Nucleoside Triphosphates — Building Blocks for the Modification of Nucleic Acids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
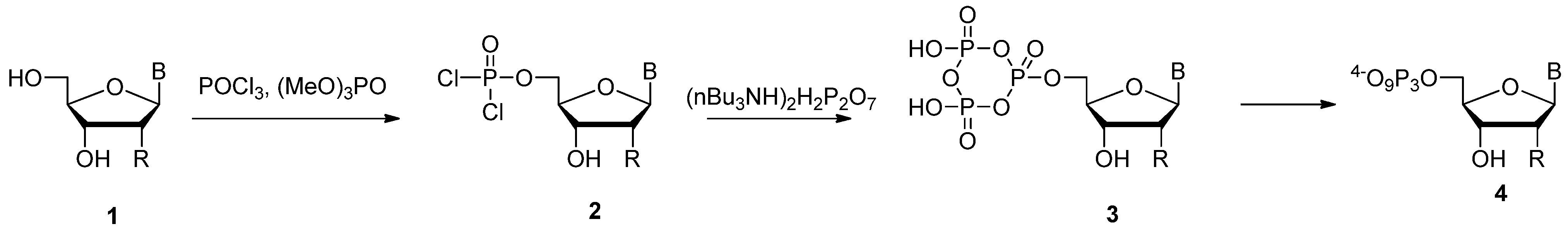{kind=link}
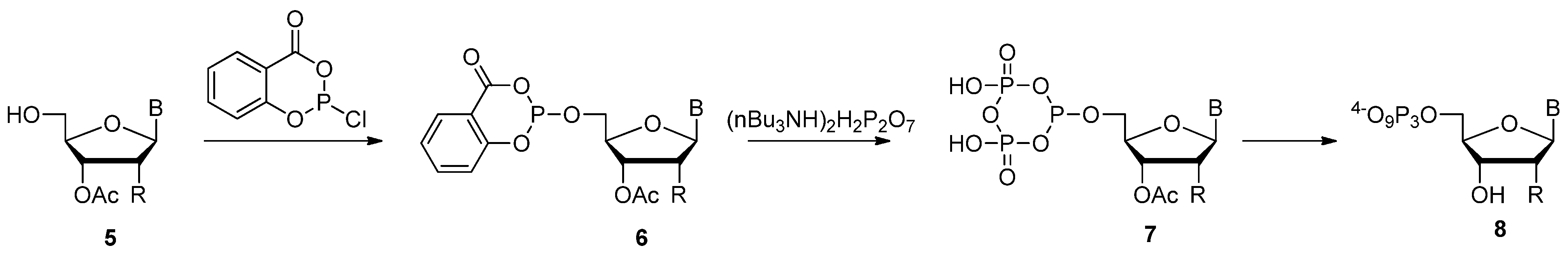{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Synthesis of Modified dNTPs
2.1. Yoshikawa Protocol

2.2. Ludwig-Eckstein Method


2.3. The Borch Approach and Other Strategies


3. Applications of Modified dNTPs
3.1. As Probes for Polymerases and Substrate Acceptance


3.2. Modified dNTPs and SELEX
3.2.1. Selections of Modified DNAzymes


3.2.2. Aptamer Selections

4. Conclusions
Acknowledgments
References
- Lewis, W.; Day, B.J.; Copeland, W.C. Mitochondrial Toxicity of NRTI Antiviral Drugs: An Integrated Cellular Perspective. Nat. Rev. Drug Discov. 2003, 2, 812–822. [Google Scholar] [CrossRef]
- Kranaster, R.; Marx, A. Engineered DNA Polymerases in Biotechnology. ChemBioChem 2010, 11, 2077–2084. [Google Scholar] [CrossRef]
- Kuwahara, M.; Sugimoto, N. Molecular Evolution of Functional Nucleic Acids with Chemical Modifications. Molecules 2010, 15, 5423–5444. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.D. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. [Google Scholar]
- Joyce, G.F. Directed Evolution of Nucleic Acid Enzymes. Annu. Rev. Biochem. 2004, 73, 791–836. [Google Scholar] [CrossRef]
- Burgess, K.; Cook, D. Syntheses of Nucleoside Triphosphates. Chem. Rev. 2000, 100, 2047–2059. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Kato, T.; Takenishi, T. A Novel Method for Phosphorylation of Nucleosides to 5'-Nucleotides. Tetrahedron Lett. 1967, 5065–5068. [Google Scholar]
- Yoshikawa, M.; Kato, T.; Takenishi, T. Studies of phosphorylation III. Selective phosphorylation of unprotected nucleosides. Bull. Chem. Soc. Jpn. 1969, 42, 3505–3508. [Google Scholar] [CrossRef]
- Cramer, F.; Schaller, H. Zur Chemie der energiereichen Phosphate, XIII. Phosphorylierungsreaktionen mit Diestern der Imidazolyl-phosphonsäure und Monoestern der Diimidazolylphosphinsäure. Chem. Ber. 1961, 94, 1634–1640. [Google Scholar] [CrossRef]
- Gillerman, I.; Fischer, B. An Improved One-Pot Synthesis of Nucleoside 5'-Triphosphate analogues. Nucleosides Nucleotides Nucleic Acids 2010, 29, 245–256. [Google Scholar] [CrossRef]
- Baccaro, A.; Marx, A. Enzymatic Synthesis of Organic-Polymer-Grafted DNA. Chem. Eur. J. 2010, 16, 218–226. [Google Scholar] [CrossRef]
- Wang, Y.; Tkachenko, B.A.; Schreiner, P.R.; Marx, A. Diamondoid-modified DNA. Org. Biomol. Chem. 2011, 9, 7482–7490. [Google Scholar] [CrossRef]
- Sakthivel, K.; Barbas, C.F. Expanding the Potential of DNA for Binding and Catalysis: Highly Functionalized dUTP Derivatives That Are Substrates for Thermostable DNA Polymerases. Angew. Chem. Int. Ed. 1998, 37, 2872–2875. [Google Scholar] [CrossRef]
- Thum, O.; Jäger, S.; Famulok, M. Functionalized DNA: A New Replicable Biopolymer. Angew. Chem. Int. Ed. 2001, 40, 3990–3993. [Google Scholar] [CrossRef]
- Jäger, S.; Famulok, M. Generation and Enzymatic Amplification of High-Density Functionalized DNA Double Strands. Angew. Chem. Int. Ed. 2004, 43, 3337–3340. [Google Scholar] [CrossRef]
- Jäger, S.; Rasched, G.; Kornreich-Leshem, H.; Engeser, M.; Thum, O.; Famulok, M. A versatile toolbox for variable DNA functionalization at high density. J. Am. Chem. Soc. 2005, 127, 15071–15082. [Google Scholar]
- Vaish, N.K.; Fraley, A.W.; Szostak, J.W.; McLaughlin, L.W. Expanding the Structural and Functional Diversity of RNA: Analog Uridine Triphosphates as Candidates for in vitro Selection of Nucleic Acids. Nucleic Acids Res. 2000, 28, 3316–3322. [Google Scholar] [CrossRef]
- Lee, S.E.; Sidorov, A.V.; Gourlain, T.; Mignet, N.; Thorpe, S.J.; Brazier, J.A.; Dickman, M.J.; Hornby, D.P.; Grasby, J.A.; Williams, D.M. Enhancing the catalytic repertoire of nucleic acids: A systematic study of linker length and rigidity. Nucleic Acids Res. 2001, 29, 1565–1573. [Google Scholar]
