Synthesis and Calcium Mobilization Activity of cADPR Analogues Which Integrate Nucleobase, Northern and Southern Ribose Modifications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

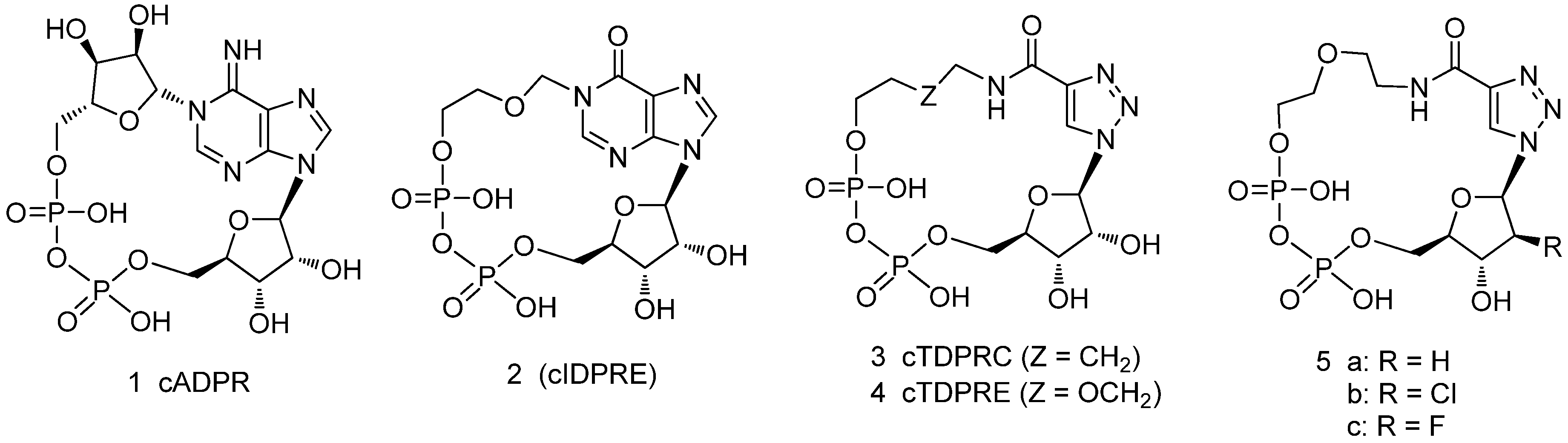
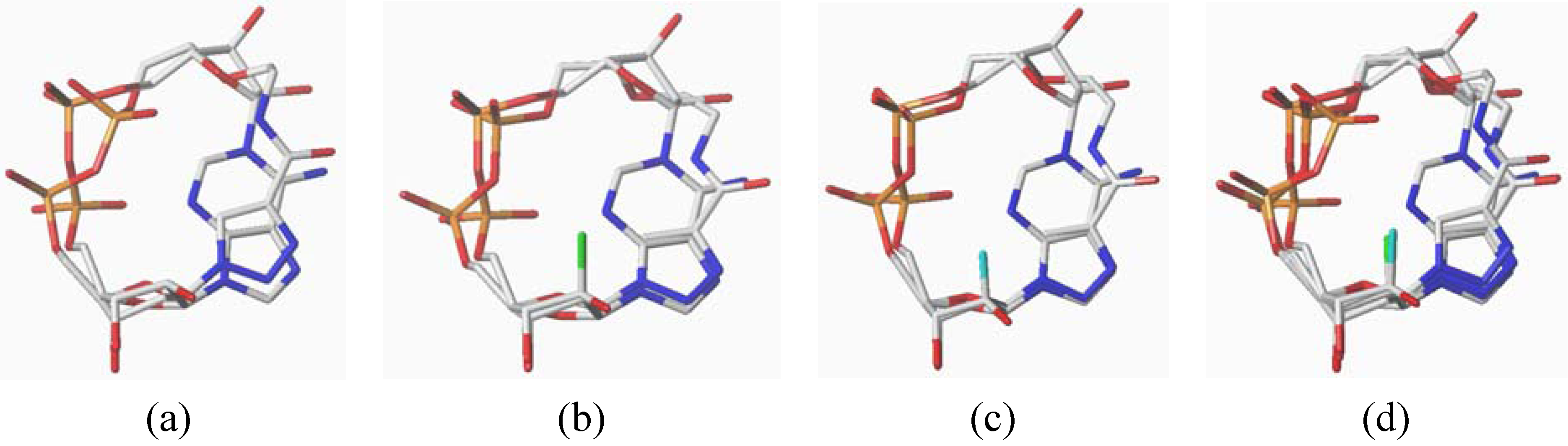
2. Results and Discussion
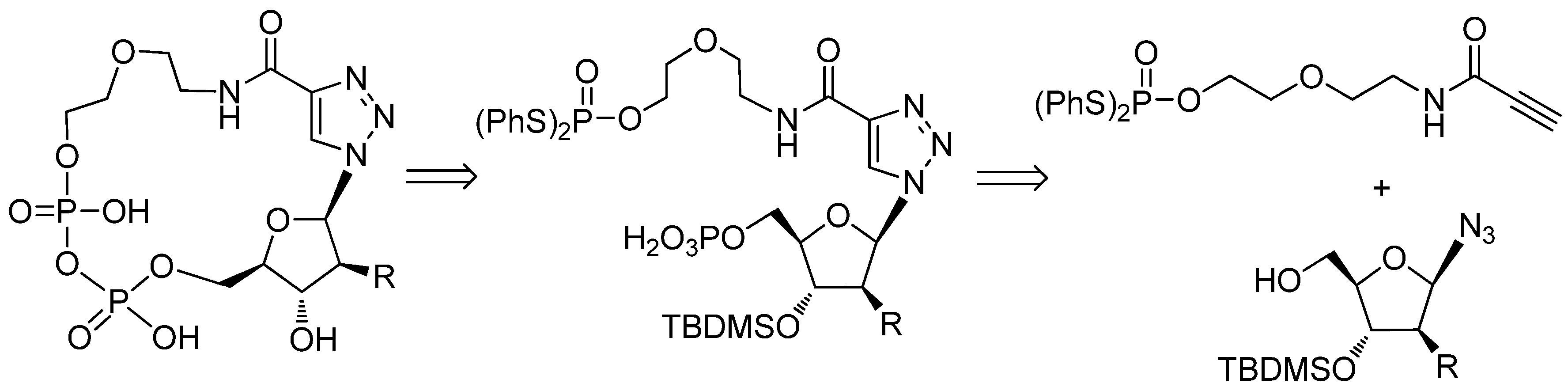
2.1. Chemistry



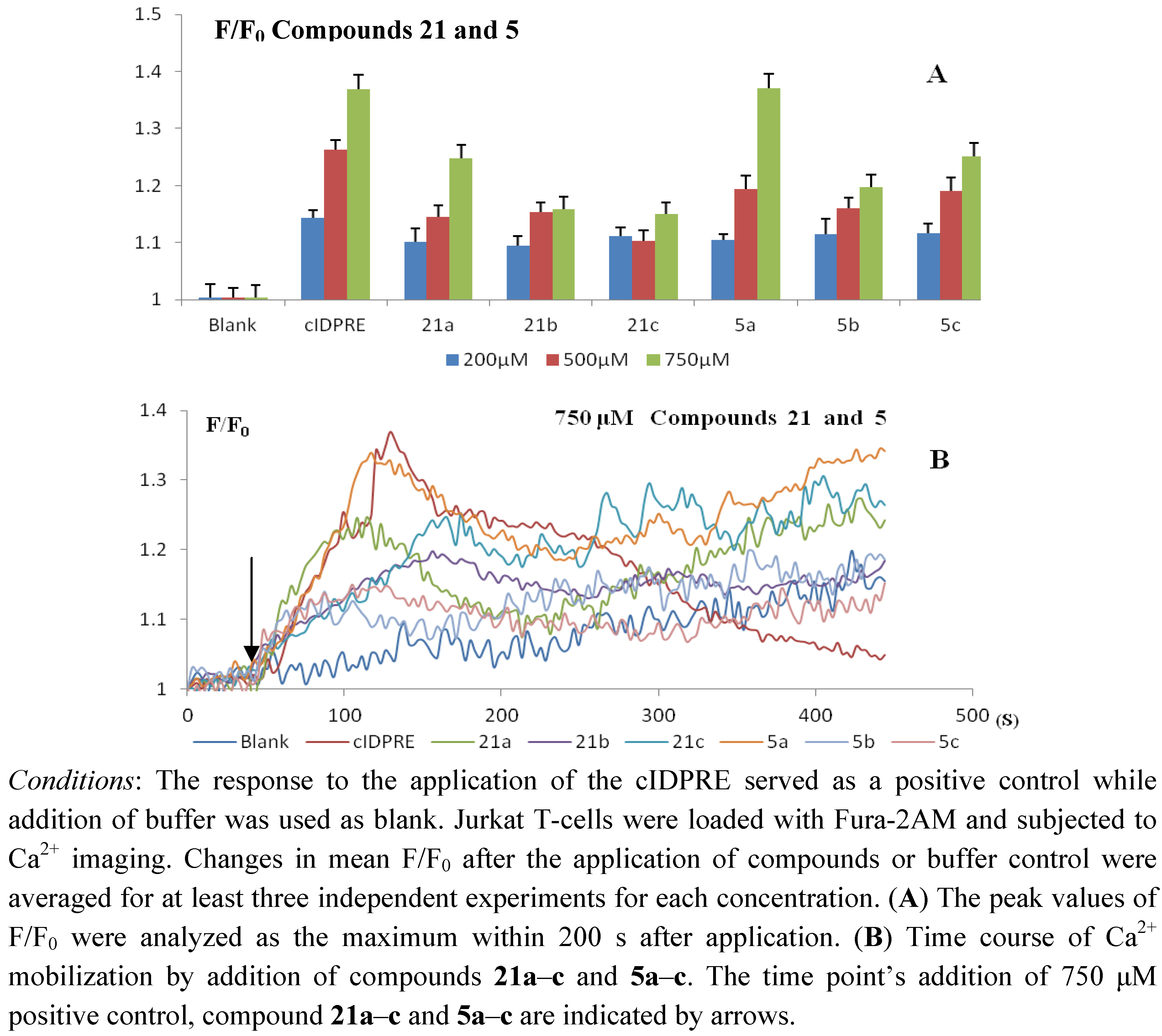
2.2. Pharmacology


3. Experimental
3.1. Chemistry
3.1.1. General
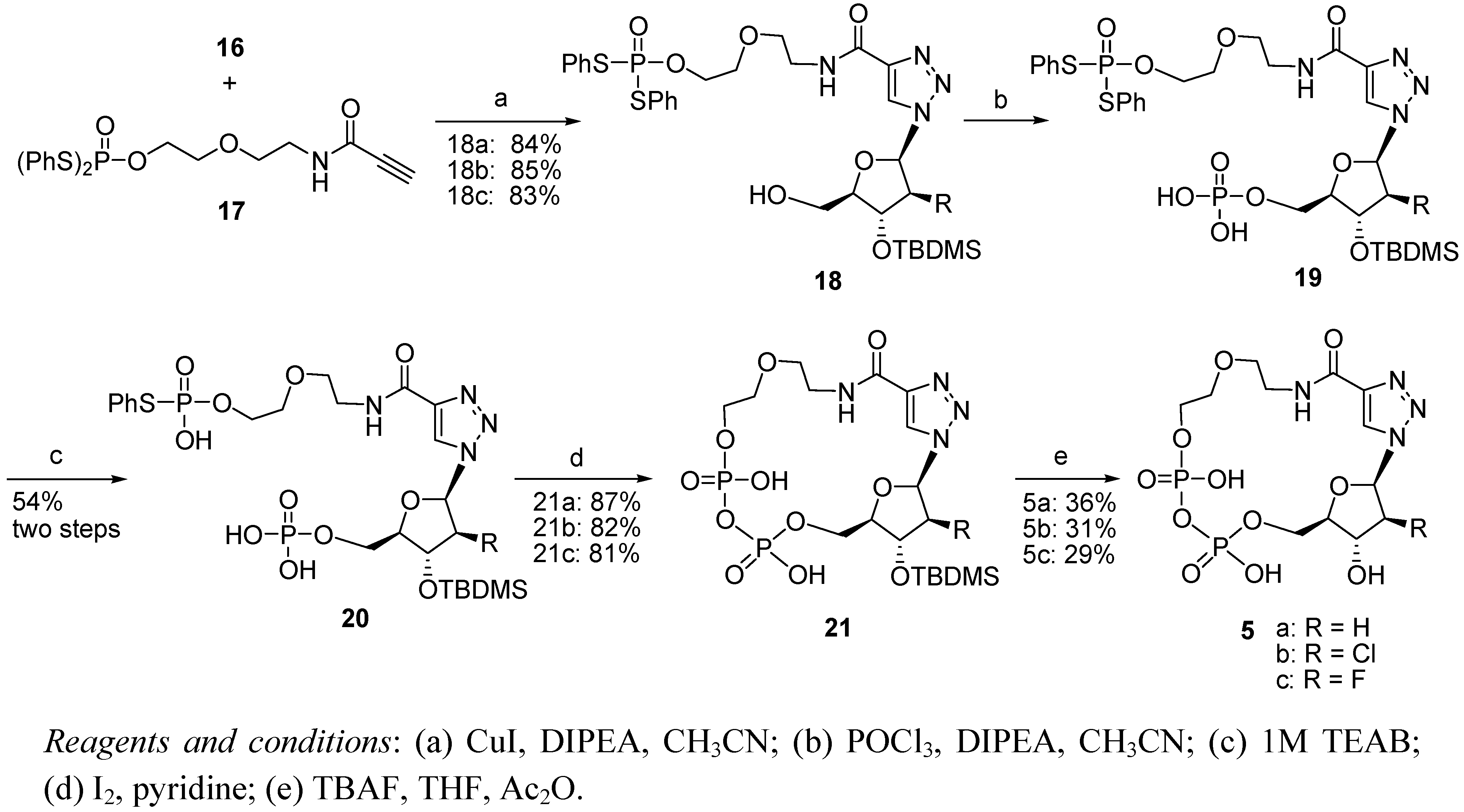
3.1.2. General Procedure for the Synthesis of 18a–c
3.1.3. General Procedure for the Synthesis of 20a–c
3.1.4. General Procedure for the Synthesis of 21a–c
3.1.5. General Procedure for the Synthesis of 5a–c
3.2. Biological Studies
3.3. Calculations
4. Conclusions
Acknowledgements
References and Notes
- Guse, A.H. Regulation of calcium signaling by the second messenger cyclic adenosine diphosphoribose (cADPR). Curr. Mol. Med. 2004, 4, 239–248. [Google Scholar]
- Lee, H.C. Cyclic ADP-Ribose and NAADP:Structures, Metabolism and Functions; Kluwer Academic Publisher: Dordrecht, The Netherland, 2002; pp. 217–444. [Google Scholar]
- Lee, H.C. Multiplicity of Ca2+ messengers and Ca2+ stores: A perspective from cyclic ADP-ribose and NAADP. Curr. Mol. Med. 2004, 4, 227–237. [Google Scholar]
- Shuto, S.; Matsuda, A. Chemistry of cyclic ADP-ribose and its analogs. Curr. Med. Chem. 2004, 11, 827–845. [Google Scholar]
