Chiral Aminophosphines as Catalysts for Enantioselective Double-Michael Indoline Syntheses

Abstract
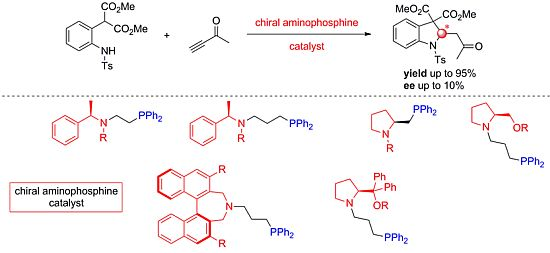
:
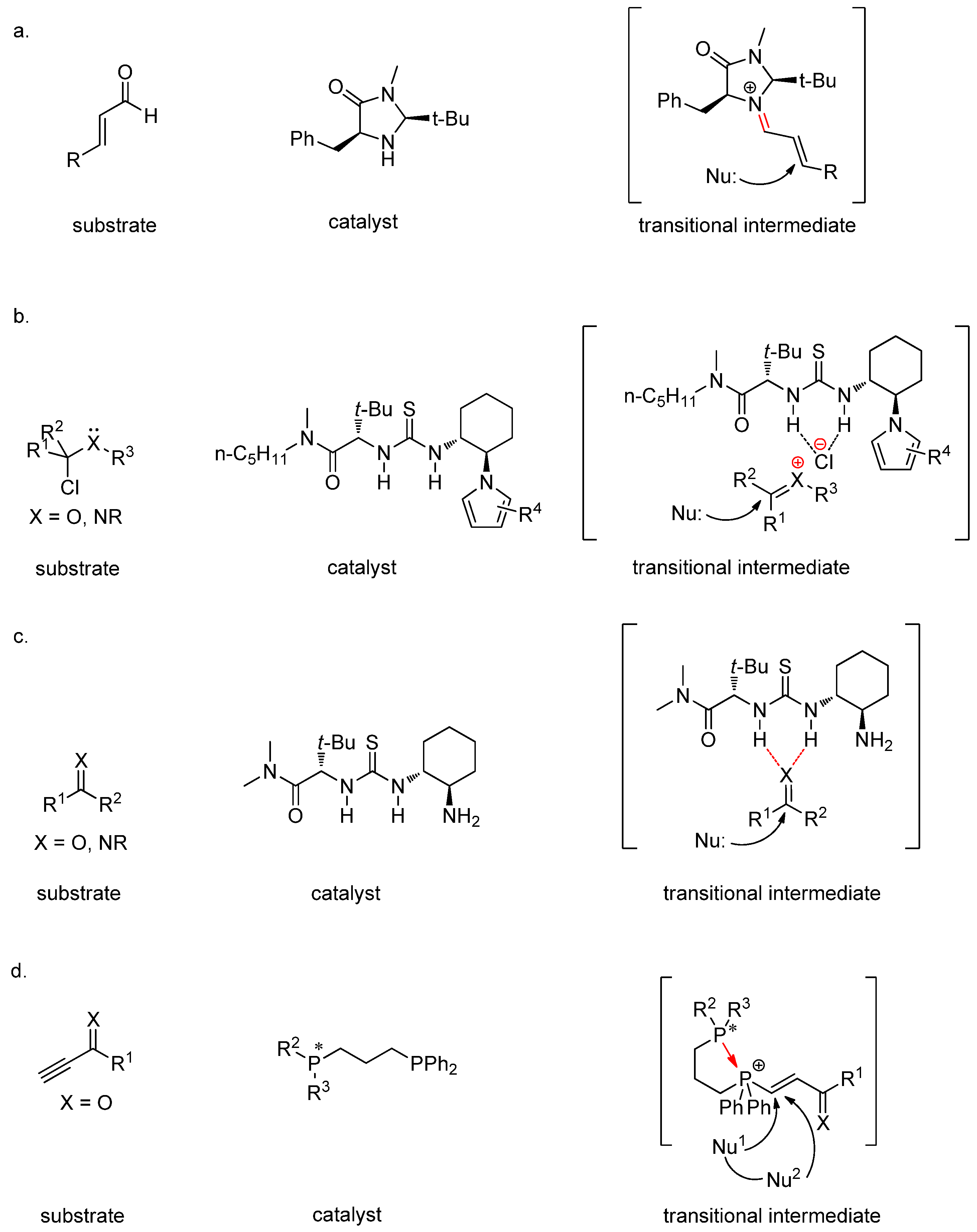
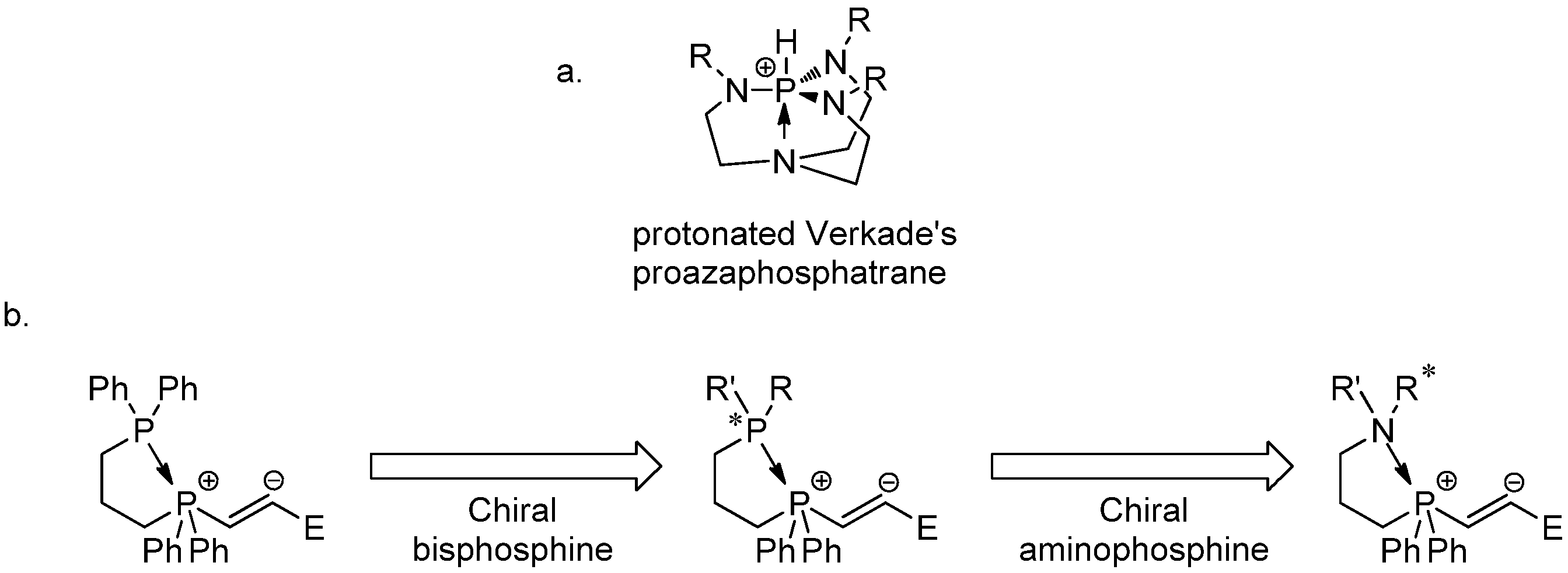
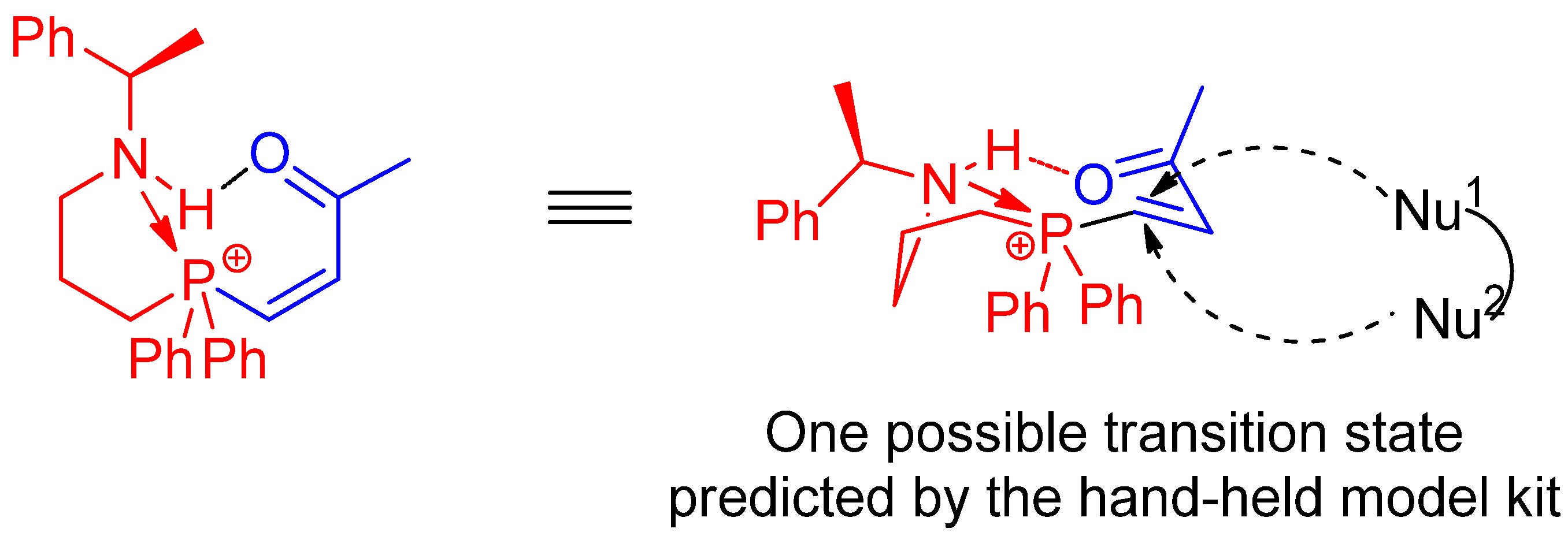
1. Introduction



2. Results and Discussion


| Entry | Catalyst | Temp (°C) | Time (h) | Yield (%) a | ee (%) | |
|---|---|---|---|---|---|---|
| 1 |  | DPPP | 80 | 9 | 81 | 0 |