- Roychowdhury, A.; Illangkoon, H.; Hendrickson, C.L.; Benner, S.A. 2'-Deoxycytidines Carrying Amino and Thiol Functionality: Synthesis and Incorporation by Vent (Exo−) Polymerase. Org. Lett. 2004, 6, 489–492. [Google Scholar] [CrossRef]
- Obayashi, T.; Masud, M.M.; Ozaki, A.N.; Ozaki, H.; Kuwahara, M.; Sawai, H. Enzymatic Synthesis of Labeled DNA by PCR Using New Fluorescent Thymidine Nucleotide Analogue and Superthermophilic KOD Dash DNA Polymerase. Bioorg. Med. Chem. Lett. 2002, 12, 1167–1170. [Google Scholar] [CrossRef]
- Sawai, H.; Ozaki, A.N.; Mine, M.; Ozaki, H. Synthesis of New Modified DNAs by Hyperthermophilic DNA Polymerase: Substrate and Template Specificity of Functionalized Thymidine Analogues Bearing an sp3-Hybridized Carbon at the C5 α-Position for Several DNA Polymerases. Bioconjug. Chem. 2002, 13, 309–316. [Google Scholar] [CrossRef]
- Kuwahara, M.; Nagashima, J.-I.; Hasegawa, M.; Tamura, T.; Kitagata, R.; Hanawa, K.; Hososhima, S.-I.; Kasamatsu, T.; Ozaki, H.; Sawai, H. Systematic characterization of 2'-deoxynucleoside-5'-triphosphate analogs as substrates for DNA polymerases by polymerase chain reaction and kinetic studies on enzymatic production of modified DNA. Nucleic Acids Res. 2006, 34, 5383–5394. [Google Scholar]
- Kuwahara, M.; Hanawa, K.; Ohsawa, K.; Kitagata, R.; Ozaki, H.; Sawai, H. Direct PCR amplification of various modified DNAs having amino acids: Convenient preparation of DNA libraries with high-potential activities for in vitro selection. Bioorg. Med. Chem. 2006, 14, 2518–2526. [Google Scholar] [CrossRef]
- Holzberger, B.; Marx, A. Enzymatic synthesis of perfluoroalkylated DNA. Bioorg. Med. Chem. 2009, 17, 3653–3658. [Google Scholar] [CrossRef]
- Hirao, I.; Harada, Y.; Kimoto, M.; Mitsui, T.; Fujiwara, T.; Yokoyama, S. A Two-Unnatural-Base-Pair System toward the Expansion of the Genetic Code. J. Am. Chem. Soc. 2004, 126, 13298–13305. [Google Scholar] [CrossRef]
- Kimoto, M.; Mitsui, T.; Yokoyama, S.; Hirao, I. A Unique Fluorescent Base Analogue for the Expansion of the Genetic Alphabet. J. Am. Chem. Soc. 2010, 132, 4988–4989. [Google Scholar] [CrossRef]
- Seo, Y.J.; Malyshev, D.A.; Lavergne, T.; Ordoukhanian, P.; Romesberg, F.E. Site-Specific Labeling of DNA and RNA Using an Efficiently Replicated and Transcribed Class of Unnatural Base Pairs. J. Am. Chem. Soc. 2011, 133, 19878–19888. [Google Scholar]
- Matray, T.J.; Kool, E.T. Aspecific partner for abasic damage in DNA. Nature 1999, 309, 704–708. [Google Scholar]
- Cheng, Y.; Dai, C.; Peng, H.; Zheng, S.; Jin, S.; Wang, B. Design, Synthesis, and Polymerase-Catalyzed Incorporation of Click-Modified Boronic Acid-TTP Analogues. Chem. Asian J. 2011, 6, 2747–2752. [Google Scholar] [CrossRef]
- Santner, T.; Siegmund, V.; Marx, A.; Micura, R. The synthesis of 2'-methylseleno adenosine and guanosine 5'-triphosphates. Bioorg. Med. Chem. 2012, 20, 2416–2418. [Google Scholar] [CrossRef]
- Siegmund, V.; Santner, T.; Micura, R.; Marx, A. Enzymatic synthesis of 2'-methylseleno-modified RNA. Chem. Sci. 2011, 2, 2224–2231. [Google Scholar] [CrossRef]
- Nawale, G.N.; Gore, K.R.; Höbartner, C.; Pradeepkumar, P.I. Incorporation of 4'-C-aminomethyl-2'-O-methylthymidine into DNA by thermophilic DNA polymerases. Chem. Commun. 2012, 48, 9619–9621. [Google Scholar] [CrossRef]
- Wu, W.; Bergstrom, D.E.; Davisson, V.J. A Combination Chemical and Enzymatic Approach for the Preparation of Azole Carboxamide Nucleoside Triphosphate. J. Org. Chem. 2003, 68, 3860–3865. [Google Scholar] [CrossRef]
- Borsenberger, V.; Kukwikila, M.; Howorka, S. Synthesis and Enzymatic Incorporation of Modified Deoxyuridine Triphosphates. Org. Biomol. Chem. 2009, 7, 3826–3835. [Google Scholar] [CrossRef]
