3.1. General
Reagents and solvents were obtained from commercial sources (Sigma-Aldrich, Alfa Aesar) and used as received. Reaction progress was monitored by thin-layer chromatography (TLC) on commercial silica gel plates (KieselGel 60 F245 on aluminum sheets, Merck) and visualized by UV-light (254 and 365 nm). Column chromatography purifications were carried out using silica gel Merck 230–400 Mesh. Melting points were determined on a Quimis Q240.23 apparatus and are uncorrected. 1H- and 13C- nuclear magnetic resonance spectra were recorded in DMSO-d6 solutions on a Bruker Avance 200 MHz or on a Varian 400 MHz instrument. Chemical shift (δ) are given in parts per million (ppm) down field from tetramethylsilane (TMS) and couple constants (J) are given in Hertz (Hz); splitting patterns are reported as follows: s, singlet; d, doublet; dd, doublet of doublets; ddd, doublet of doublets of doublets; t, triplet; q, quartet; m, multiplet; br, broad. Electrospray ionization mass spectra (ESI-MS) were acquired using a Micromass quadrupole-time-of-flight (QTOF) spectrometer operating in a positive mode. Infrared (IR) spectra were performed in bromide potassium (KBr) disks on Nicoleta Magna IR 760 spectrometer. HPLC analysis were performed on a Shimadzu 20A using a C18 column (4,6 mm × 250 mm, 5 μM) eluted with MeCN/H2O (90:10;70:30 or 45:55) over 15 or 20 min at flow rate of 1 mL·min−1, with UV detection at 254 nm.