| 2 |  | (S,S)-DIPAMP | 80 | 7 | 80 | 0 |
| 3 | rt b | 48 | 87 | 5 | ||
| 4 |  | (R,R)-Et-DuPHOS | 80 | 9 | 51 | 0 |
| 5 | rt | 9 | 58 | 5 | ||
| 6 | 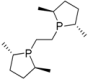 | (S,S)-Me-BPE | 80 | 7 | 72 | 0 |
| 7 | rt | 9 | 78 | 5 | ||
| 8 | 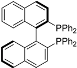 | (R)-BINAP | rt | 9 | 66 | 5 |
| 9 |  | (S,S)-DIOP | rt | 24 | n/r c | n/a d |
| 10 | 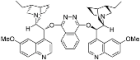 | (DHQ)2PHAL | rt | 9 | 46 | 5 |
| 11 | 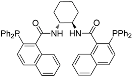 | (R,R)-DACH- napthyl Trost ligand | rt | 16 | 21 | 0 |
| 12 e | 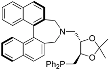 | (4S,5R)-42 | rt | 24 | 86 | 6 |
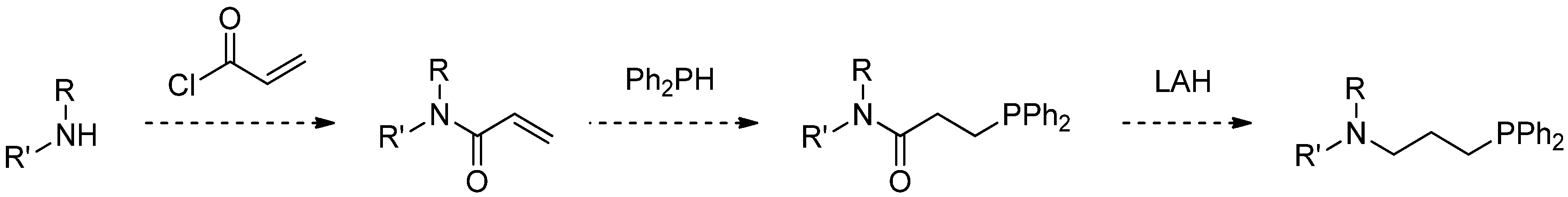
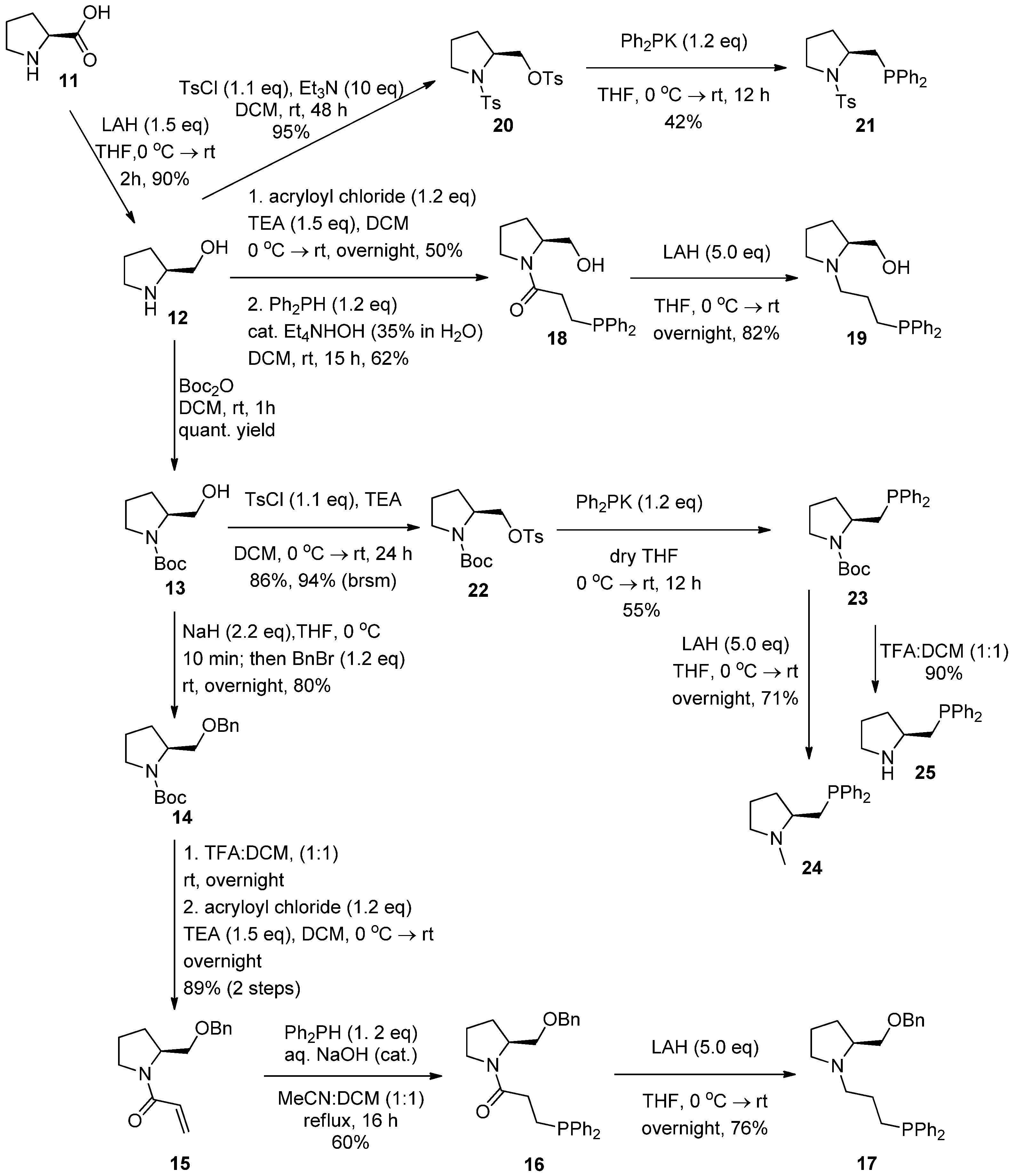
2.1. Synthesis of Aminophosphines

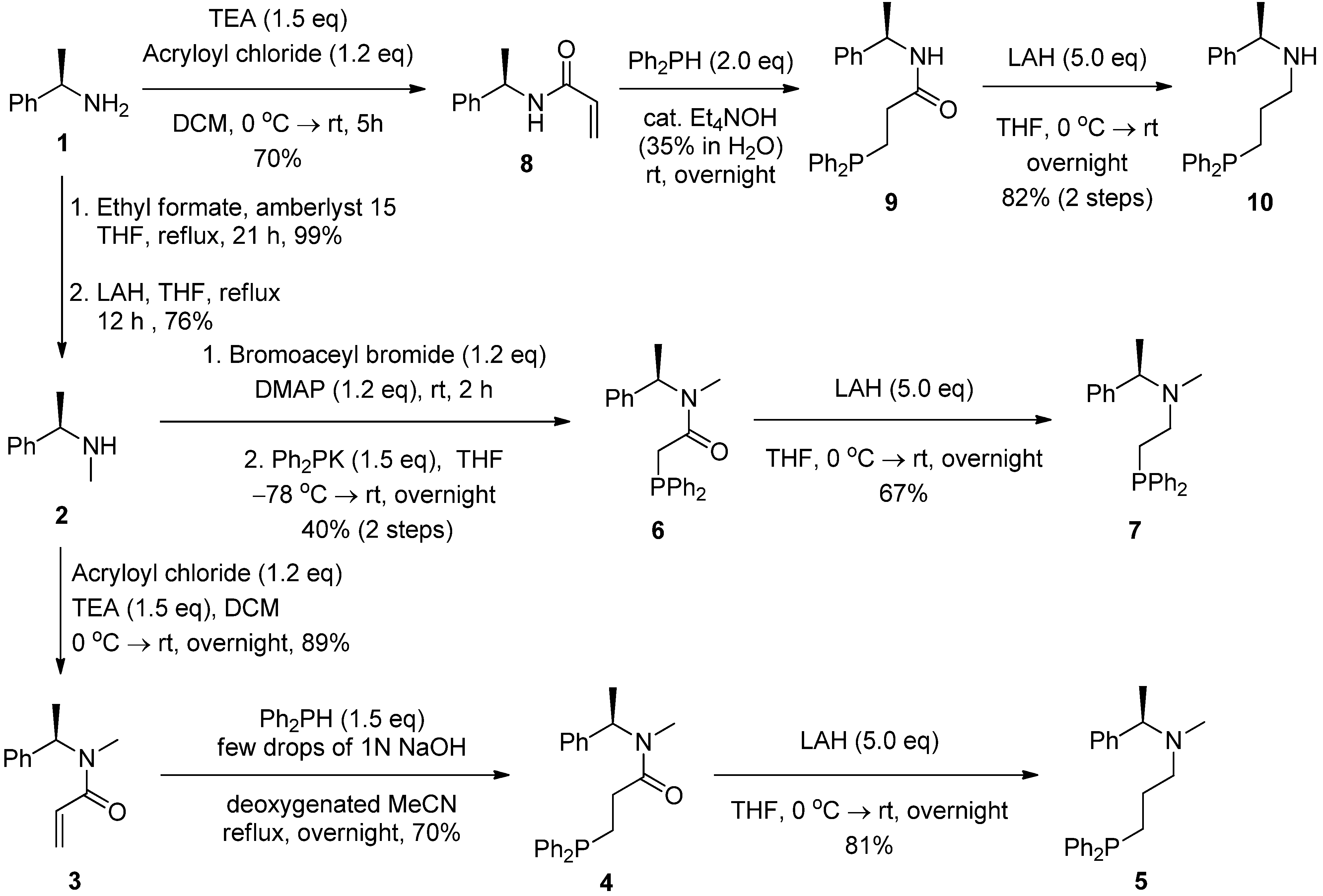
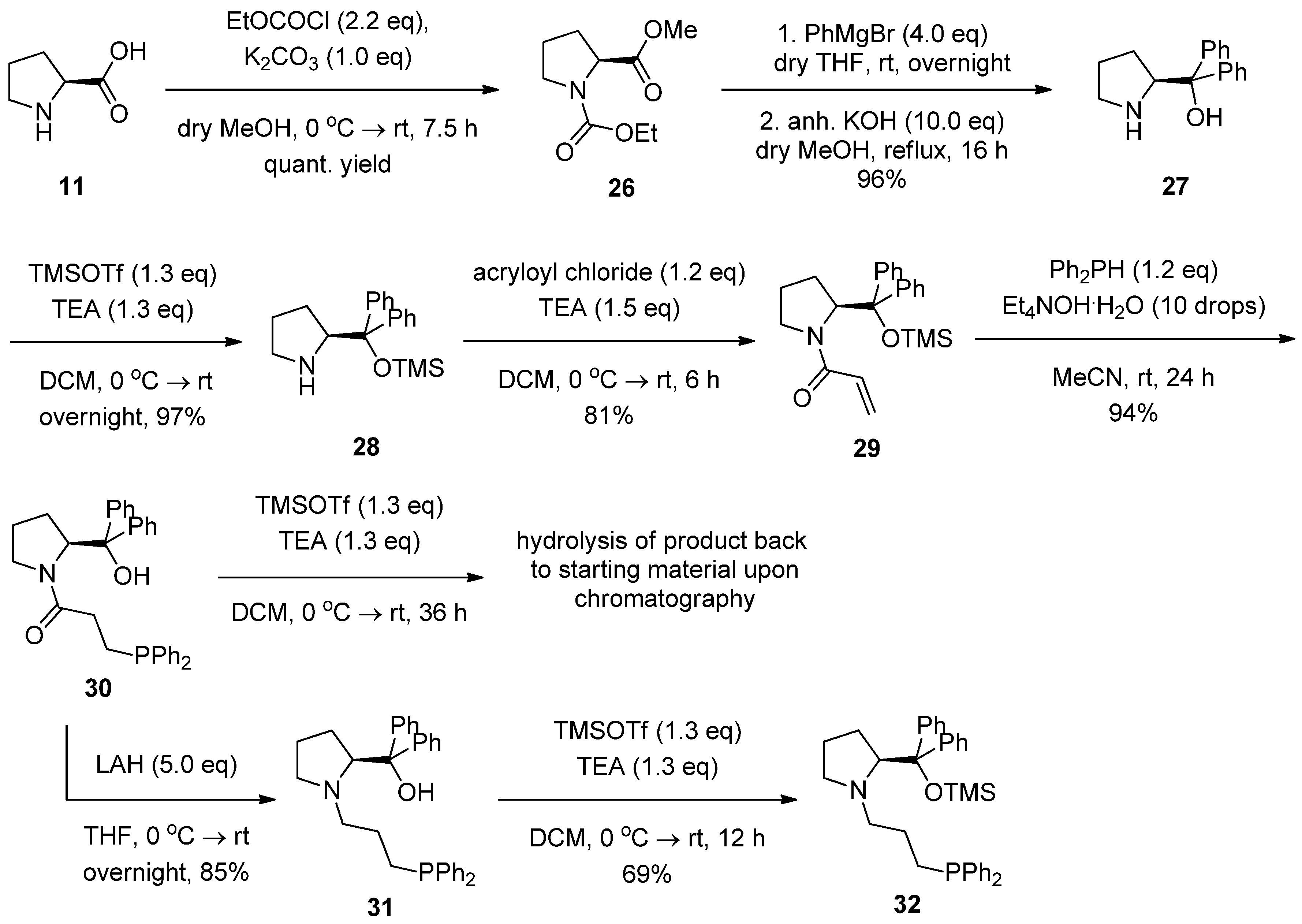
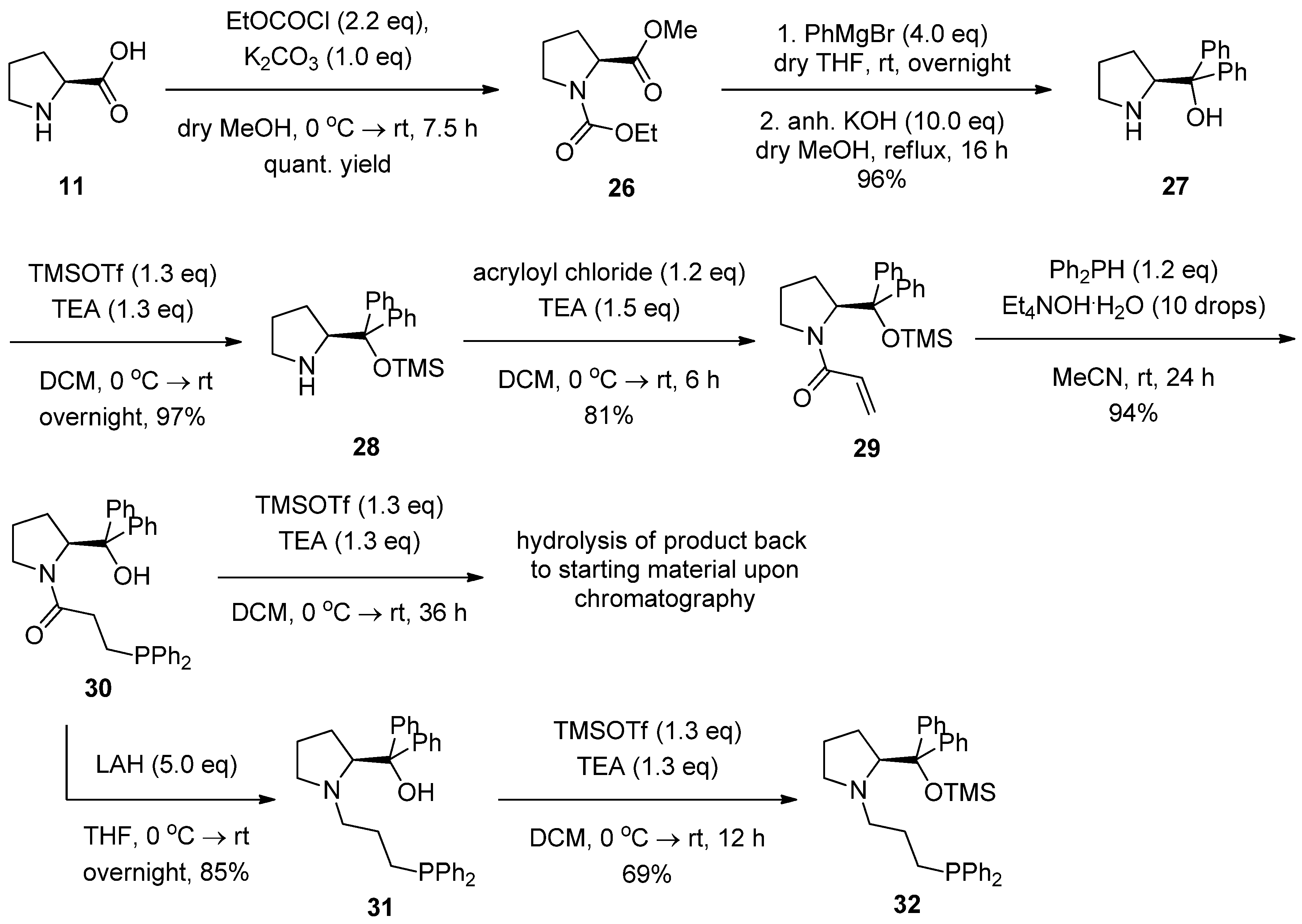