- Ludwig, J.; Eckstein, F. Rapid and efficient synthesis of nucleoside 5'-0-(1-thiotriphosphates), 5'-triphosphates and 2',3'-cyclophosphorothioates using 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one. J. Org. Chem. 1989, 54, 631–635. [Google Scholar] [CrossRef]
- Caton-Williams, J.; Lin, L.; Smith, M.; Huang, Z. Convenient synthesis of nucleoside 50-triphosphates for RNA transcription. Chem. Commun. 2011, 47, 8142–8144. [Google Scholar]
- Lam, C.; Hipolito, C.; Perrin, D.M. Synthesis and Enzymatic Incorporation of Modified Deoxyadenosine Triphosphates. Eur. J. Org. Chem. 2008, 2008, 4915–4923. [Google Scholar]
- Caton-Williams, J.; Smith, M.; Carrasco, N.; Huang, Z. Protection-Free One-Pot Synthesis of 2'-Deoxynucleoside 5'-Triphosphates and DNA Polymerization. Org. Lett. 2011, 13, 4156–4159. [Google Scholar] [CrossRef]
- Vaught, J.D.; Bock, C.; Carter, J.; Fitzwater, T.; Otis, M.; Schneider, D.; Rolando, J.; Waugh, S.; Wilcox, S.K.; Eaton, B.E. Expanding the Chemistry of DNA for in Vitro Selection. J. Am. Chem. Soc. 2010, 132, 4141–4151. [Google Scholar]
- Schoetzau, T.; Langner, J.; Moyroud, E.; Roehl, I.; Vonhoff, S.; Klussmann, S. Aminomodified Nucleobases: Functionalized Nucleoside Triphosphates Applicable for SELEX. Bioconjug. Chem. 2003, 14, 919–926. [Google Scholar] [CrossRef]
- Zou, K.; Horhota, A.; Yu, B.; Szostak, J.W.; McLaughlin, L.W. Synthesis of α-L-Threofuranosyl Nucleoside Triphosphates (tNTPs). Org. Lett. 2005, 7, 1485–1487. [Google Scholar] [CrossRef]
- Horhota, A.; Zou, K.; Ichida, J.K.; Yu, B.; McLaughlin, L.W.; Szostak, J.W.; Chaput, J.C. Kinetic Analysis of an Efficient DNA-Dependent TNA Polymerase. J. Am. Chem. Soc. 2005, 127, 7427–7434. [Google Scholar]
- Veedu, R.N.; Burri, H.V.; Kumar, P.; Sharma, P.K.; Hrdlicka, P.J.; Vester, B.; Wengel, J. Polymerase-directed synthesis of C5-ethynyl locked nucleic acids. Bioorg. Med. Chem. Lett. 2010, 20, 6565–6568. [Google Scholar] [Green Version]
- Veedu, R.N.; Vester, B.; Wengel, J. Enzymatic Incorporation of LNA Nucleotides into DNA Strands. ChemBioChem 2007, 8, 490–492. [Google Scholar] [CrossRef] [Green Version]
- Veedu, R.N.; Vester, B.; Wengel, J. Polymerase directed incorporation studies of LNA-G nucleoside 5'-triphosphate and primer extension involving all four LNA nucleotides. New J. Chem. 2010, 34, 877–879. [Google Scholar] [CrossRef]
- Johannsen, M.W.; Veedu, R.N.; Madsen, A.S.; Wengel, J. Enzymatic polymerisation involving 2'-amino-LNA nucleotides. Bioorg. Med. Chem. Lett. 2012, 22, 3522–3526. [Google Scholar] [CrossRef]
- Hirao, I.; Mitsui, T.; Kimoto, M.; Yokoyama, S. An Efficient Unnatural Base Pair for PCR Amplification. J. Am. Chem. Soc. 2007, 129, 15549–15555. [Google Scholar] [CrossRef]
- Yang, Z.; Sismour, A.M.; Sheng, P.; Puskar, N.L.; Benner, S.A. Enzymatic incorporation of a third nucleobase pair. Nucleic Acids Res. 2007, 35, 4238–4249. [Google Scholar] [CrossRef]
- Hollenstein, M.; Wojciechowski, F.; Leumann, C.J. Polymerase incorporation of pyrene-nucleoside triphosphates. Bioorg. Med. Chem. Lett. 2012, 22, 4428–4430. [Google Scholar] [CrossRef]
- Hollenstein, M. Synthesis of deoxynucleoside triphosphates that include proline, urea, or sulfamide groups and their polymerase incorporation into DNA. Chem. Eur. J. 2012, 18, 13320–13330. [Google Scholar] [CrossRef]
- Wu, W.; Freel Meyers, C.L.; Borch, R.F. A Novel Method for the Preparation of Nucleoside Triphosphates from Activated Nucleoside Phosphoramidates. Org. Lett. 2004, 6, 2257–2260. [Google Scholar] [CrossRef]
- Clark, M.K.; Scott, S.A.; Wojtkowiak, J.; Chirco, R.; Mathieu, P.; Reiners, J.J., Jr.; Mattingly, R.R.; Borch, R.F.; Gibbs, R.A. Synthesis, Biochemical, and Cellular Evaluation of Farnesyl Monophosphate Prodrugs as Farnesyltransferase Inhibitors. J. Med. Chem. 2007, 50, 3274–3282. [Google Scholar] [CrossRef]
