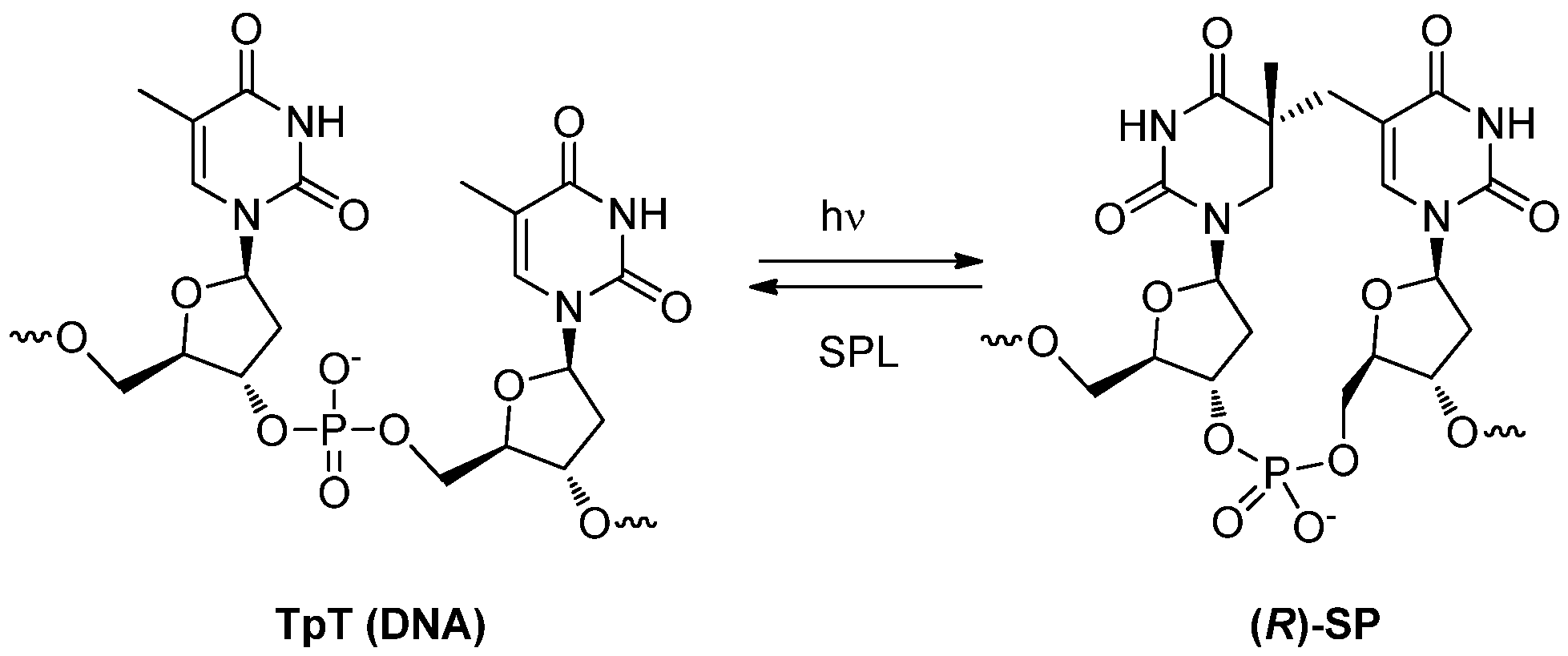
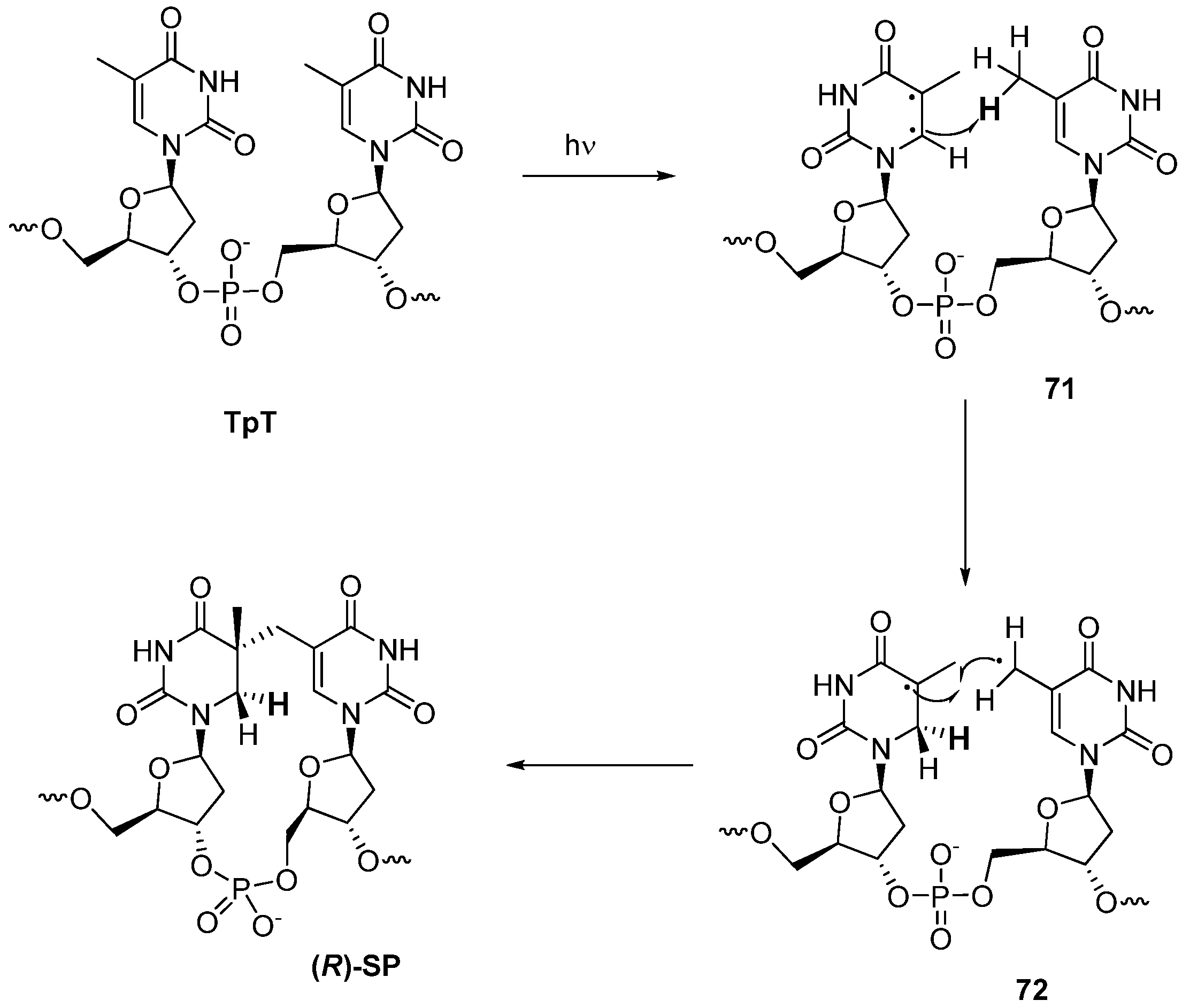
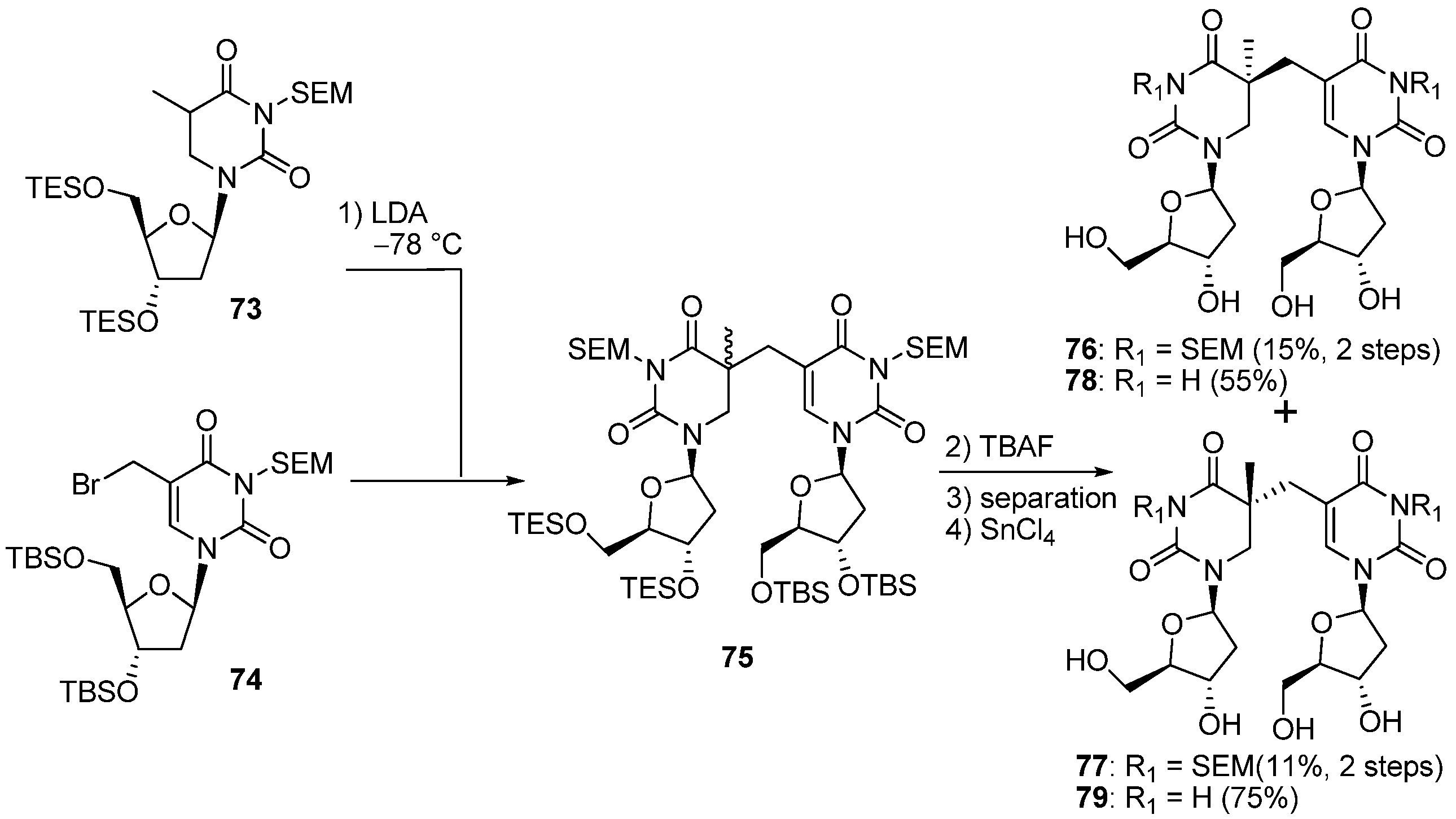
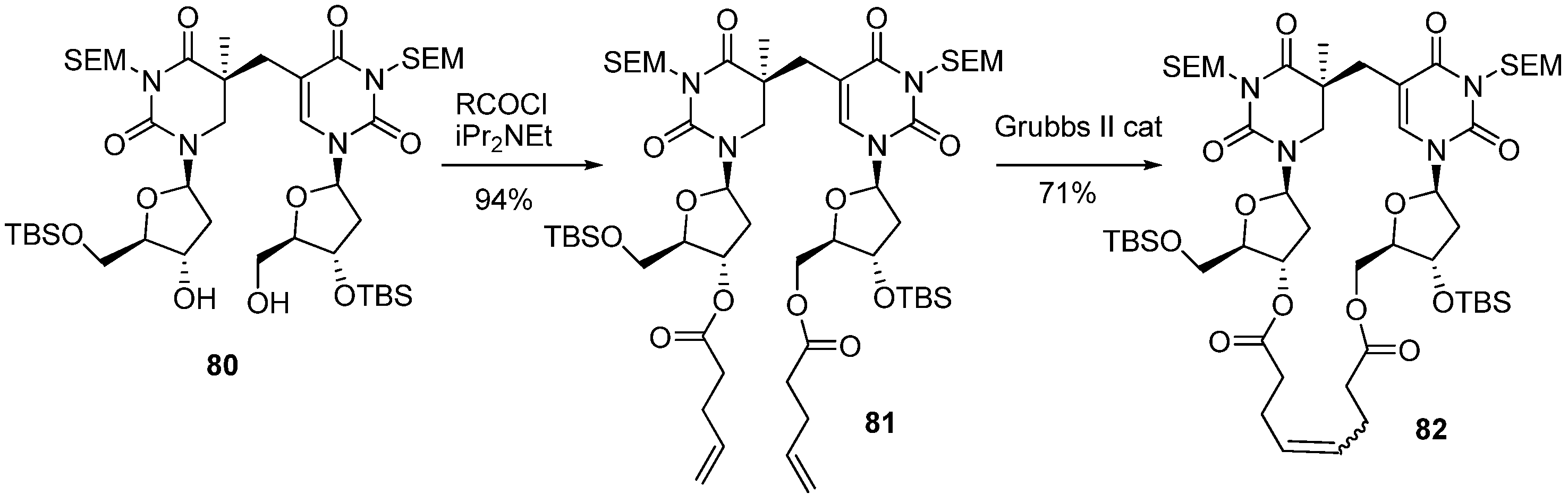
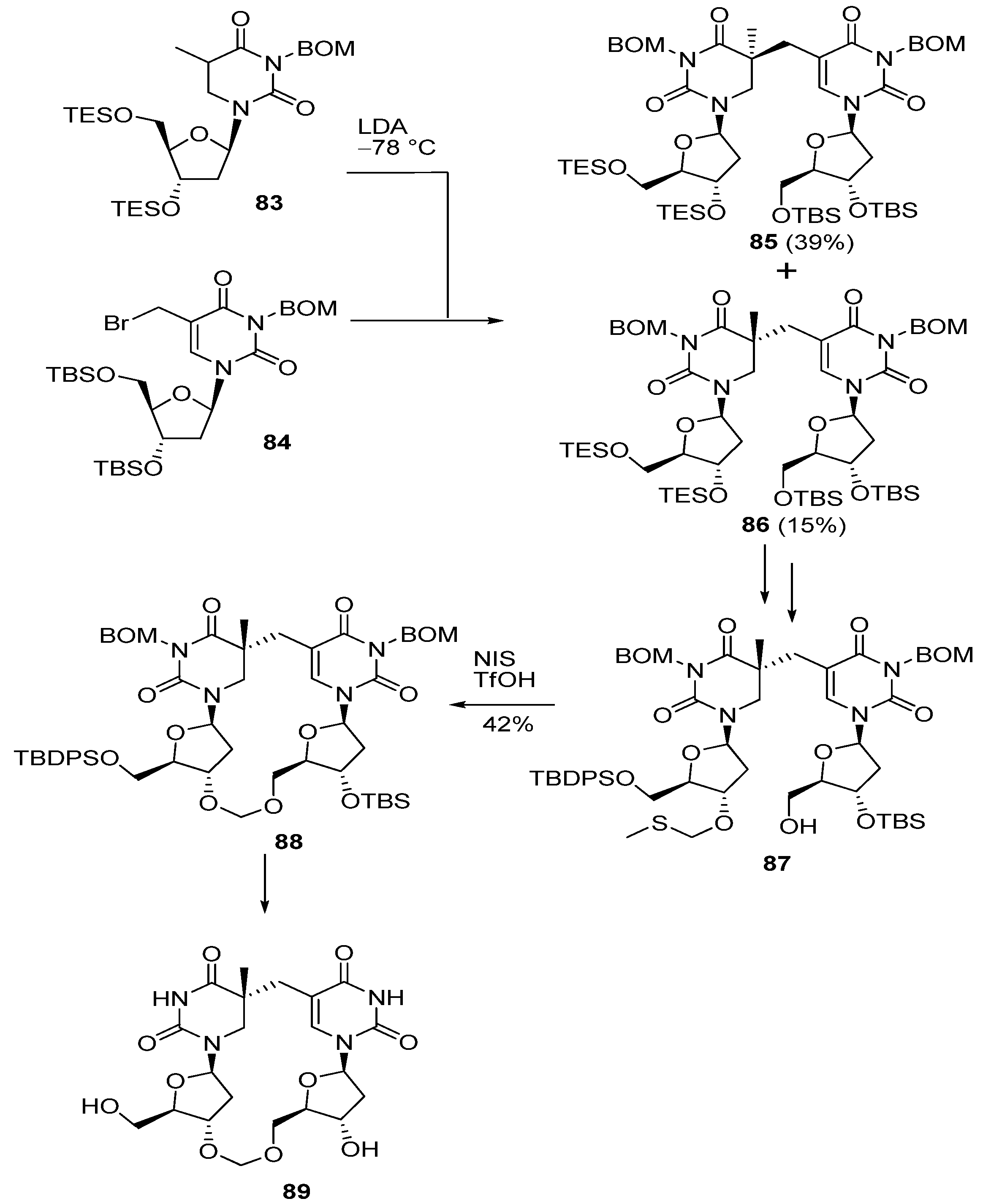
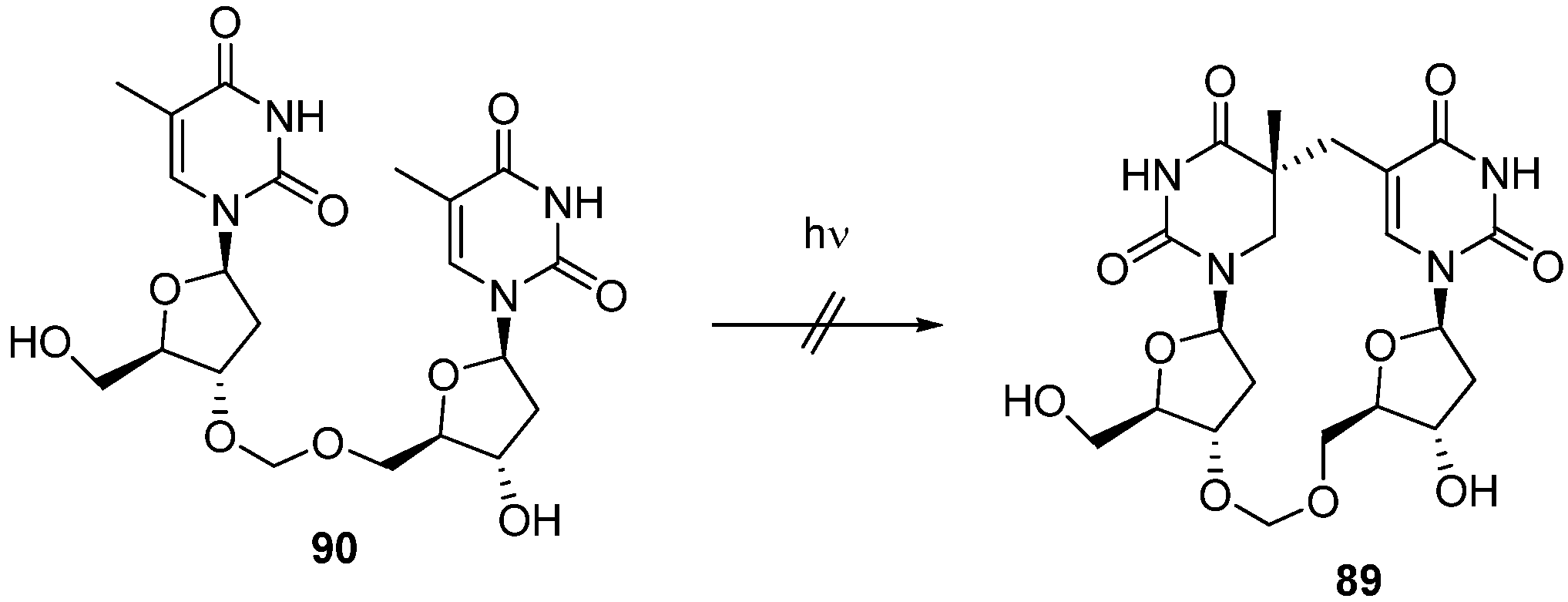
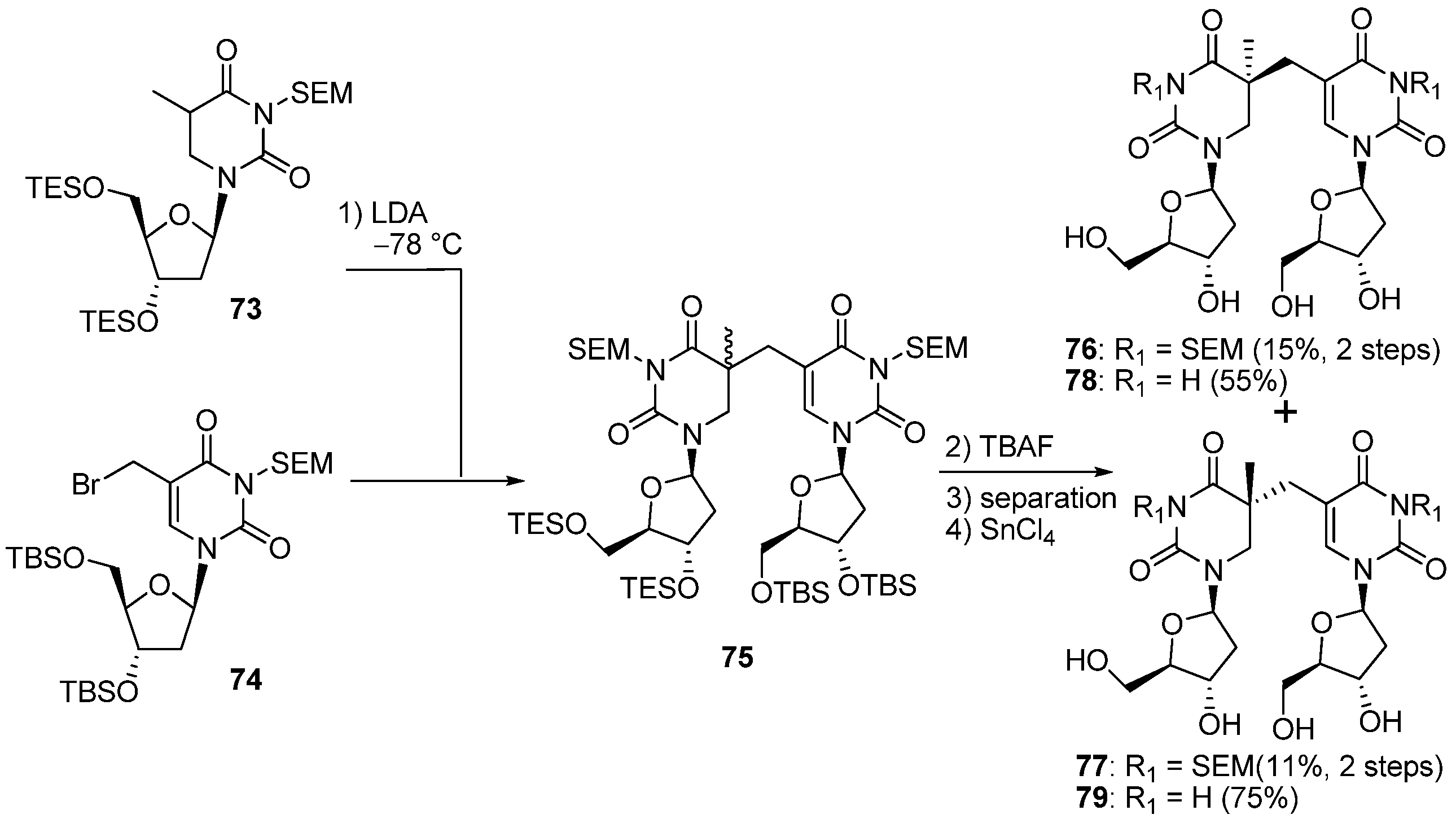
3.2. Formation of a Spore Photoproduct and Its Repair by Spore Photoproduct Lyase





4. Conclusions
References
- Elion, G.B. An overview of the role of nucleosides in chemotherapy. Adv. Enzyme Regul. 1985, 24, 323–334. [Google Scholar] [CrossRef]
- Obata, T.; Endo, Y.; Murata, D.; Sakamoto, K.; Sasaki, T. The molecular targets of antitumor 2′-deoxycytidine analogues. Curr. Drug Targets 2003, 4, 305–313. [Google Scholar] [CrossRef]
- Szafraniec, S.I.; Stachnik, K.J.; Skierski, J.S. New nucleoside analogs in the treatment of hematological disorders. Acta Pol. Pharm. 2004, 61, 223–232. [Google Scholar]
- De Clercq, E. Antiviral drugs in current clinical use. J. Clin. Virol. 2004, 30, 115–133. [Google Scholar] [CrossRef]
- Meng, W.D.; Qing, F.L. Fluorinated nucleosides as antiviral and antitumor agents. Curr. Top. Med. Chem. 2006, 6, 1499–1528. [Google Scholar] [CrossRef]
- De Clercq, E. The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 2007, 6, 1001–1018. [Google Scholar] [CrossRef]
- Cihlar, T.; Ray, A.S. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antiviral Res. 2010, 85, 39–58. [Google Scholar] [CrossRef]
- Dhermain, F.G.; Hau, P.; Lanfermann, H.; Jacobs, A.H.; van den Bent, M.J. Advanced MRI and PET imaging for assessment of treatment response in patients with gliomas. Lancet Neurol. 2010, 9, 906–920. [Google Scholar] [CrossRef]
- Alauddin, M.M.; Gelovani, J.G. Pyrimidine nucleosides in molecular pet imaging of tumor proliferation. Curr. Med. Chem. 2010, 17, 1010–1029. [Google Scholar]
- Saenger, W. Principles of Nucleic Acid Structure; Springer-Verlag: New York, NY, USA, 1984. [Google Scholar]
- Obika, S.; Nanbu, D.; Hari, Y.; Morio, K; In, Y.; Ishida, T.; Imanishi, T. Synthesis of 2′-O,4′-C-methyleneuridine and-cytidine. Novel bicyclic nucleosides having a fixed C3,-endo sugar puckering. Tetrahedron Lett. 1997, 38, 8735–8738. [Google Scholar]
- Singh, S.K.; Nielsen, P.; Koshkin, A.A.; Wengel, J. LNA (locked nucleic acids): Synthesis and high-affinity nucleic acid recognition. Chem. Commun. 1998, 455–456. [Google Scholar]