2.2. Syntheses of Chiral Aminophosphines from a Commercially Available Chiral Amine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Aminophosphine | Yield (%) a | ee (%) b | Entry | Amidophosphine | Yield (%) a | ee (%) b |
|---|---|---|---|---|---|---|---|
| 1 b | 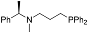 | 46 | 5 | 14 | 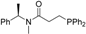 | 39 | 5 |
| 2 | 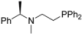 | 86 | 0 | 15 | 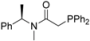 | 95 | 0 |
| 3 | 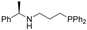 | 84 | 4 | 16 | 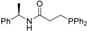 | 85 | 4 |
| 4 | 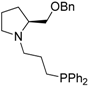 | 83 | 3 | 17 | 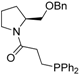 | 91 | 0 |
| 5 |  | 89 | 6 | 18 | 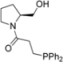 | 94 | 9 |
| 6 |  | 74 | 2 | 19 | 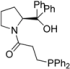 | 86 | 0 |
| 7 |  | 73 | 2 | 20 | n/a c | n/a d | n/a d |
| 8 | 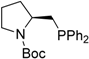 | 84 | -9 | 21 | n/a c | n/a d | n/a d |
| 9 |  | 91 | -2 | 22 | n/a c | n/a d | n/a d |
| 10 | 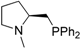 | 83 | 0 | 23 | n/a c | n/a d | n/a d |
| 11 | 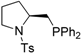 | 86 | -6 | 24 | n/a c | n/a d | n/a d |
| 12 |  | 69 | 10 | 25 |  | 81 | 0 |
| 13 |  | 90 | 3 | 26 |  | 88 | 3 |
2.3. Syntheses of l-Proline-Derived Chiral Aminophosphines


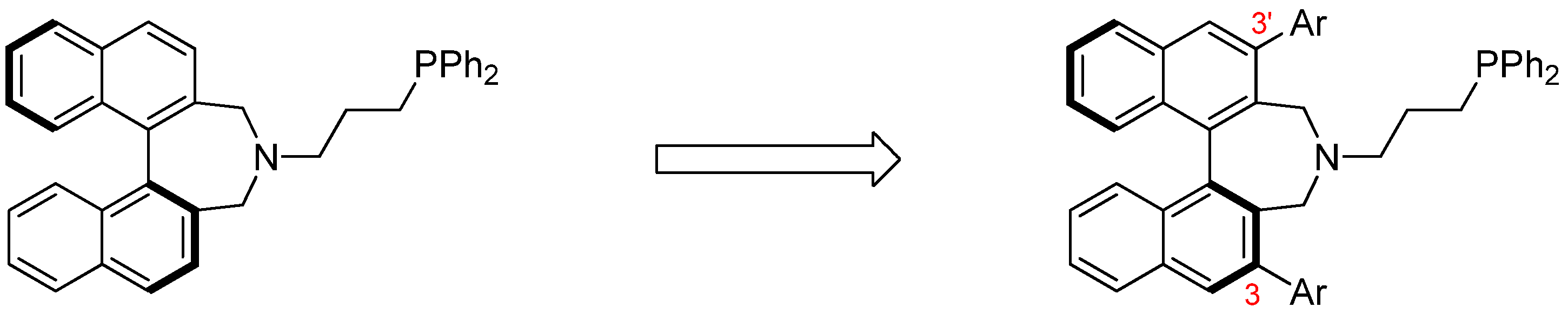