- Wu, W.; Sigmond, J.; Peters, G.J.; Borch, R.F. Synthesis and Biological Activity of a Gemcitabine Phosphoramidate Prodrug. J. Med. Chem. 2007, 50, 3743–3746. [Google Scholar] [CrossRef]
- Sun, Q.; Edathil, J.P.; Wu, R.; Smidansky, E.D.; Cameron, C.E.; Peterson, B.R. One-Pot Synthesis of Nucleoside 5'-Triphosphates from Nucleoside 5'-H-Phosphonates. Org. Lett. 2008, 10, 1703–1706. [Google Scholar] [CrossRef]
- Hocek, M.; Fojta, M. Cross-coupling reactions of nucleoside triphosphates followed by polymerase incorporation. Construction and applications of base-functionalized nucleic acids. Org. Biomol. Chem. 2008, 6, 2233–2241. [Google Scholar] [CrossRef]
- Brázdilová, P.; Vrábel, M.; Pohl, R.; Pivoňková, H.; Havran, L.; Hocek, M.; Fojta, M. Ferrocenylethynyl Derivatives of Nucleoside Triphosphates: Synthesis, Incorporation, Electrochemistry, and Bioanalytical Applications. Chem. Eur. J. 2007, 13, 9527–9533. [Google Scholar] [CrossRef]
- Vrábel, M.; Horáková, P.; Pivoňková, H.; Kalachova, L.; Černocká, H.; Cahová, H.; Pohl, R.; Šebest, P.; Havran, L.; Hocek, M.; Fojta, M. Base-modified DNA labeled by [Ru(bpy)3]2+ and [Os(bpy)3]2+ complexes: Construction by polymerase incorporation of modified nucleoside triphosphates, electrochemical and luminescent properties, and applications. Chem. Eur. J. 2009, 15, 1144–1154. [Google Scholar] [CrossRef]
- Cahová, H.; Havran, L.; Brázdilová, P.; Pivoňková, H.; Pohl, R.; Fojta, M.; Hocek, M. Aminophenyl- and Nitrophenyl-Labeled Nucleoside Triphosphates: Synthesis, Enzymatic Incorporation, and Electrochemical Detection. Angew. Chem. Int. Ed. 2008, 47, 2059–2062. [Google Scholar]
- Macíčková-Cahová, H.; Pohl, R.; Horáková, P.; Havran, L.; Špaček, J.; Fojta, M.; Hocek, M. Alkylsulfanylphenyl Derivatives of Cytosine and 7-Deazaadenine Nucleosides, Nucleotides and Nucleoside Triphosphates: Synthesis, Polymerase Incorporation to DNA and Electrochemical Study. Chem. Eur. J. 2011, 17, 5833–5841. [Google Scholar]
- Balintová, J.; Pohl, R.; Horáková, P.; Vidláková, P.; Havran, L.; Fojta, M.; Hocek, M. Anthraquinone as a Redox Label for DNA: Synthesis, Enzymatic Incorporation, and Electrochemistry of Anthraquinone-Modified Nucleosides, Nucleotides, and DNA. Chem. Eur. J. 2011, 17, 14063–14073. [Google Scholar] [CrossRef]
- Raindlová, V.; Pohl, R.; Šanda, M.; Hocek, M. Direct Polymerase Synthesis of Reactive Aldehyde-Functionalized DNA and Its Conjugation and Staining with Hydrazines. Angew. Chem. Int. Ed. 2010, 49, 1064–1066. [Google Scholar]
- Thoresen, L.H.; Jiao, G.-S.; Haaland, W.C.; Metzker, M.L.; Burgess, K. Rigid, Conjugated, Fluoresceinated Thymidine Triphosphates: Syntheses and Polymerase Mediated Incorporation into DNA Analogues. Chem. Eur. J. 2003, 9, 4603–4610. [Google Scholar] [CrossRef]
- Riedl, J.; Pohl, R.; Rulíšek, L.; Hocek, M. Synthesis and Photophysical Properties of Biaryl-Substituted Nucleos(t)ides. Polymerase Synthesis of DNA Probes Bearing Solvatochromic and pH-Sensitive Dual Fluorescent and 19F-NMR Labels. J. Org. Chem. 2012, 77, 1026–1044. [Google Scholar] [CrossRef]
- Riedl, J.; Pohl, R.; Ernsting, N.P.; Orság, P.; Fojta, M.; Hocek, M. Labelling of nucleosides and oligonucleotides by solvatochromic 4-aminophthalimide fluorophore for studying DNA-protein interactions. Chem. Sci. 2012, 3, 2797–2806. [Google Scholar] [CrossRef]
- Raindlová, V.; Pohl, R.; Klepetářová, B.; Havran, L.; Šimková, E.; Horáková, P.; Pivoňková, H.; Fojta, M.; Hocek, M. Synthesis of Hydrazone-Modified Nucleotides and Their Polymerase Incorporation onto DNA for Redox Labeling. ChemPlusChem 2012, 77, 652–662. [Google Scholar] [CrossRef]
- Ikonen, S.; Macíčková-Cahová, H.; Pohl, R.; Šanda, M.; Hocek, M. Synthesis of nucleoside and nucleotide conjugates of bile acids, and polymerase construction of bile acid-functionalized DNA. Org. Biomol. Chem. 2010, 8, 1194–1201. [Google Scholar] [CrossRef]
