Telomerase Reverse Transcriptase (TERT) is a Therapeutic Target of Oleanane Triterpenoid CDDO-Me in Prostate Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
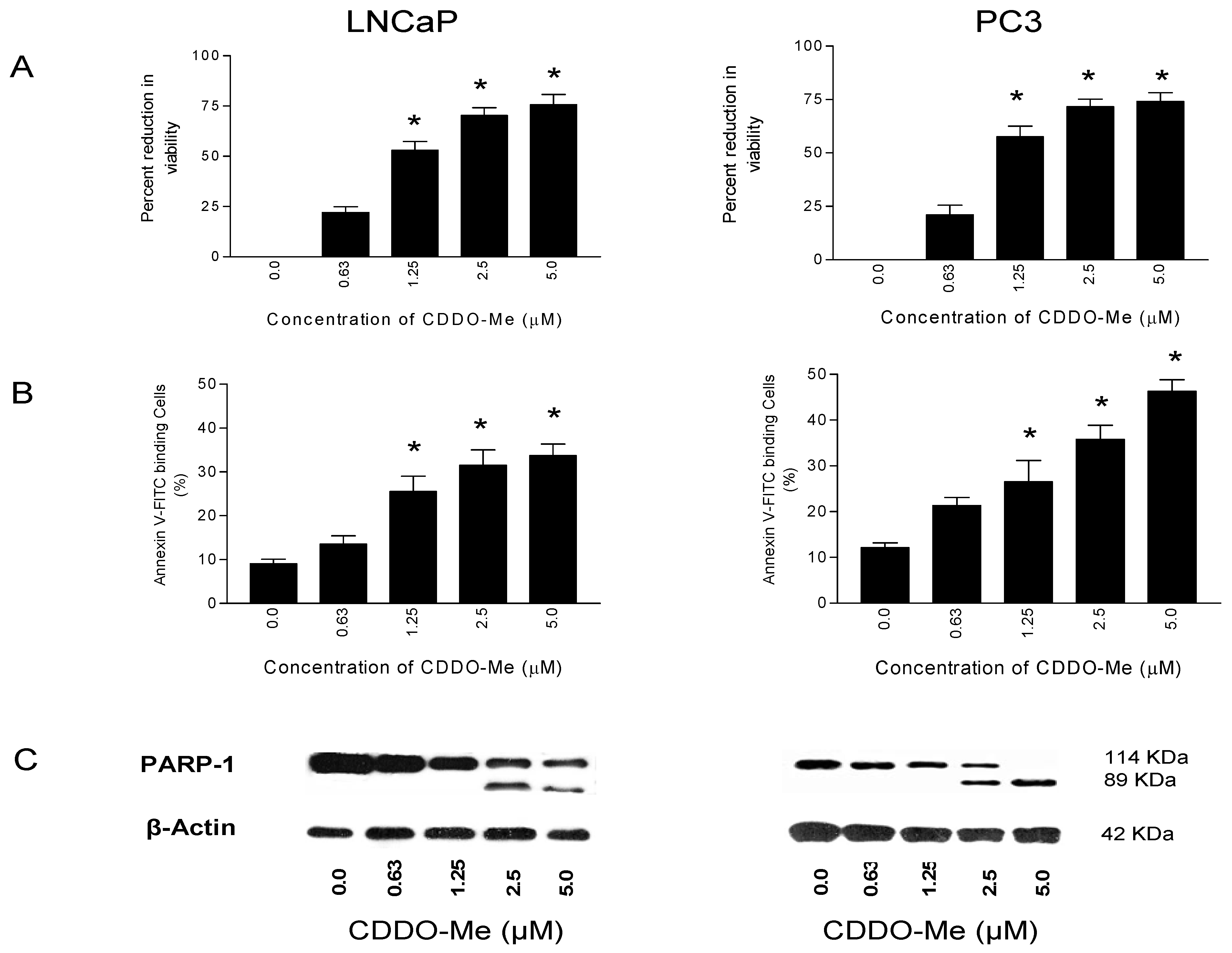
2.1. CDDO-Me Inhibits Proliferation and Induces Apoptosis in Prostate Cancer Cells
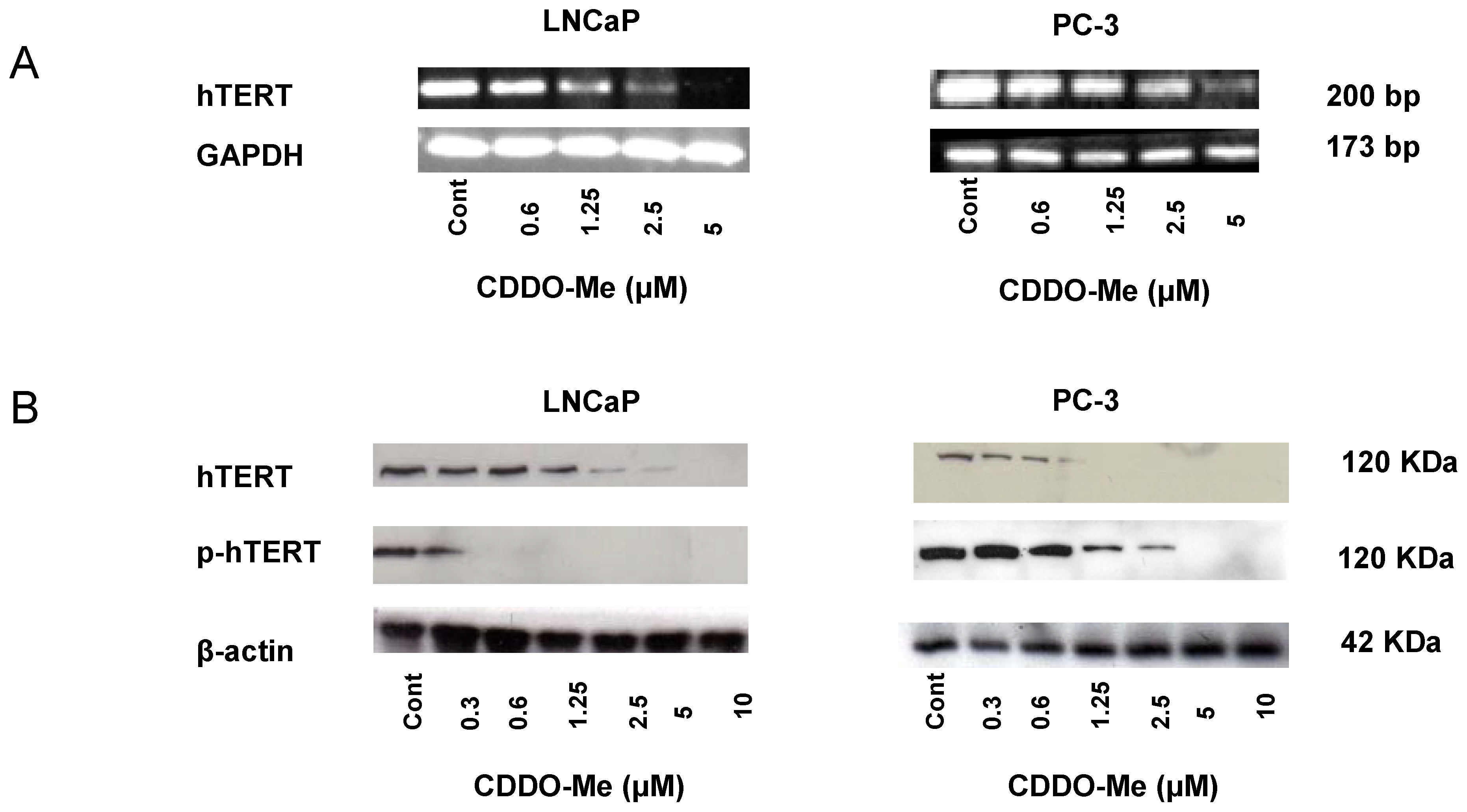
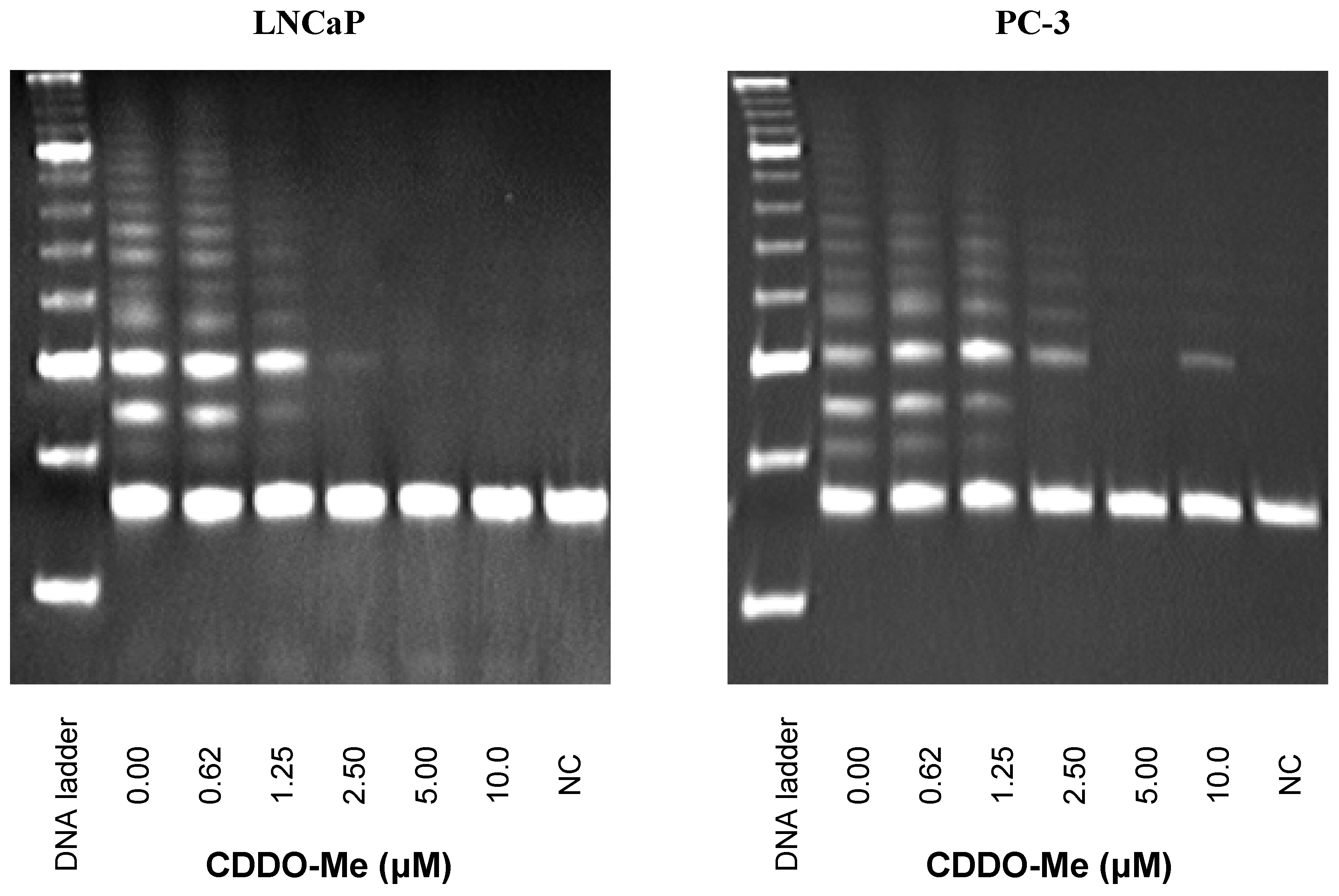
2.2. CDDO-Me Inhibits hTERT Expression and Telomerase Activity in Prostate Cancer Cells
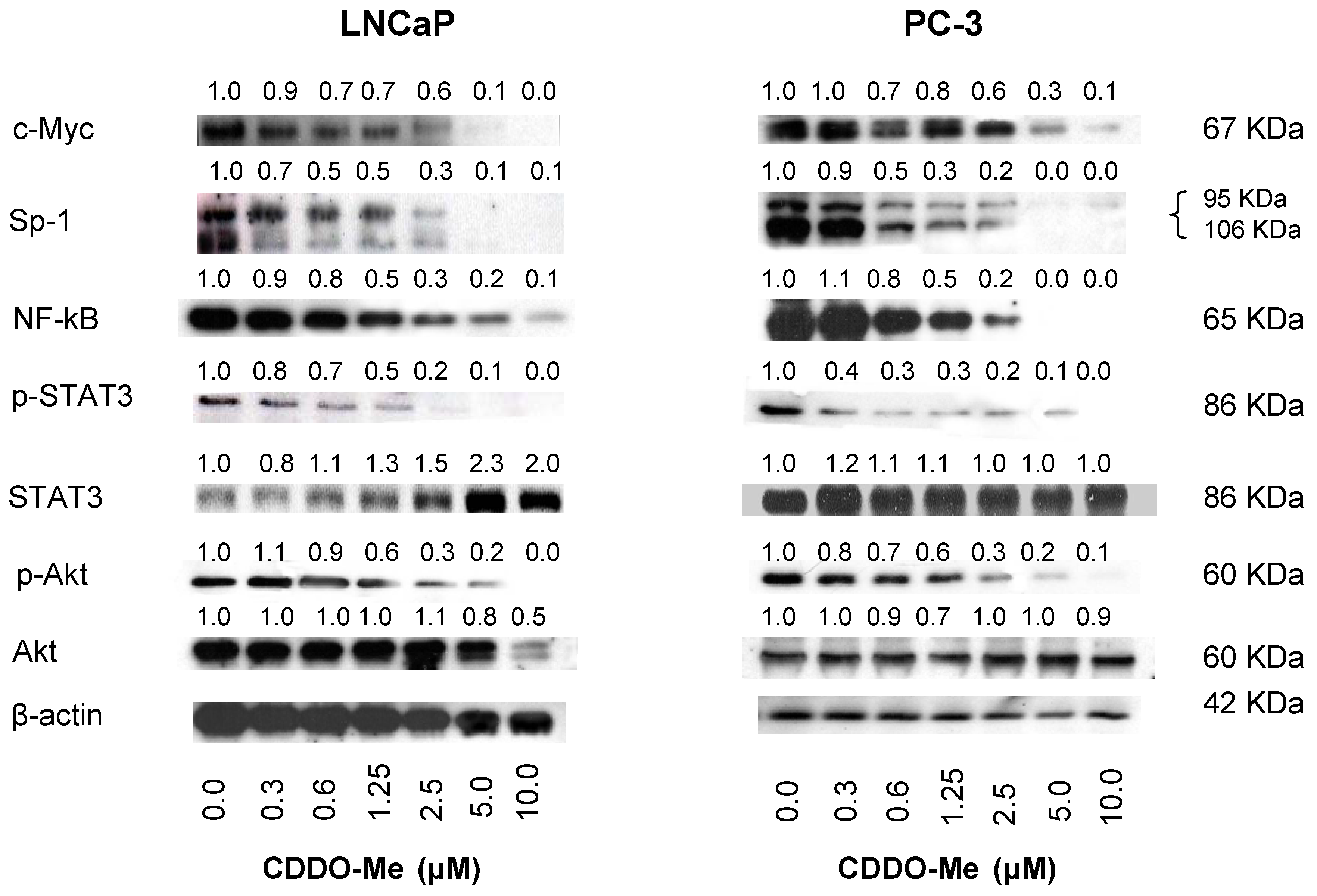
2.3. CDDO-Me Inhibits hTERT Regulatory Proteins
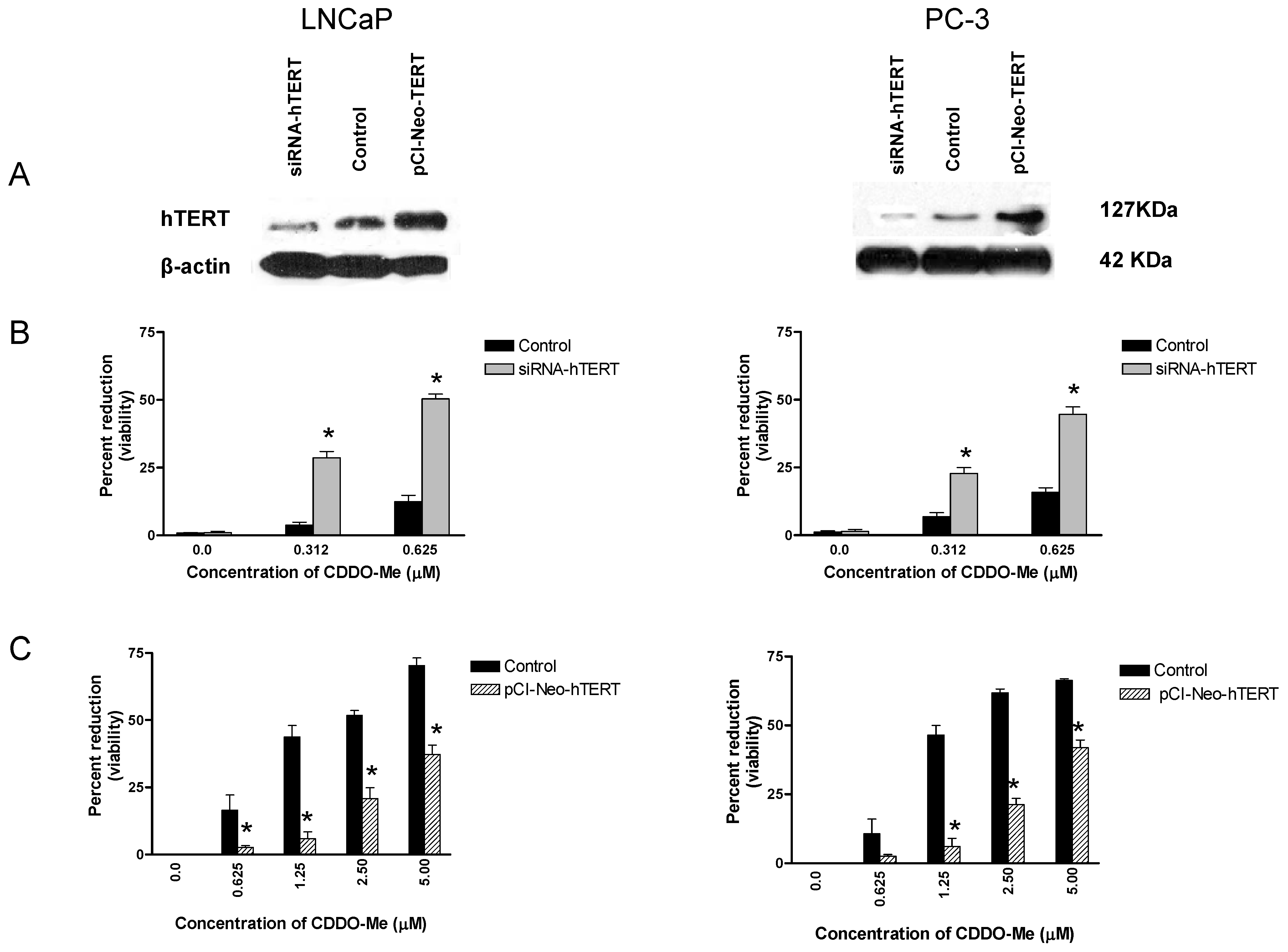
2.4. hTERT is a Target of CDDO-Me
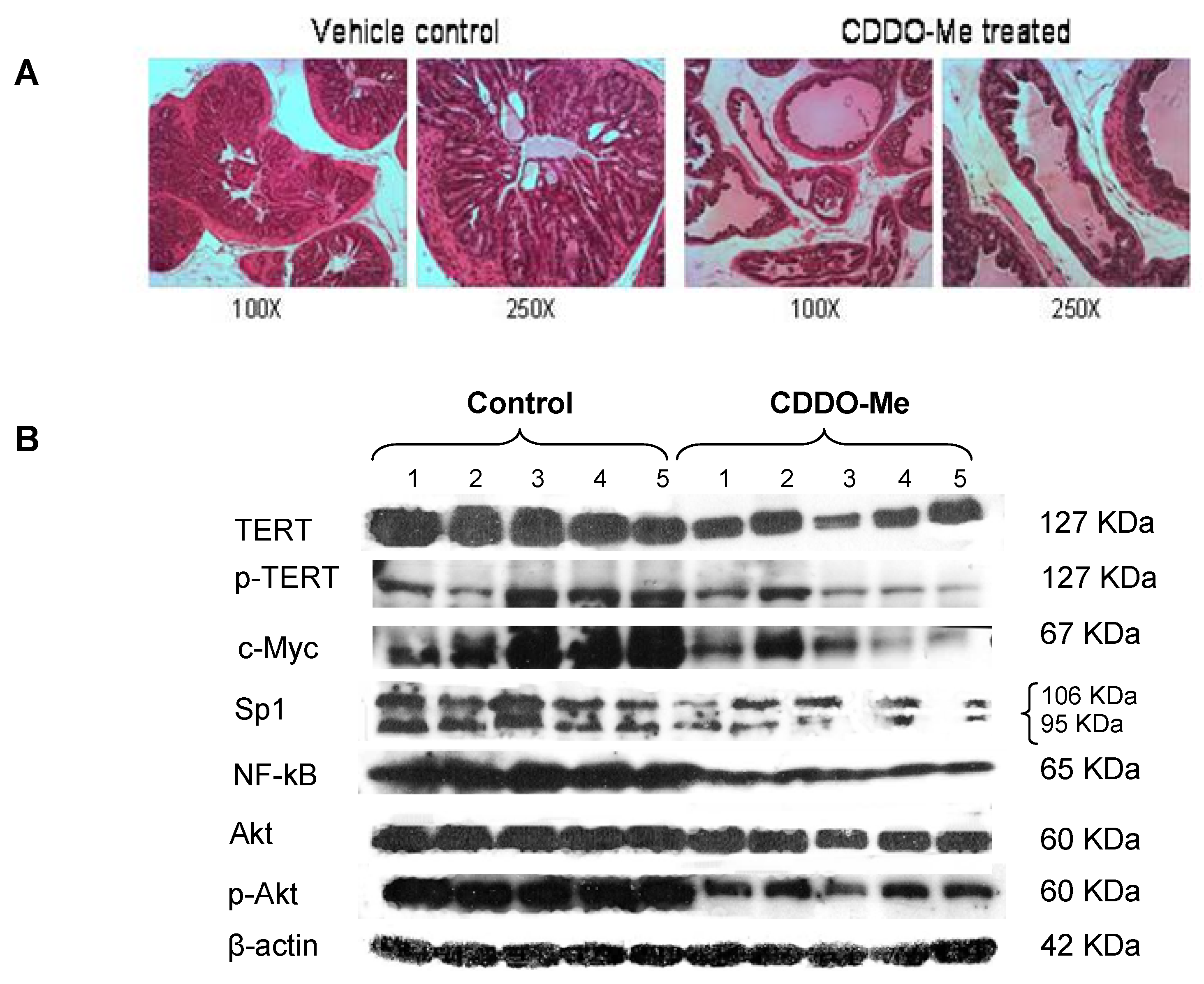
2.5. Inhibition of PINs by CDDO-Me in TRAMP Mice is Associated with Inhibition of hTERT
2.6. Discussion
3. Experimental
3.1. Reagents
3.2. Cell Culture
3.3. MTS Assay
3.4. Analysis of Apoptosis
3.5. Western Blotting
3.6. Measurement of hTERT Expression
3.7. Telomerase Activity Assay
3.8. Histology
3.9. Statistical Analysis
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Liby, K.T.; Yore, M.M.; Sporn, M.B. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat. Rev. Cancer 2007, 7, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Pandey, P.; Sporn, M.B.; Datta, R.; Kharbanda, S.; Kufe, D. The novel triterpenoid CDDO induces apoptosis and differentiation of human osteosarcoma cells by a caspase-8 dependent mechanism. Mol. Pharmacol. 