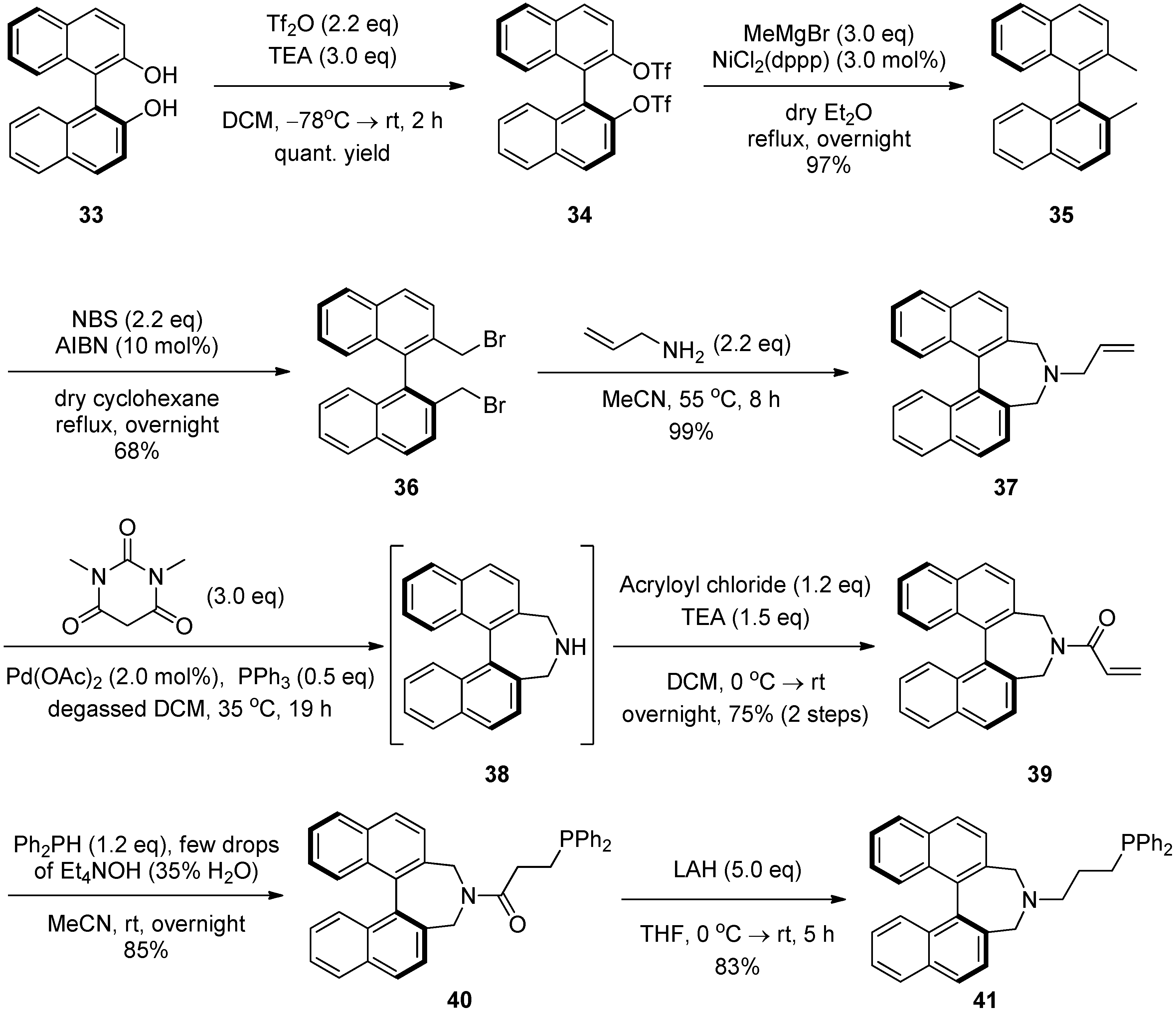
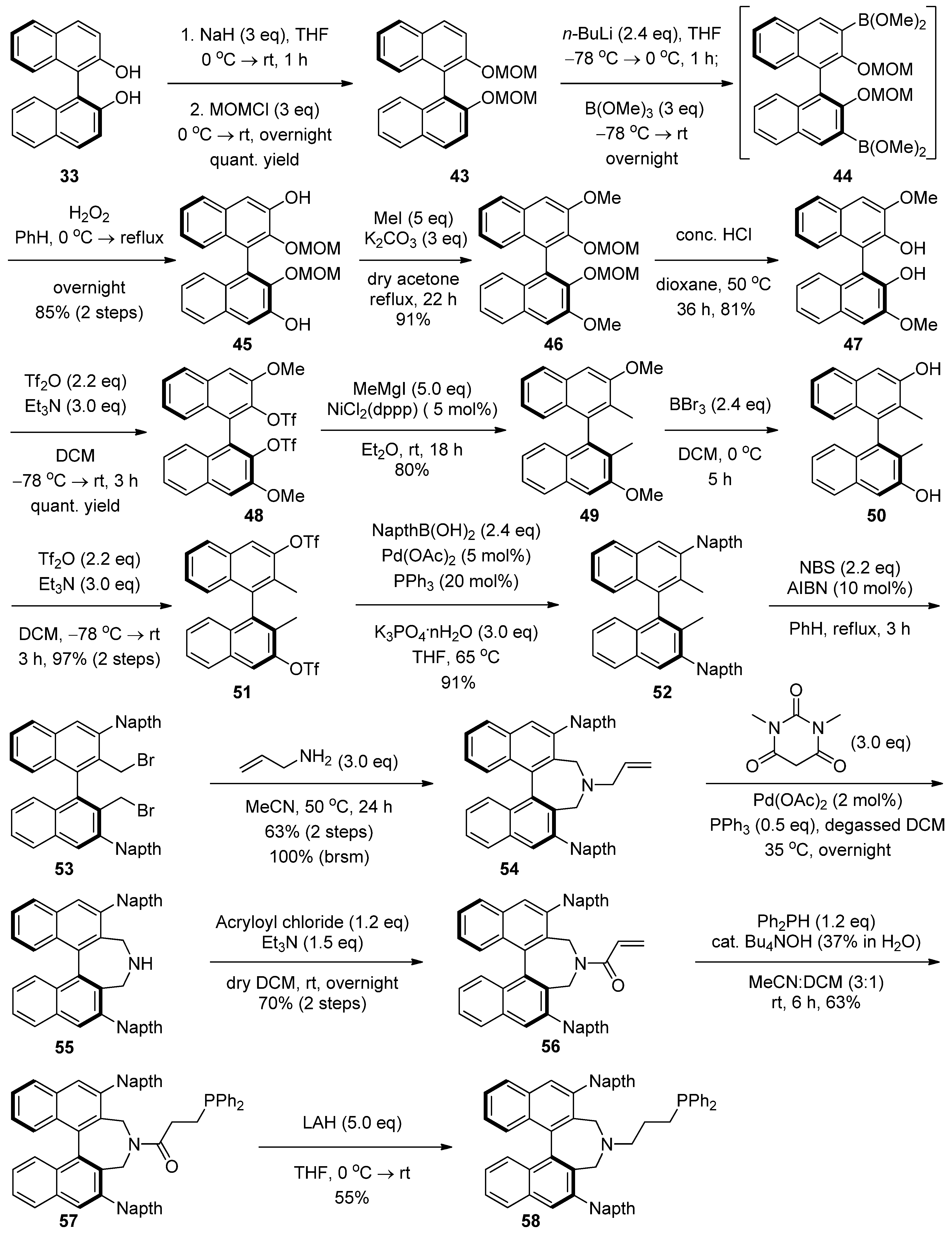
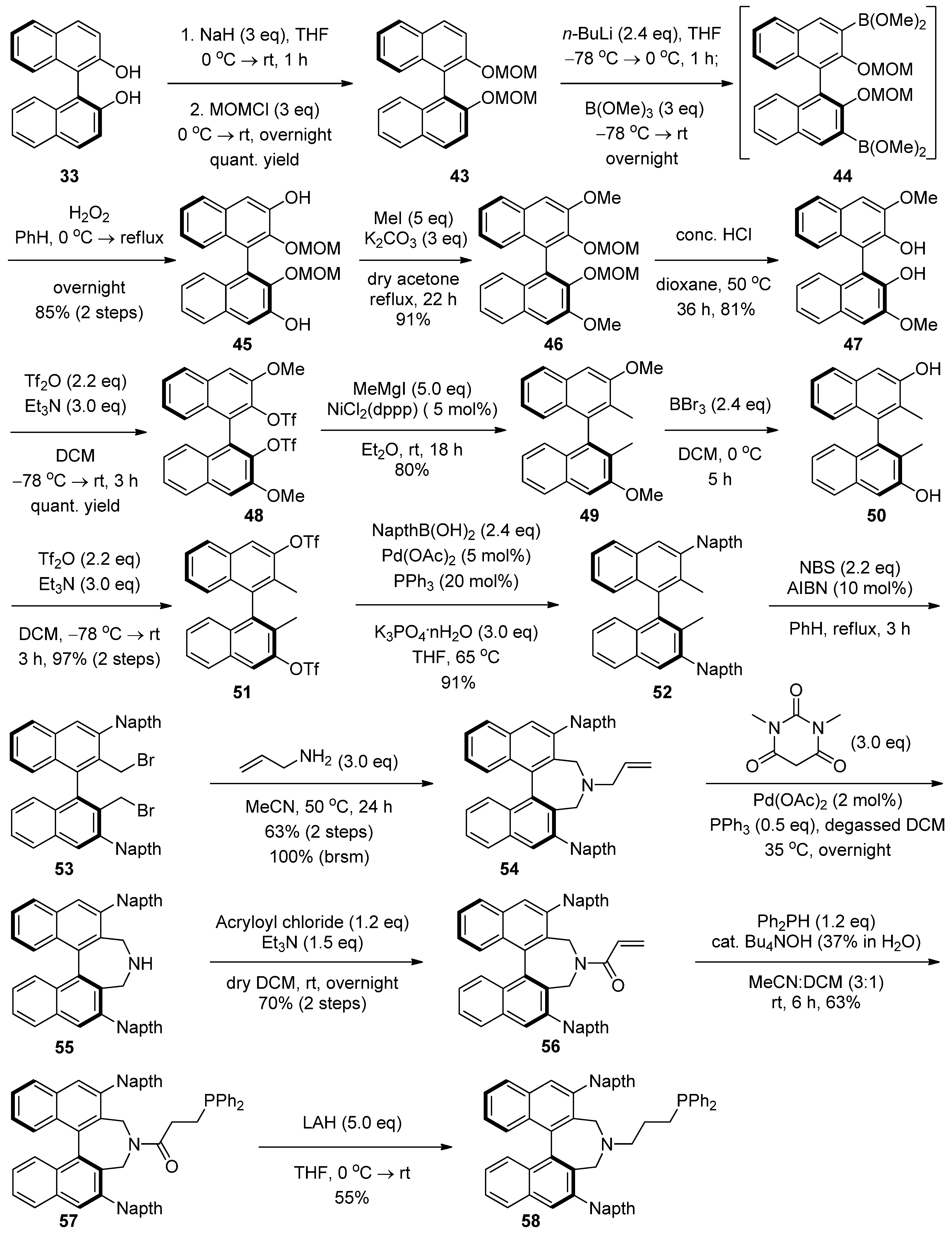
2.4. Syntheses of Binol-Derived Chiral Aminophosphines


3. Experimental
3.1. General
3.2. Materials and Reagents
3.3. Instrumentation
3.4. Procedures for Syntheses of Amidophosphines and Aminophosphines
4. Conclusion
Acknowledgements
- Sample Availability: Samples of compounds 21, 40, and 41 are available from the authors.