- Čapek, P.; Cahová, H.; Pohl, R.; Hocek, M.; Gloeckner, C.; Marx, A. An Efficient Method for the Construction of Functionalized DNA Bearing Amino Acid Groups through Cross-Coupling Reactions of Nucleoside Triphosphates Followed by Primer Extension or PCR. Chem. Eur. J. 2007, 13, 6196–6203. [Google Scholar] [CrossRef]
- Raindlová, V.; Pohl, R.; Hocek, M. Synthesis of Aldehyde-Linked Nucleotides and DNA and Their Bioconjugations with Lysine and Peptides through Reductive Amination. Chem. Eur. J. 2012, 18, 4080–4087. [Google Scholar] [CrossRef]
- Alpha-Bazin, B.; bazin, H.; Guillemer, S.; Sauvaigo, S.; Mathis, G. Europium Cryptate Labeled Deoxyuridine-Triphosphate Analog: Synthesis and Enzymatic Incorporation. Nucleos. Nucleot. 2000, 19, 1463–1474. [Google Scholar] [CrossRef]
- Weizman, H.; Tor, Y. Redox-Active Metal-Containing Nucleotides: Synthesis, Tunability, and Enzymatic Incorporation into DNA. J. Am. Chem. Soc. 2002, 124, 1568–1569. [Google Scholar] [CrossRef]
- Weisbrod, S.H.; Marx, A. Novel strategies for the site-specific covalent labelling of nucleic acids. Chem. Commun. 2008, 5675–5685. [Google Scholar] [CrossRef]
- Wicke, L.; Engels, J.W. Postsynthetic on Column RNA Labeling via Stille Coupling. Bioconjug. Chem. 2012, 23, 627–642. [Google Scholar] [CrossRef]
- Ménová, P.; Hocek, M. Preparation of short cytosine-modified oligonucleotides by nicking enzyme amplification reaction. Chem. Commun. 2012, 48, 6921–6923. [Google Scholar]
- Van Ness, J.; van Ness, L.K.; Galas, D.J. Isothermal reactions for the amplification of oligonucleotides. Proc. Natl. Acad. Sci. USA 2003, 100, 4504–4509. [Google Scholar] [CrossRef]
- Gramlich, P.M.E.; Wirges, C.T.; Manetto, A.; Carell, T. Postsynthetic DNA Modification through the Copper-Catalyzed Azide-Alkyne Cycloaddition Reaction. Angew. Chem. Int. Ed. 2008, 47, 8350–8358. [Google Scholar] [CrossRef]
- El-Sagheer, A.H.; Brown, T. Click chemistry with DNA. Chem. Soc. Rev. 2010, 39, 1388–1405. [Google Scholar] [CrossRef]
- Hocek, M.; Fojta, M. Nucleobase modification as redox DNA labelling for electrochemical detection. Chem. Soc. Rev. 2011, 40, 5802–5814. [Google Scholar] [CrossRef]
- Lauridsen, L.H.; Rothnagel, J.A.; Veedu, R.N. Enzymatic Recognition of 2'-Modified Ribonucleoside 5'-Triphosphates: Towards the Evolution of Versatile Aptamers. ChemBioChem 2012, 13, 19–25. [Google Scholar] [CrossRef]
- Gierlich, J.; Gutsmiedl, K.; Gramlich, P.M.E.; Schmidt, A.; Burley, G.A.; Carell, T. Synthesis of Highly Modified DNA by aCombina tion of PCR with Alkyne-Bearing Triphosphates and Click Chemistry. Chem. Eur. J. 2007, 13, 9486–9494. [Google Scholar]
- Baccaro, A.; Steck, A.-L.; Marx, A. Barcoded Nucleotides. Angew. Chem. Int. Ed. 2012, 51, 254–257. [Google Scholar] [CrossRef]
- Gourlain, T.; Sidorov, A.V.; Mignet, N.; Thorpe, S.J.; Lee, S.E.; Grasby, J.A.; Williams, D.M. Enhancing the catalytic repertoire of nucleic acids. II. Simultaneous incorporation of amino and imidazolyl functionalities by two modified triphosphates during PCR. Nucleic Acids Res. 2001, 29, 1898–1905. [Google Scholar] [CrossRef]
- Loakes, D.; Holliger, P. Polymerase engineering: Towards the encoded synthesis of unnatural biopolymers. Chem. Commun. 2009, 4619–4631. [Google Scholar] [CrossRef]
- Betz, K.; Streckenbach, F.; Schnur, A.; Exner, T.; Welte, W.; Diedrichs, K.; Marx, A. Structures of DNA Polymerases Caught Processing Size-Augmented Nucleotide Probes. Angew. Chem. Int. Ed. 2010, 49, 5181–5184. [Google Scholar] [CrossRef]
- Myers, T.C.; Nakamura, K.; Flesher, J.W. Phosphonic Acid Analogs of Nucleoside Phosphates. I. The Synthesis of 5'-Adenylyl Methylenediphosphonate, a Phosphonic Acid Analog of ATP. J. Am. Chem. Soc. 1963, 85, 3292–3295. [Google Scholar] [CrossRef]