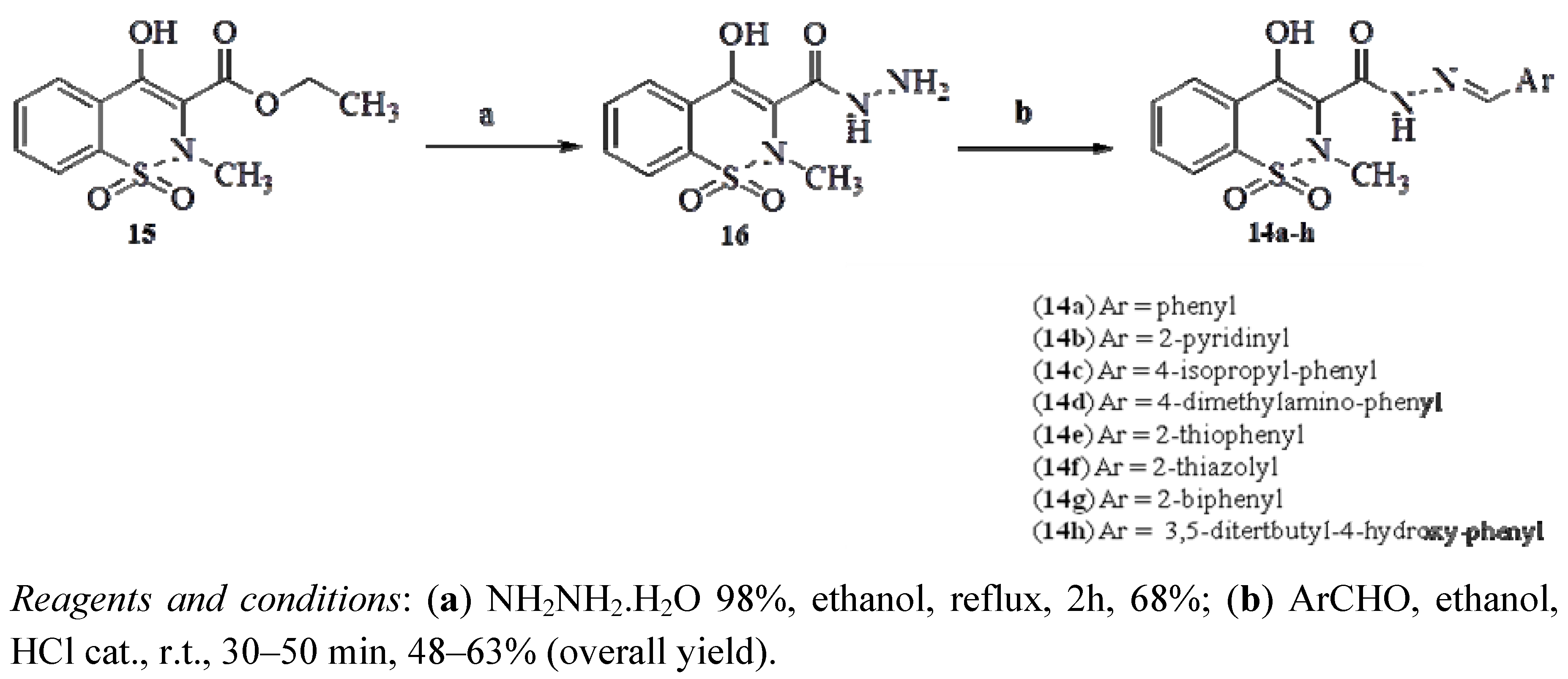
4-Hydroxy-2-methyl-2H-1,2-benzothiazine-3-carbohydrazide 1,1-dioxide (16). A mixture of compound 15 (2.000 g, 7.06 mmol), and hydrazine hydrate 80% (6.8 mL, 137.4 mmol) in ethanol (40,0 mL) was stirred under reflux for 2h, when completion of reaction was indicated by TLC. The mixture was partially concentrated under vacuum, followed by addition of water and HCl 37% until precipitation. The solid was filtered and washed with water and cold ethanol to furnish compound 22 (1.256 g, 68%), which had the following properites: Mp 198–199 °C; Rf = 0.40 (CH2Cl2/MeOH, 9:1). IR (KBr): 3335, 3282, 1621, 1344, 1041 cm−1. 1H-NMR (DMSO-d6): δ = 2.74 (s, 3H, CH3), 7.85–7.91 (m, 3H, ArH), 8.00–7.95 (m, 1H, ArH), 10.15 ppm (br, 1H, CONH). 13C-NMR (DMSO-d6): δ = 110.97, 124.49, 126.44, 128.94, 132.92, 133.85, 134.59, 155.87, 166.44. ESI-HRMS m/z = 270.04543 [M+H]+. Anal. calcd for C10H11N3O4S: C 44.60, H 4.12, N 15.60; found: C 44.62, H 4.11, N 15.44.
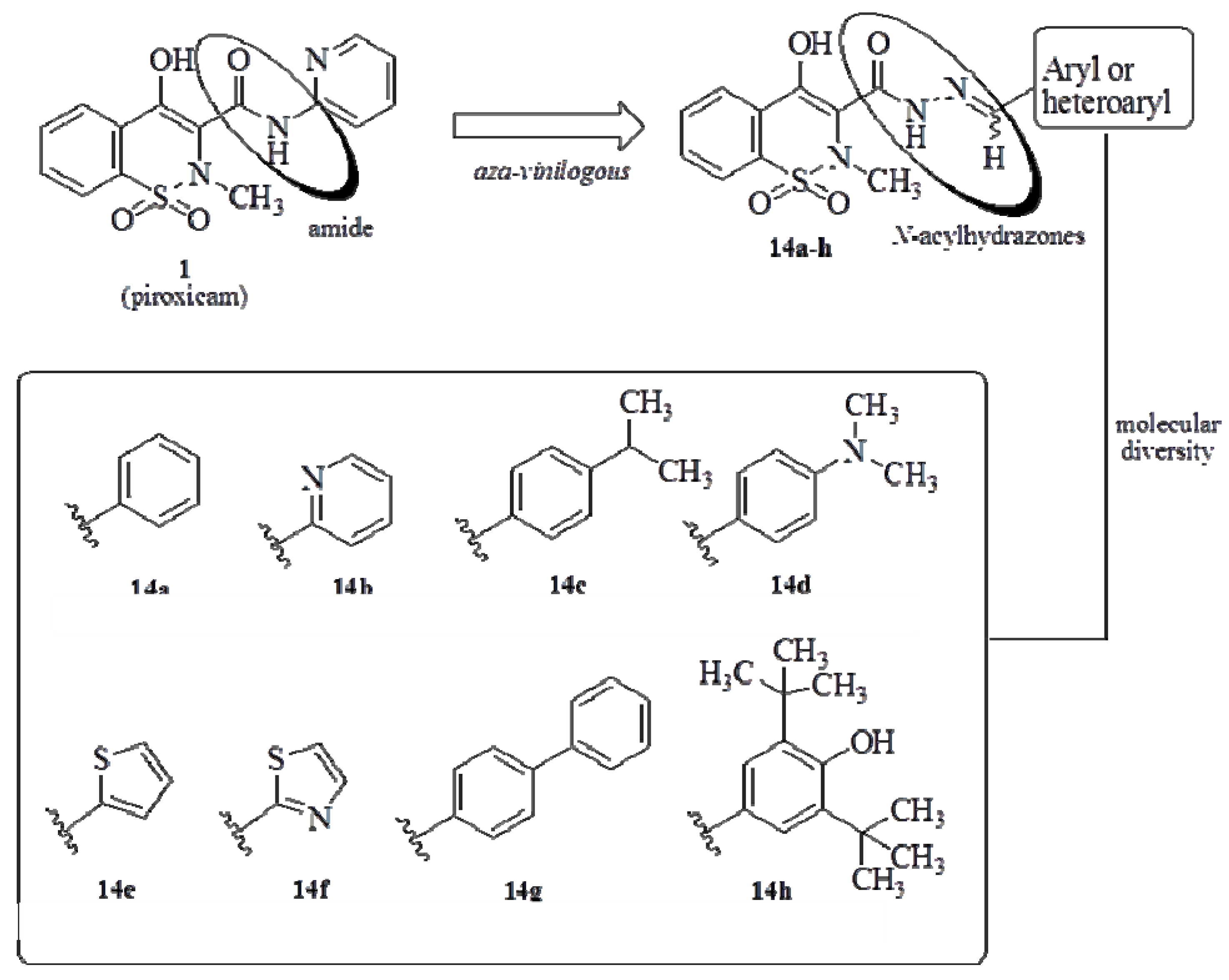
General Procedure for Preparation of Compounds 14a–h