2001, 59, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Tsao, T.; Estrov, Z.; Lee, R.M.; Wang, R.Y.; Jackson, C.E.; McQueen, T.; Monaco, G.; Munsell, M.; Belmont, J.; et al. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Res. 2004, 64, 7927–7935. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Deeb, D.; Jiang, H.; Liu, Y.; Dulchavsky, S.A.; Gautam, S.C. Synthetic triterpenoids inhibit growth and induce apoptosis in human glioblastoma and neuroblastoma cells through inhibition of prosurvival Akt, NF-kappaB and Notch1 signaling. J. Neurooncol. 2007, 84, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Kurinna, S.M.; Chen, W.; Andreeff, M.; Ruvolo, P.P. The novel triterpenoid CDDO-Me suppresses MAPK pathways and promotes p38 activation in acute myeloid leukemia cells. Leukemia 2005, 19, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Raina, D.; Meyer, C.; Kharbanda, S.; Kufe, D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J. Biol. Chem. 2006, 281, 35764–35769. [Google Scholar] [CrossRef] [PubMed]
- Suh, N.; Roberts, A.B.; Birkey Reffey, S.; Miyazono, K.; Itoh, S.; ten Dijke, P.; Heiss, E.H.; Place, A.E.; Risingsong, R.; Williams, C.R.; et al. Synthetic triterpenoids enhance transforming growth factor beta/Smad signaling. Cancer Res. 2003, 63, 1371–1376. [Google Scholar] [PubMed]
- Greider, C.W. Chromosome first aid. Cell 1991, 67, 645–647. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Kilian, A.; Bowtell, D.D.; Abud, H.E.; Hime, G.R.; Venter, D.J.; Keese, P.K.; Duncan, E.L.; Reddel, R.R.; Jefferson, R.A. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum. Mol. Genet. 1997, 6, 2011–2019. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, G.; Forsyth, R.G.; Praet, M.; Hogendoorn, P.C. Telomere-associated proteins: Cross-talk between telomere maintenance and telomere-lengthening mechanisms. J. Pathol. 2009, 217, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Holt, S.E.; Wright, W.E.; Shay, J.W. Regulation of telomerase activity in immortal cell lines. Mol. Cell. Biol. 1996, 16, 2932–2939. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A.; Hahn, W.C. Evolving views of telomerase and cancer. Trends Cell. Biol. 2003, 13, 289–294. [Google Scholar] [CrossRef]
- Newbold, R.F. The significance of telomerase activation and cellular immortalization in human cancer. Mutagenesis 2002, 17, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Janknecht, R. On the road to immortality: hTERT upregulation in cancer cells. FEBS Lett. 2004, 564, 9–13. [Google Scholar] [CrossRef]