- Herdewijn, P.; Marlière, P. Redesigning the leaving group in nucleic acid polymerization. FEBS Lett. 2012, 586, 2049–2056. [Google Scholar] [CrossRef]
- Sucato, C.A.; Upton, T.G.; Kashemirov, B.A.; Batra, V.K.; Martínek, V.; Xiang, Y.; Beard, W.A.; Pedersen, L.C.; Wilson, S.H.; McKenna, C.E.; et al. Modifying the β,γ Leaving-Group Bridging Oxygen Alters Nucleotide Incorporation Efficiency, Fidelity, and the Catalytic Mechanism of DNA Polymerase β. Biochemistry 2007, 46, 461–471. [Google Scholar]
- Mohamady, S.; Jakeman, D.L. An Improved Method for the Synthesis of Nucleoside Triphosphate Analogues. J. Org. Chem. 2005, 70, 10588–10591. [Google Scholar] [CrossRef]
- Batra, V.K.; Pedersen, L.C.; Beard, W.A.; Wilson, S.H.; Kashemirov, B.A.; Upton, T.G.; Goodman, M.F.; McKenna, C.E. Halogenated β,γ-Methylene- and Ethylidene-dGTP-DNA Ternary Complexes with DNA Polymerase β: Structural Evidence for Stereospecific Binding of the Fluoromethylene Analogues. J. Am. Chem. Soc. 2010, 132, 7617–7625. [Google Scholar]
- Wu, Y.; Zakharova, V.M.; Kashemirov, B.A.; Goodman, M.F.; Batra, V.K.; Wilson, S.H.; McKenna, C.E. β,γ-CHF- and β,γ-CHCl-dGTP Diastereomers: Synthesis, Discrete 31P-NMR Signatures, and Absolute Configurations of New Stereochemical Probes for DNA Polymerases. J. Am. Chem. Soc. 2012, 134, 8734–8737. [Google Scholar]
- Chamberlain, B.T.; Upton, T.G.; Kashemirov, B.A.; McKenna, C.E. α-Azido Bisphosphonates: Synthesis and Nucleotide Analogues. J. Org. Chem. 2011, 76, 5132–5136. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Zibinsky, M.; Upton, T.G.; Kashemirov, B.A.; McKenna, C.E.; Oertell, K.; Goodman, M.F.; Batra, V.K.; Pedersen, L.C.; Beard, W.A.; et al. Synthesis and biological evaluation of fluorinated deoxynucleotide analogs based on bis-(difluoromethylene)triphosphoric acid. Proc. Natl. Acad. Sci. USA 2010, 107, 15693–15698. [Google Scholar]
- Berman, A.J.; Kamtekar, S.; Goodman, J.L.; Lázaro, J.M.; de Vega, M.; Blanco, L.; Salas, M.; Steitz, T.A. Structures of phi29 DNA polymerase complexed with substrate: The mechanism of translocation in B-family polymerases. EMBO J. 2007, 26, 3494–3505. [Google Scholar] [CrossRef] [Green Version]
- Betz, K.; Malyshev, D.A.; Lavergne, T.; Welte, W.; Diedrichs, K.; Dwyer, T.J.; Ordoukhanian, P.; Romesberg, F.E.; Marx, A. KlenTaq polymerase replicates unnatural base pairs by inducing a Watson-Crick geometry. Nat. Chem. Biol. 2012, 8, 612–614. [Google Scholar] [CrossRef]
- Obeid, S.; Yulikov, M.; Jeschke, G.; Marx, A. Enzymatic Synthesis of Multiple Spin-Labeled DNA. Angew. Chem. Int. Ed. 2008, 47, 6782–6785. [Google Scholar]
- Obeid, S.; Baccaro, A.; Welte, W.; Diedrichs, K.; Marx, A. Structural basis for the synthesis of nucleobase modified DNA by Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA 2010, 107, 21327–21331. [Google Scholar]
- Bergen, K.; Steck, A.-L.; Strütt, S.; Baccaro, A.; Welte, W.; Diedrichs, K.; Marx, A. Structures of KlenTaq DNA Polymerase Caught While Incorporating C5-Modified Pyrimidine and C7-Modified 7-Deazapurine Nucleoside Triphosphates. J. Am. Chem. Soc. 2012, 134, 11840–11843. [Google Scholar]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef]
- Joyce, G.F. Forty Years of In Vitro Evolution. Angew. Chem. Int. Ed. 2007, 46, 6420–6436. [Google Scholar] [CrossRef]
- Mayer, G. The Chemical Biology of Aptamers. Angew. Chem. Int. Ed. 2009, 48, 2672–2689. [Google Scholar] [CrossRef]
- Breaker, R.R. Catalytic DNA: In training and seeking employment. Nat. Biotechnol. 1999, 17, 422–423. [Google Scholar] [CrossRef]
- Fiammengo, R.; Jäschke, A. Nucleic acid enzymes. Curr. Opin. Biotechnol. 2005, 16, 614–621. [Google Scholar]
- Dass, C.R. Deoxyribozymes: Cleaving a path to clinical trials. Trends Pharmacol. Sci. 2004, 25, 395–397. [Google Scholar] [CrossRef]