- Paquette, L.A.; Owen, D.R.; Bibart, R.T.; Seekamp, C.K.; Kahane, A.L. Spirocyclic restriction of nucleosides. An analysis of protecting group feasibility while accessing prototype anti-1-oxaspiro[4.4]nonanyl mimics. Org. Lett. 2001, 3, 4043–4045. [Google Scholar] [CrossRef]
- Marquez, V.E.; Ben-Kasus, T.; Barchi, J.J., Jr.; Green, K.M.; Nicklaus, M.C.; Agbaria, R. Experimental and structural evidence that herpes 1 kinase and cellular DNA polymerase(s) discriminate on the basis of sugar pucker. J. Am. Chem. Soc. 2004, 126, 543–549. [Google Scholar]
- Mieczkowski, A.; Roy, V.; Agrofoglio, L.A. Preparation of cyclonucleosides. Chem. Rev. 2010, 110, 1828–1856. [Google Scholar]
- Len, C.; Mondon, M.; Lebreton, J. Synthesis of cyclonucleosides having a C-C bridge. Tetrahedron 2008, 64, 7453–7475. [Google Scholar] [CrossRef]
- Keck, K. Formation of cyclonucleotides during irradiation of aqueous solutions of purine nucleotides. Z. Naturforsch. B 1968, 23, 1034–1043. [Google Scholar]
- Matsuda, A.; Muneyama, K.; Nishida, T.; Sato, T.; Ueda, T. A new synthesis of 5’-deoxy-8,5′-cyclo-adenosine and -inosine: Conformationally-fixed purine nucleosides (nucleosides and nucleotides. XVI). Nucleic Acids Res. 1976, 3, 3349–3357. [Google Scholar]
- Brooks, P.J. DNA repair in neural cells: Basic science and clinical implications. Mutat. Res. 2002, 509, 93–108. [Google Scholar] [CrossRef]
- Brooks, P.J. The case for 8,5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience 2007, 145, 1407–1417. [Google Scholar] [CrossRef]
- D'Errico, M.; Parlanti, E.; Teson, M.; Degan, P.; Lemma, T.; Calcagnile, A.; Iavarone, I.; Jaruga, P.; Ropolo, M.; Pedrini, A.M.; et al. The role of CSA in the response to oxidative DNA damage in human cells. Oncogene 2007, 26, 4336–4343. [Google Scholar]
- Anderson, K.M.; Jaruga, P.; Ramsey, C.R.; Gilman, N.K.; Green, V.M.; Rostad, S.W.; Emerman, J.T.; Dizdaroglu, M.; Malins, D.C. Structural alterations in breast stromal and epithelial DNA: The influence of 8,5′-cyclo-2′-deoxyadenosine. Cell Cycle 2006, 5, 1240–1244. [Google Scholar] [CrossRef]
- Nyaga, S.G.; Jaruga, P.; Lohani, A.; Dizdaroglu, M.; Evans, M.K. Accumulation of oxidatively induced DNA damage in human breast cancer cell lines following treatment with hydrogen peroxide. Cell Cycle 2007, 6, 1472–1478. [Google Scholar]
- Wang, J.; Clauson, C.L.; Robbins, P.D.; Niedernhofer, L.J.; Wang, Y. The oxidative DNA lesions 8,5'-cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell 2012, 11, 714–716. [Google Scholar] [CrossRef]