A mixture of compound 16 (0.37 mmol) and the corresponding aromatic or heteroaromatic aldehydes (0.37 mmol) in absolute ethanol (10 mL) containing one drop of 37% hydrochloric acid was stirred at room temperature for ca 30 min, until reaction completion (as indicated by TLC). Then the mixture was poured in cold water and filtered. The residue was washed with water and hot hexane and dried under vacuum to produce the desired N-acylhydrazone derivatives 14a–h. When necessary, further purification was performed by silica gel column chromatography to give compounds 14a–h with purity of over 98% by HPLC.
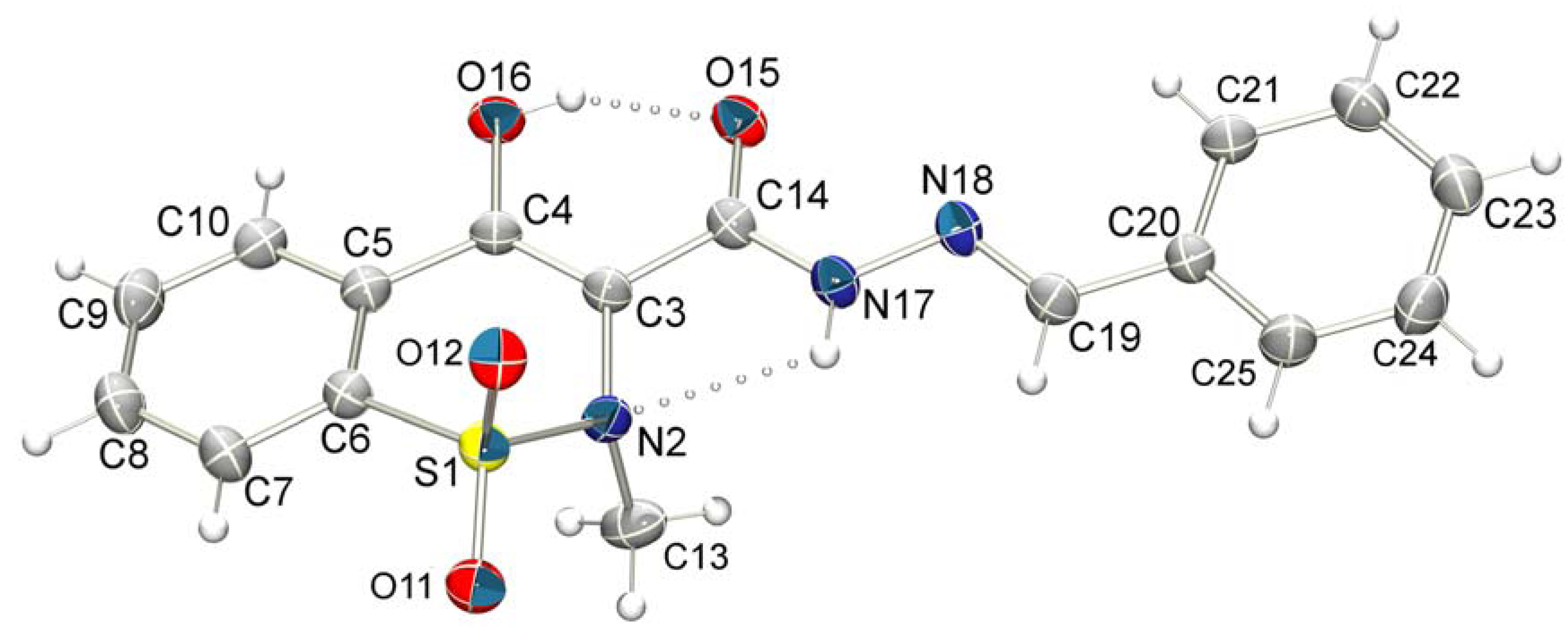
4-Hydroxy-2-methyl-N’-[(E)-phenylmethylidene]-2H-1,2-benzothiazine-3-carbohydrazide 1,1-dioxide (14a). The title compound was obtained by condensation of 16 with benzaldehyde as a white powder (116 mg, 88%). Mp 223–224 °C; Rf = 0.71 (CH2Cl2/MeOH, 9:1). IR (KBr): 3281, 1638, 1618, 1341, 1181, 956 cm−1. 1H-NMR (DMSO-d6): δ = 2.85 (s, 3H, CH3), 7.48-7.49 (m, 3H, ArH), 7.74 (d, J = 2 Hz, 2H, ArH), 7.90–7.92 (m, 3H, ArH), 8.02-8.05 (m, 1H, ArH), 8.68 (s, 1H, N=CH), 11.95 (s, 1H, CONH), 14.23 (br, 1H, OH). 