- Dass, C.R.; Choong, P.F.M.; Khachigian, L.M. DNAzyme technology and cancer therapy: Cleave and let die. Mol. Cancer Ther. 2008, 7, 243–251. [Google Scholar]
- Keefe, A.D.; Cload, S.T. SELEX with modified nucleotides. Curr. Opin. Chem. Biol. 2008, 12, 448–456. [Google Scholar] [CrossRef]
- Hollenstein, M. Expanding the Catalytic Repertoire of DNAzymes by Modified Nucleosides. Chimia 2011, 65, 770–775. [Google Scholar] [CrossRef]
- Breaker, R.R.; Joyce, G.F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223–229. [Google Scholar] [CrossRef]
- Santoro, S.W.; Joyce, G.F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 4262–4266. [Google Scholar] [CrossRef]
- Santoro, S.W.; Joyce, G.F.; Sakthivel, K.; Gramatikova, S.; Barbas, C.F. RNA cleavage by a DNA enzyme with extended chemical functionality. J. Am. Chem. Soc. 2000, 122, 2433–2439. [Google Scholar] [CrossRef]
- Perrin, D.M.; Garestier, T.; Hélène, C. Bridging the gap between proteins and nucleic acids: A metal-independent RNAseA mimic with two protein-like functionalities. J. Am. Chem. Soc. 2001, 123, 1556–1563. [Google Scholar] [CrossRef]
- Sidorov, A.V.; Grasby, J.A.; Williams, D.M. Sequence-specific cleavage of RNA in the absence of divalent metal ions by a DNAzyme incorporating imidazolyl and amino functionalities. Nucleic Acids Res. 2004, 32, 1591–1601. [Google Scholar] [CrossRef]
- Hollenstein, M.; Hipolito, C.J.; Lam, C.H.; Perrin, D.M. A self-cleaving DNA enzyme modified with amines, guanidines and imidazoles operates independently of divalent metal cations (M2+). Nucleic Acids Res. 2009, 37, 1638–1649. [Google Scholar] [CrossRef]
- Hollenstein, M.; Hipolito, C.J.; Lam, C.H.; Perrin, D.M. A DNAzyme with Three Protein-Like Functional Groups: Enhancing Catalytic Efficiency of M2+-Independent RNA Cleavage. ChemBioChem 2009, 10, 1988–1992. [Google Scholar] [CrossRef]
- Hipolito, C.J.; Hollenstein, M.; Lam, C.H.; Perrin, D.M. Protein-inspired modified DNAzymes: Dramatic effects of shortening side-chain length of 8-imidazolyl modified deoxyadenosines in selecting RNaseA mimicking DNAzymes. Org. Biomol. Chem. 2011, 9, 2266–2273. [Google Scholar] [CrossRef]
- Perrin, D.M.; Garestier, T.; Hélène, C. Expanding the catalytic repertoire of nucleic acid catalysts: Simultaneous incorporation of two modified deoxyribonucleoside triphosphates bearing ammonium and imidazolyl functionalities. Nucleos. Nucleot. 1999, 18, 377–391. [Google Scholar] [CrossRef]
- Lermer, L.; Roupioz, Y.; Ting, R.; Perrin, D.M. Toward an RNaseA mimic: A DNAzyme with imidazoles and cationic amines. J. Am. Chem. Soc. 2002, 124, 9960–9961. [Google Scholar] [CrossRef]
- Ting, R.; Thomas, J.M.; Lermer, L.; Perrin, D.M. Substrate specificity and kinetic framework of a DNAzyme with an expanded chemical repertoire: A putative RNaseA mimic that catalyzes RNA hydrolysis independent of a divalent metal cation. Nucleic Acids Res. 2004, 32, 6660–6672. [Google Scholar] [CrossRef]
- Ting, R.; Thomas, J.M.; Perrin, D.M. Kinetic characterization of a cis- and trans-acting M2+-independent DNAzyme that depends on synthetic RNaseA-like functionality—Burst-phase kinetics from the coalescence of two active DNAzyme folds. Can. J. Chem. 2007, 85, 313–329. [Google Scholar] [CrossRef]
- Geyer, C.R.; Sen, D. Evidence for the metal-cofactor independence of an RNA phosphodiester-cleaving DNA enzyme. Chem. Biol. 1997, 4, 579–593. [Google Scholar] [CrossRef]
- Li, Y.; Breaker, R.R. Kinetics of RNA Degradation by Specific Base Catalysis of Transesterification Involving the 2'-Hydroxyl Group. J. Am. Chem. Soc. 1999, 121, 5364–5372. [Google Scholar] [CrossRef]
- Schlosser, K.; Li, Y. Biologically Inspired Synthetic Enzymes Made from DNA. Chem. Biol. 2009, 16, 311–322. [Google Scholar] [CrossRef]