- Desnous, C.; Guillaume, D.; Clivio, P. Spore photoproduct: A key to bacterial eternal life. Chem. Rev. 2010, 110, 1213–1232. [Google Scholar]
- Douki, T.; Cadet, J. Formation of the spore photoproduct and other dimeric lesions between adjacent pyrimidines in uvc-irradiated dry DNA. Photochem. Photobiol. Sci. 2003, 2, 433–436. [Google Scholar] [CrossRef]
- Donnellan, J.E.; Setlow, J.R.B. Thymine photoproducts but not thymine dimers found in ultraviolet-irradiated bacterial spores. Science 1965, 149, 308–310. [Google Scholar]
- Ueda, T.; Usui, H.; Shuto, S.; Inoue, H. Synthesis of 6,5’-cyclo-5’-deoxyuridines by radical cyclization: Nucleosides and nucleotides L. Chem. Pharm. Bull. 1984, 32, 3410–3416. [Google Scholar] [CrossRef]
- Matsuda, A.; Tezuka, M.; Ueda, T. Photochemical cyclization of 2′,3′-O-isopropylidene-8-phenylthioadenosine to the 8,5′(R)- and 8,5′(S)-cycloadenosines. Tetrahedron 1978, 34, 2449–2452. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Sano, T.; Matsuda, A.; Ueda, T. Nucleosides and nucleotides. LXXVII. Synthesis of 6,3′-methanocytidine, 6,3′-methanouridine, and their 2′-deoxyribonucleosides. Chem. Pharm. Bull. 1988, 36, 162–167. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Matsuda, A.; Ueda, T. Nucleosides and nucleotides. Part 79. Synthesis of 6,3′-methanothymidine from a ribofuranos-3-ulose and 2,4-dimethoxy-5,6-dimethylpyrimidine. Nucleosides Nucleotides 1988, 7, 409–416. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Matsuda, A.; Ueda, T. Synthesis of 6,6′-Cyclo-5′,6′-dideoxy-1-(β-D-allofuranosyl)cytosine and Related Nucleosides (Nucleosides Nucleotides LXXVIII). Chem. Pharm. Bull. 1989, 37, 660–664. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Matsuda, A.; Ueda, T. Alternative synthesis of 2'-deoxy-6,2'-methano-pyrimidine nucleosides (nucleosides and nucleotides. XC). Chem. Pharm. Bull. 1990, 38, 389–392. [Google Scholar] [CrossRef]
- Tanaka, H.; Hayakawa, H.; Miyasaka, T. “Umpulong” of reactivity at the C-6 position of uridine: a simple and general method for 6-substituted uredines. Tetrahedron 1982, 38, 2635–2642. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Kumamoto, H.; Baba, A.; Takeda, S.; Tanaka, H. Lithiation at the 6-position of uridine with lithium hexamethyldisilazide: Crucial role of temporary silylation. Org. Lett. 2004, 6, 1793–1795. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Yamazaki, Y.; Wachi, K.; Satoh, S.; Takahata, H. Synthesis of 6,5′-C-cyclouridine by a novel tandem radical 1,6-hydrogen transfer and cyclization reaction. Synlett 2007, 111–114. [Google Scholar]