- Guse, A.H. Biochemistry, biology, and pharmacology of cyclic adenosine diphosphoribose (cADPR. Curr. Med. Chem. 2004, 11, 847–855. [Google Scholar]
- Potter, B.V.L.; Walseth, T.F. Medicinal chemistry and pharmacology of cyclic ADP-ribose. Curr. Mol. Med. 2004, 4, 303–311. [Google Scholar]
- Kudoh, T.; Fukuoka, M.; Ichikawa, S.; Murayama, T.; Ogawa, Y.; Hashii, M.; Higashida, H.; Kunerth, S.; Weber, K.; Guse, A.H.; et al. Synthesis of stable and cell-type selective analogues of cyclic ADP-ribose, a Ca2+-mobilizing second messenger. Structure-activity relationship of the N1-ribose moiety. J. Am. Chem. Soc. 2005, 127, 8846–8855. [Google Scholar]
- Shuto, S.; Fukuoka, M.; Manikowsky, A.; Ueno, Y.; Nakano, T.; Kuroda, R.; Kuroda, H.; Matsuda, A. A total synthesis of cyclic ADP-carbocyclic-ribose, a stable mimic of Ca2+-mobilizing second messenger cyclic ADP-ribose. J. Am. Chem. Soc. 2001, 123, 8750–8759. [Google Scholar]
- Xu, L.; Walseth, T.; Slama, J. Cyclic ADP-ribose analogues containing the methylenebisphosphonate linkage: Effect of pyrophosphate modifications on Ca2+ release activity. J. Med. Chem. 2005, 48, 4177–4181. [Google Scholar]
- Aarhus, R.; Gee, K.; Lee, H.C. Caged cyclic ADP-ribose. Synthesis and use. J. Biol. Chem. 1995, 270, 7745–7749. [Google Scholar]
- Zhang, F.; Yamada, S.; Gu, Q.M.; Jing, P.; Sih, C.J. Synthesis and characterization of cyclic ATP-ribose: A potent mediator of calcium release. Bioorg. Med. Chem. Lett. 1996, 6, 1203–1208. [Google Scholar]