13C-NMR (DMSO-d6): δ = 111.14, 124.75, 126.79, 127.96, 128.32, 129.48, 131.18, 133.62, 134.10, 134.45, 134.76, 151.47, 158.10, 165.63. ESI-HRMS m/z = 358.0856 [M+H]+. Anal. calcd. For C17H15N3O4S: C 57.13, H 4.23, N 11.76; found: C 57.25, H 4.24, N 11.44. HPLC (C18, acetonitrile-water, 7:3, 254 nm): ret. time: 5.868 min; peak area = 98,299%.
4-Hydroxy-2-methyl-N'-[(E)-pyridinyl-2-methylidene]-2H-1,2-benzothiazine-3-carbohydrazide 1,1-dioxide (14b). The title compound was obtained by condensation of 16 with 2-pyridinecarboxaldehyde as a light yellow powder (90 mg, 71%). Mp 253–254 °C; Rf = 0.33 (CH2Cl2/MeOH, 9:1). IR (KBr): 3344, 1666, 1597, 1342, 1177, 962 cm−1. 1H-NMR (DMSO-d6): δ = 2.85 (s, 3H, CH3), 7.44 (m,1H, ArH), 7.85–7.88 (m, 1H, ArH), 7.89–7.90 (t, J = 4, 1H, ArH), 7.90–7.94 (m, 1H, ArH), 7.97 (d, J = 8, 1H, ArH), 8.02–8.05 (m, 1H, ArH), 8.64 (ddd, J = 8 Hz,J = 4 Hz, J = 2 Hz, 1H, ArH), 8.69 (s, 1H, N=CH), 12.14 (s, 1H, CONH), 14.09 (br, 1H, OH). 13C-NMR (DMSO-d6): δ = 111.13, 120.81, 124.78, 125.35, 126.86, 128.23, 133.75, 134.14, 134.78, 137.53, 150.22, 151.40, 153.44, 158.47, 165.98; ESI-HRMS m/z = 359.0809 [M+H]+. Anal. calcd for C16H14N4O4S: C 53.62, H 3.94, N 15.63; found: C 53.48, H 3.92, N 15.59. HPLC (C18, acetonitrile-water, 45:55, 254 nm): ret. time: 3.916 min; peak area = 98,484%.