- Theile, C.S.; McLaughlin, L.W. An efficient synthetic approach to 6,5′-(S)- and 6,5′-(R)-cyclouridine. Chem. Commun. 2012, 48, 5587–5589. [Google Scholar]
- Chatgilialoglu, C.; Ferreri, C.; Terzidis, M.A. Purine 5′,8-cyclonucleoside lesions: Chemistry and biology. Chem. Soc. Rev. 2011, 40, 1368–1382. [Google Scholar]
- Flyunt, R.; Bazzanini, R.; Chatgilialoglu, C.; Mulazzani, Q.G. Fate of the 2′-deoxyadenosin-5′-yl radical under anaerobic conditions. J. Am. Chem. Soc. 2000, 122, 4225–4226. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Guerra, M.; Mulazzani, Q.G. Model studies of DNA C5′ radicals. Selective generation and reactivity of 2′-deoxyadenosin-5′-yl radical. J. Am. Chem. Soc. 2003, 125, 3839–3848. [Google Scholar]
- Navacchia, M.L.; Chatgilialoglu, C.; Monteveechi, P.C. C5′-Adenosinyl radical cyclization. A stereochemical investigation. J. Org. Chem. 2006, 71, 4445–4452. [Google Scholar] [CrossRef]
- Sugawara, T.; Otter, B.A.; Ueda, T. A novel stereospecific radical cyclization of 2′,3′-O-isopropylideneuridine and -adenosine 5′-aldehyde to the corresponding 6,5′-cyclodihydrouridine and 8,5′-cycloadenosine derivatives. Tetrahedron Lett. 1988, 29, 75–78. [Google Scholar] [CrossRef]
- Romieu, A.; Gasparutto, D.; Cadet, J. Synthesis and characterization of oligodeoxynucleotides containing the two 5R and 5S diastereomers of (5′S,6S)-5′,6-cyclo-5,6-dihydrothymidine; radiation-induced tandem lesions of thymidine. J. Chem. Soc. Perkin Trans. 1 1999, 1257–1263. [Google Scholar]
- Navacchia, M.L.; Manetto, A.; Montevecchi, P.C.; Chatgilialoglu, C. Radical cyclization approach to cyclonucleosides. Eur. J. Org. Chem. 2005, 4640–4648. [Google Scholar]
- Muller, E.; Gasparutto, D.; Jaquinod, M.; Romieu, A.; Cadet, J. Chemical and biochemical properties of oligonucleotides that contain (5′S,6S)-cyclo-5,6-dihydro-2′-deoxyuridine and (5′S,6S)-cyclo-5,6-dihydrothymidine, two main radiation-induced degradation products of pyrimidine 2′-deoxyribonucleosides. Tetrahedron 2000, 56, 8689–8701. [Google Scholar] [CrossRef]