- Deeb, D.; Gao, X.; Liu, Y.; Kim, S.H.; Pindolia, K.R.; Arbab, A.S.; Gautam, S.C. Inhibition of cell proliferation and induction of apoptosis by oleanane triterpenoid (CDDO-Me) in pancreatic cancer cells is associated with the suppression of hTERT gene expression and its telomerase activity. Biochem. Biophys. Res. Commun. 2012, 422, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Deeb, D.; Gao, X.; Liu, Y.; Jiang, D.; Divine, G.W.; Arbab, A.S.; Dulchavsky, S.A.; Gautam, S.C. Synthetic triterpenoid CDDO prevents the progression and metastasis of prostate cancer in TRAMP mice by inhibiting survival signaling. Carcinogenesis 2011, 32, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Deeb, D.; Liu, Y.; Arbab, A.S.; Divine, G.W.; Dulchavsky, S.A.; Gautam, S.C. Prevention of Prostate Cancer with Oleanane Synthetic Triterpenoid CDDO-Me in the TRAMP Mouse Model of Prostate Cancer. Cancers (Basel) 2011, 3, 3353–3369. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Kwon, T.; Kwon, D.Y.; Do, S.I. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J. Biol. Chem. 1999, 274, 13085–13090. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Khadka, P.; Chung, I.K. Nuclear import of hTERT requires a bipartite nuclear localization signal and Akt-mediated phosphorylation. J. Cell Sci. 2012, 125, 2684–2697. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Coller, H.A.; Roberts, J.M. Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat. Cell. Biol. 2003, 5, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Takakura, M.; Taira, T.; Kanaya, T.; Itoh, H.; Yutsudo, M.; Ariga, H.; Inoue, M. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res. 2000, 28, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Hsin, I.L.; Sheu, G.T.; Chen, H.H.; Chiu, L.Y.; Wang, H.D.; Chan, H.W.; Hsu, C.P.; Ko, J.L. N-acetyl cysteine mitigates curcumin-mediated telomerase inhibition through rescuing of Sp1 reduction in A549 cells. Mutat. Res. 2010, 688, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Marconett, C.N.; Sundar, S.N.; Tseng, M.; Tin, A.S.; Tran, K.Q.; Mahuron, K.M.; Bjeldanes, L.F.; Firestone, G.L. Indole-3-carbinol downregulation of telomerase gene expression requires the inhibition of estrogen receptor-alpha and Sp1 transcription factor interactions within the hTERT promoter and mediates the G1 cell cycle arrest of human breast cancer cells. Carcinogenesis 2011, 32, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Konnikova, L.; Simeone, M.C.; Kruger, M.M.; Kotecki, M.; Cochran, B.H. Signal transducer and activator of transcription 3 (STAT3) regulates human telomerase reverse transcriptase (hTERT) expression in human cancer and primary cells. Cancer Res. 2005, 65, 6516–6520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mar, V.; Zhou, W.; Harrington, L.; Robinson, M.O. Telomere shortening and apoptosis in telomerase-inhibited human tumor cells. Genes Dev. 1999, 13, 2388–2399. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Patel, S.N.; Tollefsbol, T.O. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS One 2010, 5, e11457. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, S.; Kyo, S.; Banerjee, P.P. Genistein represses telomerase activity via both transcriptional and posttranslational mechanisms in human prostate cancer cells. Cancer Res. 2006, 66, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Jutooru, I.; Chadalapaka, G.; Abdelrahim, M.; Basha, M.R.; Samudio, I.; Konopleva, M.; Andreeff, M.; Safe, S. Methyl 2-cyano-3,12-dioxooleana-1,9-dien-28-oate decreases specificity protein transcription factors and inhibits pancreatic tumor growth: role of microRNA-27a. Mol. Pharmacol. 2010, 78, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Chintharlapalli, S.; Papineni, S.; Ramaiah, S.K.; Safe, S. Betulinic acid inhibits prostate cancer growth through inhibition of specificity protein transcription factors. Cancer Res. 2007, 67, 2816–2823. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Chadalapaka, G.; Pathi, S.S.; Jin, U.H.; Lee, J.S.; Park, Y.Y.; Cho, S.G.; Chintharlapalli, S.; Safe, S. Induction of the transcriptional repressor ZBTB4 in prostate cancer cells by drug-induced targeting of microRNA-17–92/106b-25 clusters. Mol. Cancer Ther. 2012, 11, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Jutooru, I.; Chadalapaka, G.; Lei, P.; Safe, S. Inhibition of NFkappaB and pancreatic cancer cell and tumor growth by curcumin is dependent on specificity protein down-regulation. J. Biol. Chem. 2010, 285, 25332–25344. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, X.; Deeb, D.; Gautam, S.C. Oleanane triterpenoid CDDO-Me inhibits Akt activity without affecting PDK1 kinase or PP2A phosphatase activity in cancer cells. Biochem. Biophys. Res. Commun. 2012, 417, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Chadalapaka, G.; Jutooru, I.; Burghardt, R.; Safe, S. Drugs that target specificity proteins downregulate epidermal growth factor receptor in bladder cancer cells. Mol. Cancer Res. 2010, 8, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Chintharlapalli, S.; Papineni, S.; Lee, S.O.; Lei, P.; Jin, U.H.; Sherman, S.I.; Santarpia, L.; Safe, S. Inhibition