- Lam, C.H.; Hipolito, C.J.; Hollenstein, M.; Perrin, D.M. A divalent metal-dependent self-cleaving DNAzyme with a tyrosine side chain. Org. Biomol. Chem. 2011, 9, 6949–6954. [Google Scholar] [CrossRef]
- Wiegand, T.W.; Janssen, R.C.; Eaton, B.E. Selection of RNA amide synthase. Chem. Biol. 1997, 4, 675–683. [Google Scholar] [CrossRef]
- Tarasow, T.M.; Tarasow, S.L.; Eaton, B.E. RNA-catalysed carbon-carbon bond formation. Nature 1997, 389, 54–57. [Google Scholar]
- Liu, J.; Cao, Z.; Lu, Y. Functional Nucleic Acid Sensors. Chem. Rev. 2009, 109, 1948–1998. [Google Scholar] [CrossRef]
- Shamah, S.M.; Healy, J.M.; Cload, S.T. Complex Target SELEX. Acc. Chem. Res. 2008, 41, 130–138. [Google Scholar] [CrossRef]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef]
- Famulok, M.; Hartig, J.S.; Mayer, G. Functional Aptamers and Aptazymes in Biotechnology, Diagnostics, and Therapy. Chem. Rev. 2007, 107, 3715–3743. [Google Scholar] [CrossRef]
- Battersby, T.R.; Ang, D.N.; Burgstaller, P.; Jurczyk, S.C.; Bowser, M.T.; Buchanan, D.D.; Kennedy, R.T.; Benner, S.A. Quantitative Analysis of Receptors for Adenosine Nucleotides Obtained via In Vitro Selection from a Library Incorporating a Cationic Nucleotide Analog. J. Am. Chem. Soc. 1999, 121, 9781–9789. [Google Scholar]
- Huizenga, D.E.; Szostak, J.W. A DNA Aptamer That Binds Adenosine and ATP. Biochemistry 1995, 34, 656–665. [Google Scholar] [CrossRef]
- Lin, C.H.; Patel, D.J. Structural basis of DNA folding and recognition in an AMP-DNA aptamer complex: Distinct architectures but common recognition motifs for DNA and RNA aptamers complexed to AMP. Chem. Biol. 1997, 4, 817–832. [Google Scholar] [CrossRef]
- Hollenstein, M.; Hipolito, C.; Lam, C.; Dietrich, D.; Perrin, D.M. A highly selective DNAzyme sensor for mercuric ions. Angew. Chem. Int. Ed. 2008, 47, 4346–4350. [Google Scholar] [CrossRef]
- Sawai, H.; Ozaki, A.N.; Satoh, F.; Ohbayashi, T.; Masud, M.M.; Ozaki, H. Expansion of structural and functional diversities of DNA using new 5-substituted deoxyuridine derivatives by PCR with superthermophilic KOD Dash DNA polymerase. Chem. Commun. 2001, 2604–2605. [Google Scholar]
- Shoji, A.; Kuwahara, M.; Ozaki, H.; Sawai, H. Modified DNA aptamer that binds the (R)-Isomer of a thalidomide derivative with high enantioselectivity. J. Am. Chem. Soc. 2007, 129, 1456–1464. [Google Scholar] [CrossRef]
- Latham, J.A.; Johnson, R.; Toole, J.J. The application of a modified nucleotide in aptamer selection: Novel thrombin aptamers containing -(1-pentynyl)-2'-deoxyuridine. Nucleic Acids Res. 1994, 22, 2817–2822. [Google Scholar] [CrossRef]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef]
- Schubert, S.; Gül, D.C.; Grunert, H.-P.; Zeichhardt, H.; Erdmann, V.A.; Kurreck, J. RNA cleaving “10-23” DNAzymes with enhanced stability and activity. Nucleic Acids Res. 2003, 31, 5982–5992. [Google Scholar] [CrossRef]
- Fahmy, R.G.; Khachigian, L.M. Locked nucleic acid modified DNA enzymes targeting early growth response-1 inhibit human vascular smooth muscle cell growth. Nucleic Acids Res. 2004, 32, 2281–2285. [Google Scholar] [CrossRef]
- Doessing, H.; Vester, B. Locked and Unlocked Nucleosides in Functional Nucleic Acids. Molecules 2011, 16, 4511–4526. [Google Scholar] [CrossRef] [Green Version]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hollenstein, M. Nucleoside Triphosphates — Building Blocks for the Modification of Nucleic Acids. Molecules 2012, 17, 13569-13591. https://doi.org/10.3390/molecules171113569
Hollenstein M. Nucleoside Triphosphates — Building Blocks for the Modification of Nucleic Acids. Molecules. 2012; 17(11):13569-13591. https://doi.org/10.3390/molecules171113569
Chicago/Turabian StyleHollenstein, Marcel. 2012. "Nucleoside Triphosphates — Building Blocks for the Modification of Nucleic Acids" Molecules 17, no. 11: 13569-13591. https://doi.org/10.3390/molecules171113569