- Qi, N.; Jung, K.; Wang, M.; Na, L.X.; Yang, Z.J.; Zhang, L.R.; Guse, A.H.; Zhang, L.H. A novel membrane-permeant cADPR antagonist modified in the pyrophosphate bridge. Chem. Commun. 2011, 47, 9462–9464. [Google Scholar]
- Moreau, C.; Wagner, G.K.; Weber, K.; Guse, A.H.; Potter, B.V.L. Strutural determinants for N1/N7 cyclization of nicotinamide hypoxanthine 5'-Dinucleotide (NHD+) derivatives by ADP-ribosyl cyclase from Aplysia californica: Ca2+-Mobilising activity of 8-substituted cyclic inosine 5-diphosphoribose analogues in T-lymphocytes. J. Med. Chem. 2006, 49, 5162–5176. [Google Scholar]
- Graeff, R.M.; Walseth, T.F.; Hill, T.K.; Lee, H.C. Flourescent analogos of cyclic ADP-ribose: Synthesis, spectral characterization and use. Biochemistry 1996, 35, 379–386. [Google Scholar]
- Bailey, V.C.; Sethi, J.K.; Fortl, S.M.; Galione, A.; Potter, B.V.L. 7-Deaza cyclic adenosine 5'-diphosphate ribose first example of a Ca2+-mobilizing partial agonist related to cyclic adenosine 5'-diphosphate ribose. Chem. Biol. 1997, 4, 51–56. [Google Scholar]
- Huang, X.C.; Dong, M.; Liu, J.; Zhang, K.H.; Yang, Z.J.; Zhang, L.R.; Zhang, L.H. Concise syntheses of trifluoromethylated cyclic and acyclic analogues of cADPR. Molecules 2010, 15, 8689–8701. [Google Scholar]
- Gu, X.F.; Yang, Z.J.; Zhang, L.R.; Kunerth, S.; Fliegert, R.; Weber, K.; Guse, A.H.; Zhang, L.H. Synthesis and biological evaluation of novel membrane-permeant cyclic ADP-ribose mimics: N1-[(5"-O-phosphoryl-ethoxy)-methyl]-5'-O-phosphoryl-inosine 5',5"-cyclicpyrophosphate (cIDPRE) and 8-substituted derivatives. J. Med. Chem. 2004, 47, 5674–5682. [Google Scholar]