of pituitary tumor-transforming gene-1 in thyroid cancer cells by drugs that decrease specificity proteins. Mol. Carcinogenesis 2011, 50, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. Genistein depletes telomerase activity through cross-talk between genetic and epigenetic mechanisms. Int. J. Cancer 2009, 125, 286–296. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |






© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Y.; Gao, X.; Deeb, D.; Arbab, A.S.; Gautam, S.C. Telomerase Reverse Transcriptase (TERT) is a Therapeutic Target of Oleanane Triterpenoid CDDO-Me in Prostate Cancer. Molecules 2012, 17, 14795-14809. https://doi.org/10.3390/molecules171214795
Liu Y, Gao X, Deeb D, Arbab AS, Gautam SC. Telomerase Reverse Transcriptase (TERT) is a Therapeutic Target of Oleanane Triterpenoid CDDO-Me in Prostate Cancer. Molecules. 2012; 17(12):14795-14809. https://doi.org/10.3390/molecules171214795
Chicago/Turabian StyleLiu, Yongbo, Xiaohua Gao, Dorrah Deeb, Ali S. Arbab, and Subhash C. Gautam. 2012. "Telomerase Reverse Transcriptase (TERT) is a Therapeutic Target of Oleanane Triterpenoid CDDO-Me in Prostate Cancer" Molecules 17, no. 12: 14795-14809. https://doi.org/10.3390/molecules171214795