- Zhong, S.; Mondon, M.; Pilard, S.; Len, C. Synthesis and structure of novel cyclonucleoside analogues of uridine. Tetrahedron 2008, 64, 7828–7836. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Ueda, T.; Matsuda, A. Synthesis of 6,1′-propanouridine, fixed in syn-conformation by a spiro-carbon bridge. Tetrahedron Lett. 1991, 32, 4549–4552. [Google Scholar]
- Yoshimura, Y.; Otter, B.A.; Ueda, T.; Matsuda, A. Nucleosides and nucleotides. 108. Synthesis and optical properties of syn-fixed carbon-bridged pyrimidine cyclonucleosides. Chem. Pharm. Bull. 1992, 40, 1761–1769. [Google Scholar] [CrossRef]
- Gimisis, T.; Chatgilialoglu, C. 1,5-Radical translocation protocol for the generation of C-1′ radicals in nucleosides. Synthesis of spiro nucleosides through a rare 5-endo-trig cyclization. J. Org. Chem. 1996, 61, 1908–1909. [Google Scholar] [CrossRef]
- Kittaka, A.; Asakura, A.; Kuze, T.; Tanaka, H.; Yamada, N.; Nakamura, K. T.; Miyasaka, T. Cyclization reactions of nucleoside anomeric radical with olefin tethered on base: Factors that induce anomeric stereochemistry. J. Org. Chem. 1999, 64, 7081–7093. [Google Scholar]
- Matsuda, A.; Fujisawa, Y.; Yamanaka, M.; Tanaka, H.; Miyasaka, T.; Ueda, T. Synthesis and properties of 2’-deoxy-8,2′-methylene-cycloadenosine. Tetrahedron 1985, 41, 6013–6017. [Google Scholar] [CrossRef]
- Sukuru, S.C.; Crepin, T.; Milev, Y.; Marsh, L.C.; Hill, J.B.; Anderson, R.J.; Morris, J.C.; Rohatgi, A.; O'Mahony, G.; Grotli, M.; Danel, F.; Page, M.G.; Hartlein, M.; Cusack, S.; Kron, M. A.; Kuhn, L.A. Discovering new classes of Brugia malayi asparaginyl-tRNAsynthetase inhibitors and relating specificity to conformational change. J. Comput. Aided Mol. Des. 2006, 20, 159–178. [Google Scholar] [CrossRef]
- Mahony, G.; Ehrman, E.; Grøtli, M. Synthesis and photophysical properties of novel cyclonucleosides—substituent effects on fluorescence emission. Tetrahedron 2008, 64, 7151–7158. [Google Scholar] [CrossRef]
- Jaruga, P.; Dizdaroglu, M. 8,5′-Cyclopurine-2′-deoxynucleosides in DNA: Mechanisms of formation, measurement, repair and biological effects. DNA Repair 2008, 7, 1413–1425. [Google Scholar] [CrossRef]
- Boussicault, F.; Kaloudis, P.; Caminal, C.; Mulazzani, Q.G.; Chatgilialoglu, C. The fate of C5′radicals of purine nucleosides under oxidative conditions. J. Am. Chem. Soc. 2008, 130, 8377–8385. [Google Scholar] [CrossRef]
- Belmadoui, N.; Boussicault, F.; Guerra, M.; Ravanat, J.L.; Chatgilialoglu, C.; Cadet, J. Radiation-induced formation of purine 5',8-cyclonucleosides in isolated and cellular DNA: High stereospecificity and modulating effect of oxygen. Org. Biomol. Chem. 2010, 8, 3211–3219. [Google Scholar] [CrossRef]