- Xu, J.F.; Yang, Z.J.; Dammermann, W.; Zhang, L.R.; Guse, A.H.; Zhang, L.H. Synthesis and agonist activity of cyclic ADP-ribose analogues with substitution of the northern ribose by ether or alkane chains. J. Med. Chem. 2006, 49, 5001–5012. [Google Scholar]
- Guse, A.H.; Gu, X.F.; Zhang, L.R.; Weber, K.; Zhang, L.H. A minimal structural analogue of cyclic ADP-ribose. J. Biol. Chem. 2005, 280, 15952–15959. [Google Scholar]
- Huang, L.J.; Zhao, Y.Y.; Yuan, L.; Min, J.M.; Zhang, L.H. Syntheses and calcium mobilizing evaluations of N1-glycosyl substituted stable mimics of cyclic ADP-ribose. J. Med. Chem. 2002, 45, 5340–5352. [Google Scholar]
- Kudoh, T.; Fukuoka, M.; Shuto, S.; Matsuda, A. Synthesis and biological activity of cyclic ADP-carbocyclic-ribose analogues:Structure-activity relationship and conformational analysis of N-1-carbocyclic-ribose moiety. Nucleos. Nucleot. Nucleic Acids 2005, 24, 655–658. [Google Scholar]
- Kudoh, T.; Fukuoka, M.; Ichikawa, S.; Murayama, T.; Ogawa, Y.; Hashii, M.; Higashida, H.; Kunerth, S.; Weber, K.; Guse, A.H.; et al. Synthesis of stable and cell-type selective analogues of cyclic ADP-ribose, a Ca2+-mobilizing second messenger. Structure-activity relationship of the N1-ribose moiety. J. Am. Chem. Soc. 2005, 127, 8846–8855. [Google Scholar]