4-Hydroxy-2-methyl-N'-{(E)-[4-(2-propanyl)phenyl]phenylmethylidene}-2H-1,2-benzothiazine-3-carbohydrazide 1,1-dioxide (14c). The title compound was obtained by condensation of 16 with 4-isopropylbenzaldehyde as a white powder (131 mg, 89%). Mp 228–229 °C; Rf = 0.82 (CH2Cl2/MeOH, 9:1). IR (KBr): 3283, 1638, 1615, 1348, 1182, 958 cm−1. 1H-NMR (DMSO-d6): δ = 1.23 (d, J = 6 Hz, 6H, CH(CH3)2), 2.84 (s, 3H, CH3), 2.96 (h, 1H, CH(CH3)2 ), 7.36 (d, J = 8 Hz, 2H, ArH), 7.67 (d, J = 8 Hz, 2H, ArH), 7.91 (m, 3H, ArH), 8.02–8.06 (m, 1H, ArH), 8.64 (s, 1H, N=CH), 11.89 (s, 1H, CONH), 14.24 (br, 1H, OH). 13C-NMR (DMSO-d6): δ = 24.17, 33.97, 111.14, 124.75, 126.77, 127.47, 128.09, 128.34, 132.14, 133.61, 134.12, 134.74, 151.51, 151.89, 158.01, 165.51. ESI-HRMS m/z = 400.1309 [M+H]+. Anal. calcd for C20H21N3O4S: C 60.13, H 5.30, N 10.52; found: C 60.30, H 5.27, N 10.39. HPLC (C18, acetonitrile-water, 7:3, 254 nm): ret. time: 10.977 min; peak area = 98,127%.
4-Hydroxy-2-methyl-N'-{(E)-[4-(dimethylamino) phenyl] phenylmethylidene}-2H-1,2-benzothiazine-3-carbohydrazide 1,1-dioxide (14d). The title compound was obtained by condensation of 16 with 4-dimethylaminobenzaldehyde as an orange powder (137 mg, 89%). Mp 217–218 °C; Rf = 0.70 (CH2Cl2/MeOH, 9:1). IR (KBr): 3287, 1598, 1344, 1181, 956 cm−1. 1H-NMR (DMSO-d6): δ = 2.83 (s, 3H, CH3), 2.99 (s, 6H, N(CH3)2), 6.78 (d, J = 8 Hz, 2H, ArH), 7.56 (d, J = 8 Hz, 2H, ArH), 7.89-7.93 (m, 3H, ArH), 8.00–8.05 (m, 1H, ArH), 8.52 (s, 1H, N=CH), 11.65 (s, 1H, CONH), 14.42 (br, 1H, OH); 13C-NMR (DMSO-d6): δ = 111.22, 112.43, 121.67, 124.72, 126.65, 128.47, 129.42, 132.14, 133.42, 134.06, 134.68, 152.27, 152.37, 157.56, 164.90. ESI-HRMS m/z = 401.1276 [M+H]+. Anal. calcd. for C19H20N4O4S: C 56.99, H 5.03, N 13.99; found: C 56.69, H 5.03, N 13.99. HPLC (C18, acetonitrile-water, 7:3, 254 nm): ret. time: 4.420 min; peak area = 98,349%.
4-Hydroxy-2-methyl-N'-[(E)-thiophenyl-2-methylidene]-2H-1,2-benzothiazine-3-carbohydrazide 1,1-dioxide (14e). The title compound was obtained by condensation of 16 with thiophenecarboxaldehyde as a white powder (124 mg, 92%). Mp 236–237 °C; Rf = 0.63 (CH2Cl2/MeOH, 9:1). IR (KBr): 3267, 1636, 1614, 1338, 1181, 957 cm−1. 1H-NMR (DMSO-d6): δ = 2.83 (s, 3H, CH3), 7.17 (t,J = 4 Hz, 1H, ArH), 7.52 (d, J = 2 Hz, 1H, ArH), 7.73 (d, J = 4, 1H, ArH), 7.90–7.97 (m, 3H, ArH), 8.01–8.06 (m, 1H, ArH), 8.84 (s, 1H, N=CH), 11.91 (s, 1H, CONH), 14.16 ppm (br, 1H, OH); 13C-NMR (DMSO-d6): δ = 111.12, 124.74, 126.77, 128.31, 128.59, 130.41, 132.53, 133.61, 134.12, 134.70, 139.11, 146.30, 157.98, 165.31; ESI-HRMS m/z = 364.0417 [M+H]+. Anal. calcd. for C15H13N3O4S2: C 49.57, H 3.61, N 11.56; found: C 49.58, H 3.61, 11.27. HPLC (C18, acetonitrile-water, 7:3, 254 nm): ret. time: 5.378 min; peak area = 98,866%.