- Karwowski, B.T.; Gaillard, J.; Grand, A.; Cadet, J. Effect of (5′S)-5′,8-cyclo-2′-deoxyadenosine on the conformation of di and trinucleotides. A NMR and DFT study. Org. Biomol. Chem. 2008, 6, 3408–3413. [Google Scholar] [CrossRef]
- Kuraoka, I.; Bender, C.; Romieu, A.; Cadet, J.; Wood, R.D.; Lindahl, T. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3832–3837. [Google Scholar]
- Kuraoka, I.; Robins, P.; Masutani, C.; Hanaoka, F.; Gasparutto, D.; Cadet, J.; Wood, R.D.; Lindahl, T. Oxygen free radical damage to DNA. Translesion synthesis by human DNA polymerase eta and resistance to exonuclease action at cyclopurine deoxynucleoside residues. J. Biol. Chem. 2001, 276, 49283–49288. [Google Scholar]
- Marietta, C.; Gulam, H.; Brooks, P.J. A single 8,5′-cyclo-2′-deoxyadenosine lesion in a TATA box prevents binding of the TATA binding protein and strongly reduces transcription in vivo. DNA Repair 2002, 1, 967–975. [Google Scholar] [CrossRef]
- Jasti, V.P.; Das, R.S.; Hilton, B.A.; Weerasooriya, S.; Zou, Y.; Basu, A.K. (5′S)-8,5′-cyclo-2′-deoxyguanosine is a strong block to replication, a potent pol V-dependent mutagenic lesion, and is inefficiently repaired in escherichia coli. Biochemistry 2011, 50, 3862–3865. [Google Scholar]
- Karwowski, B.T. (5′R)-5′,8-Cyclo-2′-deoxyadenosine: NMR and DFT study of its influence on TPOcdA structure. Tetrahedron Asymm. 2008, 19, 2390–2395. [Google Scholar] [CrossRef]
- Huang, H.; Das, R.S.; Basu, A.K.; Stone, M.P. Structure of (5′S)-8,5′-cyclo-2′-deoxyguanosine in DNA. J. Am. Chem. Soc. 2011, 133, 20357–20368. [Google Scholar]
- Schärer, O.D. Chemistry and biology of DNA repair. Angew. Chem. Int. Ed. Engl. 2003, 42, 2946–2974. [Google Scholar] [CrossRef]
- Bohr, V.A.; Dianov, G.L. Oxidative DNA damage processing in nuclear and mitochondrial DNA. Biochimie 1999, 81, 155–160. [Google Scholar] [CrossRef]
- Theruvathu, J.A.; Jaruga, P.; Dizdaroglu, M.; Brooks, P.J. The oxidatively induced DNA lesions 8,5′-cyclo-2′-deoxyadenosine and 8-hydroxy-2′-deoxyadenosine are strongly resistant to acid-induced hydrolysis of the glycosidic bond. Mech. Ageing Dev. 2007, 128, 494–502. [Google Scholar] [CrossRef]