- Zhang, B.; Wagner, G.K.; Weber, K.; Garnham, C.; Morgan, A.; Galione, A.; Guse, A.H.; Potter, B.V.L. 2'-Deoxy cyclic adenosine 5'-diphosphate ribose derivatives: Importance of the 2'-hydroxyl motif for the antagonistic activity of 8-substituted cADPR derivatives. J. Med.Chem. 2008, 51, 1623–1636. [Google Scholar]
- Moreau, C.; Ashamu, G.A.; Bailey, V.C.; Galione, A.; Gusec, A.H.; Potter, B.V.L. Synthesis of cyclic adenosine 5'-diphosphate ribose analogues: A C2' endo/syn “southern” ribose conformation underlies activity at the sea urchin cADPR receptor. Org. Biomol. Chem. 2011, 9, 278–290. [Google Scholar]
- Li, L.J.; Lin, B.C.; Yang, Z.J.; Zhang, L.R.; Zhang, L.H. A concise route for the preparation of nucleobase-simplified cADPR mimics by click chemistry. Tetrahedron Lett. 2008, 49, 4491–4493. [Google Scholar]
- Kazuhiro, H.M.G. Synthesis and characterization of oligonucleotides containing formadidopyrimidine lesions and nonhydrolyzable analogues. J. Am. Chem. Soc. 2001, 123, 8638–8637. [Google Scholar]
- Du, J.F.; Choi, Y.; Chu, C.K. A practical synthesis of L-FMAU from L-arabinose. Nucleos. Nucleot. 1999, 18, 187–195. [Google Scholar]
- Bruce, G.; Anderson, D.P.L. Isolation, synthesis, and characterization of impurities and degradants from theclofarabine process. Org. Proc. Res. Dev. 2008, 12, 1229–1237. [Google Scholar] [CrossRef]
- Li, L.J.; Guse, A.H.; Zhang, L.H. Novel nucleobase-simplified cyclic ADP-ribose analogue: A concise synthesis and Ca2+-mobilizing activity in T-lymphocytes. Org. Biol. Chem. 2010, 8, 1843–1848. [Google Scholar]