4-Hydroxy-2-methyl-N'-[(E)-1,3-thiazolyl-2-methylidene]-2H-1,2-benzothiazine-3-carbohydrazide 1,1-dioxide (14f). The title compound was obtained by condensation of 16 with 2-thiazolecarboxaldehyde (0.04 mL, 0.37 mmol) as a white powder (93 mg, 63%). Mp 224–225 °C; Rf = 0.36 (CH2Cl2/-MeOH, 9:1). IR (KBr) 3223, 1624, 1355, 1182, 948 cm−1.1H-NMR (DMSO-d6): δ = 2.85 (s, 3H, CH3), 7.89 (dd,J = 4 Hz, J = 2 Hz, 1H, ArH), 7.90–1.95 (m, 3H, ArH), 7.99 (d, J = 4, 1H, ArH), 8.03-8.05 (m, 1H, ArH), 8.85 (s, 1H, N=CH), 12.23 (s, 1H, CONH), 13.93 (br, 1H, OH). 13C-NMR (DMSO-d6): δ = 110.55, 122.63, 124.25, 125.35, 127.58, 133.28, 133.62, 134.18, 144.28, 144.70, 158.01, 163.76, 165.23. ESI-HRMS m/z = 365.0378 [M+H]+. Anal. calcd. for C14H12N4O4S2: C 46.14, H 3.32, N 15.38; found C 46.03, H 3.39, N 15.42. HPLC (C18, acetonitrile-water, 45:55, 254 nm): ret. time: 11.831 min; peak area = 99,751%.
4-Hydroxy-2-methyl-N'-[(E)-4-biphenylphenylmethylidene]-2H-1,2-benzothiazine-3-carbohydrazide 1,1-dioxide (14g). The title compound was obtained by condensation of 16 with 4-biphenylcarboxaldehyde as a light green powder (146 mg, 91%). Mp 215–216 °C; Rf = 0.72 (CH2Cl2/MeOH, 9:1). IR (KBr): 3271, 1613, 1343, 1182, 959 cm−1. 1H-NMR (DMSO-d6): δ = 2.86 (s, 3H, CH3), 7.36–7.53 (m, 3H, ArH), 7.74 (d, J = 8 Hz, 2H, ArH), 7.81–7.82 (m, 3H, ArH), 7.87–7.93 (m, 4H, ArH), 8.03–8.06 (m, 1H, ArH), 8.72 (s, 1H, N=CH), 12.01 (s, 1H, CONH), 14.24 (br, 1H, OH). 13C-NMR (DMSO-d6): δ = 111.15, 124.76, 126.80, 127.28, 128.33, 128.59, 129.60, 133.54, 133.65, 134.13, 134.74, 139.79, 142.66, 150.99, 158.13, 165.62.; ESI-HRMS m/z = 434.1165 [M+H]+. Anal. calcd. for C23H19N3O4S: C 63.73, H 4.42, N 9.69; found: C 63.87, H 4.39, N 9.65. HPLC (C18, acetonitrile-water, 9:1, 254 nm): ret. time: 4.653 min; peak area = 99,145%.
4-Hydroxy-2-methyl-N'-{(E)-[3,5-di-terc-buthyl-4-hydroxy]phenylmethylidene}-2H-1,2-benzothiazine-3-carbohydrazide 1,1-dioxide (14h). The title compound was obtained by condensation of 16 with 3,5-di-tert-buthyl-4-hydroxyphenylcarboxaldehyde as a light green powder (161 mg, 90%). Mp 249–251 °C; Rf = 0.61 (CH2Cl2/MeOH, 9:1). IR (KBr): 3287, 1645, 1617, 1348, 1183, 954 cm−1. 1H-NMR (DMSO-d6): δ = 1.42 (s, 18H, C(CH3)3), 2.83 (s, 3H, CH3), 7.49 (s, 2H, ArH), 7.53 (s, 1H, OH), 7.91–8.02 (m, 4H, ArH), 8.59 (s, 1H, N=CH), 11.71 (s, 1H, CONH), 14.25 (br, 1H, OH); 13C-NMR (DMSO-d6): δ = 30.11, 34.50, 110.64, 124.30, 125.11, 126.16, 134.16, 139.24, 152.42, 156.66, 157.09, 164.51. ESI-HRMS m/z = 486.2056 [M+H]+. Anal. calcd for C25H31N3O5S: C 61.83, H 6.43, N 8.65; found: C 61.93, H 6.39, N 8.61. HPLC (C18, acetonitrile-water, 9:1, 254 nm): ret. time: 5.333 min; peak area = 99,697%.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}