- Brooks, P.J.; Wise, D.S.; Berry, D.A.; Kosmoski, J.V.; Smerdon, M.J.; Somers, R.L.; Mackie, H.; Spoonde, A.Y.; Ackerman, E.J.; Coleman, K.; Tarone, R.E.; Robbins, J.H. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J. Biol. Chem. 2000, 275, 22355–22362. [Google Scholar]
- Jaruga, P.; Xiao, Y.; Vartanian, V.; Lloyd, R.S.; Dizdaroglu, M. Evidence for the involvement of DNA repair enzyme neil1 in nucleotide excision repair of (5′R)- and (5′S)-8,5′-cyclo-2′-deoxyadenosines. Biochemistry 2010, 49, 1053–1055. [Google Scholar]
- Belmadoui, N.; Encinas, S.; Climent, M.J.; Gil, S.; Miranda, M.A. Intramolecular interactions in the triplet excited states of benzophenone-thymine dyads. Chem. Eur. J. 2006, 12, 553–561. [Google Scholar] [CrossRef]
- Lin, G.; Li, L. Elucidation of Spore-photoproduct formation by isotope labeling. Angew. Chem. Int. Ed. Engl. 2010, 49, 9926–9929. [Google Scholar]
- Rebeil, R.; Nicholson, W.L. The subunit structure and catalytic mechanism of the bacillus subtilis DNA repair enzyme spore photoproduct lyase. Proc. Natl. Acad. Sci. USA 2001, 98, 9038–9043. [Google Scholar] [CrossRef]
- Setlow, P. DNA in dormant spores of Bacillus species is in an A-like conformation. Mol. Microbiol. 1992, 6, 563–567. [Google Scholar] [CrossRef]
- Frenkiel-Krispin, D.; Sack, R.; Englander, J.; Shimoni, E.; Eisenstein, M.; Bullitt, E.; Horowitz-Scherer, R.; Hayes, C.S.; Setlow, P.; Minsky, A.; et al. Structure of the DNA-SSPC complex: Implications for DNA packaging, protection, and repair in bacterial spores. J. Bacteriol. 2004, 186, 3525–3530. [Google Scholar]
- Friedel, M.G.; Pieck, J.C.; Klages, J.; Dauth, C.; Kessler, H.; Carell, T. Synthesis and stereochemical assignment of DNA Spore photoproduct analogues. Chem. Eur. J. 2006, 12, 6081–6094. [Google Scholar] [CrossRef]
- Kim, S.J.; Lester, C.; Begley, T.P. Synthesis of the dinucleotide Spore photoproduct. J. Org. Chem. 1995, 60, 6256–6257. [Google Scholar]
- Friedel, M.G.; Berteau, O.; Pieck, J.C.; Atta, M.; Ollagnier-de-Choudens, S.; Fontecave, M.; Carell, T. The spore photoproduct lyase repairs the 5S- and not the 5R-configured Spore photoproduct DNA lesion. Chem. Commun. 2006, 445–447. [Google Scholar]
- Mantel, C.; Chandor, A.; Gasparutto, D.; Douki, T.; Atta, M.; Fontecave, M.; Bayle, P.A.; Mouesca, J.M.; Bardet, M. Combined NMR and DFT studies for the absolute configuration elucidation of the Spore photoproduct, a UV-induced DNA lesion. J. Am. Chem. Soc. 2008, 130, 16978–16984. [Google Scholar]