- Polshettiwar, V.; Varma, R.S. Aqueous microwave chemistry: A clean and green synthetic tool for rapid drug discovery. Chem. Soc. Rev. 2008, 37, 1546–1557. [Google Scholar]
- Kappe, C.O. Microwave dielectric heating in synthetic organic chemistry. Chem. Soc. Rev. 2008, 37, 1127–1139. [Google Scholar]
- Guse, A.H. Cyclic ADP-ribose. J. Mol. Med. 2000, 78, 26–35. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision B.01, Gaussian Inc.: Pittsburgh, PA, USA, 2010.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 93, 5648. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 5a–c are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, Y.; Yu, P.; Jin, H.; Yang, Z.; Yue, J.; Zhang, L.; Zhang, L. Synthesis and Calcium Mobilization Activity of cADPR Analogues Which Integrate Nucleobase, Northern and Southern Ribose Modifications. Molecules 2012, 17, 4343-4356. https://doi.org/10.3390/molecules17044343
Zhou Y, Yu P, Jin H, Yang Z, Yue J, Zhang L, Zhang L. Synthesis and Calcium Mobilization Activity of cADPR Analogues Which Integrate Nucleobase, Northern and Southern Ribose Modifications. Molecules. 2012; 17(4):4343-4356. https://doi.org/10.3390/molecules17044343
Chicago/Turabian StyleZhou, Yue, Peilin Yu, Hongwei Jin, Zhenjun Yang, Jianbo Yue, Liangren Zhang, and Lihe Zhang. 2012. "Synthesis and Calcium Mobilization Activity of cADPR Analogues Which Integrate Nucleobase, Northern and Southern Ribose Modifications" Molecules 17, no. 4: 4343-4356. https://doi.org/10.3390/molecules17044343