- Chandra, T.; Silver, S.C.; Zilinskas, E.; Shepard, E.M.; Broderick, W.E.; Broderick, J.B. Spore photoproduct lyase catalyzes specific repair of the 5R but not the 5S spore photoproduct. J. Am. Chem. Soc. 2009, 131, 2420–2421. [Google Scholar]
- Lin, G.; Chen, C.H.; Pink, M.; Pu, J.; Li, L. Chemical synthesis, crystal structure and enzymatic evaluation of a dinucleotide Spore photoproduct analogue containing a formacetal linker. Chem. Eur. J. 2011, 17, 9658–9668. [Google Scholar] [CrossRef]
- Butenandt, J.; Eker, A.P.M.; Carell, T. Synthesis, crystal structure, and enzymatic evaluation of a DNA-photolesion isostere. Chem. Eur. J. 1998, 4, 642–654. [Google Scholar] [CrossRef]
- Heil, K.; Kneuttinger, A.C.; Schneider, S.; Lischke, U.; Carell, T. Crystal structures and repair studies reveal the identity and the base-pairing properties of the UV-induced Spore photoproduct DNA lesion. Chem. Eur. J. 2011, 17, 9651–9657. [Google Scholar] [CrossRef]
- Benjdia, A.; Heil, K.; Barends, T.R.M.; Carell, T.; Schlichting, I. Structural insights into repair of UV-DNA damage by Spore photoproduct lyase, a radical SAM enzyme. Nucleic Acids Res. 2012. [Google Scholar] [CrossRef]
- Hampton, A.; Harper, P.J.; Sasaki, T. Substrate and inhibitor properties of 8,5′-cycloadenosine 5′-phosphate with adenylate kinase, adenylate aminohydrolase, adenylosuccinate lyase, and 5′-nucleotidase. Biochemistry 1972, 11, 4965–4969. [Google Scholar] [CrossRef]
- Stolarski, R.; Dudycz, L.; Shugar, D. NMR studies in the syn-anti dynamic equilibrium in purine nucleosides and nucleotides. Eur. J. Biochem. 1980, 108, 111–121. [Google Scholar] [CrossRef]
- Ueda, T. Synthesis and some biological properties of carbon-bridged cyclonucleosides and their phosphates. Nucleosides Nucleotides 1985, 4, 67–75. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yoshimura, Y.; Takahata, H. Recent Advances in Cyclonucleosides: C-Cyclonucleosides and Spore Photoproducts in Damaged DNA. Molecules 2012, 17, 11630-11654. https://doi.org/10.3390/molecules171011630
Yoshimura Y, Takahata H. Recent Advances in Cyclonucleosides: C-Cyclonucleosides and Spore Photoproducts in Damaged DNA. Molecules. 2012; 17(10):11630-11654. https://doi.org/10.3390/molecules171011630
Chicago/Turabian StyleYoshimura, Yuichi, and Hiroki Takahata. 2012. "Recent Advances in Cyclonucleosides: C-Cyclonucleosides and Spore Photoproducts in Damaged DNA" Molecules 17, no. 10: 11630-11654. https://doi.org/10.3390/molecules171011630