References
- The Microbial Production of Amino Acids; Yamada, K.; Kinoshita, S.; Tsunoda, T.; Aida, K. (Eds.) John Wiley & Son: New York, NY, USA, 1972.
- Dalko, P.I. Asymmetric Organocatalysis: A New Stream in Organic Synthesis. In Enantioselective Organocatalysis: Reactions and Experimental Procedures; Dalko, P.I., Ed.; Wiley-VCH: Darmstadt, Germany, 2007; pp. 1–13. [Google Scholar]
- Kagan, H.B.; Gopalaiah, K. Early history of asymmetric synthesis: Who are the scientists who set up the basic principles and the first experiments? New J. Chem 2011, 35, 1933–1937. [Google Scholar] [CrossRef]
- Bredig, G.; Balcom, R.W. Kinetik der Kohlendioxyd-abspaltung aus Camphocarbonsäure. Ber. Deutsch. Chem. Ger. 1908, 41, 740–751. [Google Scholar] [CrossRef]
- Bredig, G.; Fajans, K. Zur Stereochemie der Katalyse. Ber. Deutsch. Chem. Ger. 1908, 41, 752–763. [Google Scholar] [CrossRef]
- Bredig, G.; Fiske, P.S. Durch Katalysatoren bewirkte Asymmetrische Synthese. Biochem. Zeits. 1912, 46, 7–23. [Google Scholar]
- Pracejus, H. Organische Katalysatoren, LXI. Asymmetrische Synthesen mit Ketenen, I. Alkaloid-katalysierte asymmetrische Synthesen von α-Phenyl-propionsäureestern. Justus Liebigs Ann. Chem 1960, 634, 9–22. [Google Scholar] [CrossRef]
- Knowles, W.S.; Sabacky, M.J. Catalytic asymmetric hydrogenation employing a soluble, optically active, rhodium complex. Chem. Commun. 1968, 22, 1445–1446. [Google Scholar]
- Knowles, W.S.; Sabacky, M.J.; Vineyard, B.D. Catalytic asymmetric hydrogenation using soluble, optically active phosphine complexes. Ann. NY Acad. Sci. 1970, 172, 232–237. [Google Scholar] [CrossRef]
- Knowles, W.S.; Sabacky, M.J.; Vineyard, B.D. Catalytic asymmetric hydrogenation. J. Chem. Soc. Chem. Commun. 1972, 1, 10–11. [Google Scholar]
- Knowles, W.S.; Sabacky, M.J.; Vineyard, B.D.; Weinkauff, D.J. Asymmetric hydrogenation with a complex of rhodium and a chiral bisphosphine. J. Am. Chem. Soc. 1975, 97, 2567–2568. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric hydrogenation. Acc. Chem. Res. 1983, 16, 106–112. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric hydrogenations. Adv. Synth. Cat. 2003, 345, 3–13. [Google Scholar] [CrossRef]
- Komarov, I.V.; Börner, A. Highly enantioselective or not? Chiral monodentate monophosphorus ligands in the asymmetric hydrogenation. Angew. Chem. Int. Ed. Engl. 2001, 40, 1197–1200. [Google Scholar] [CrossRef]
- Crépy, K.V.L.; Imamoto, T. Recent developments in catalytic asymmetric hydrogenation employing P-chirogenic diphosphine ligands. Adv. Synth. Catal. 2003, 345, 79–101. [Google Scholar] [CrossRef]
- Hajos, Z.G.; Parrish, D.R.W. Werkwijze Voor de Bereiding van 1,3-Dioxycycloackanen. German Patent DE 2102623, 1971. [Google Scholar]
- Hajos, Z.G.; Parrish, D.R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Eder, U.; Sauer, G.; Wiechert, R. Process for the Manufacture of Optically Active Bicycloalkane Derivatives. German Patent DE 2014757, 1971. [Google Scholar]
- Eder, U.; Sauer, G.; Wiechert, R. New type of asymmetric cyclization to optically active steroid CD partial structures. Angew. Chem. Int. Ed. Engl. 1971, 10, 496–497. [Google Scholar]
- List, B.; Yang, J.W. The organic approach to asymmetric catalysis. Science 2006, 313, 1584–1586. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- List, B. Asymmetric Organocatalysis, 1st ed; Springer: Heidelberg, Germany, 2010; Volume 291, pp. 1–458. [Google Scholar]
- Sriramurthy, V.; Barcan, G.A.; Kwon, O. Bisphosphine-catalyzed mixed double-michael reactions: Asymmetric synthesis of oxazolidines, thiazolidines, and pyrrolidines. J. Am. Chem. Soc. 2007, 129, 12928–12929. [Google Scholar]
- Sriramurthy, V.; Kwon, O. Diphosphine-catalyzed mixed double-Michael reaction: A unified synthesis of indolines, dihydropyrrolopyridines, benzimidazolines, tetrahydroquinolines, tetrahydroisoquinolines, dihydrobenzo-1,4-oxazines, and dihydrobenzo-3,1-oxazines. Org. Lett. 2010, 12, 1084–1087. [Google Scholar] [CrossRef]
- Fan, Y.C.; Kwon, O. Diversity-oriented synthesis based on the DPPP-catalyzed mixed double-Michael reactions of electron-deficient acetylenes and β-amino alcohols. Molecules 2011, 16, 3802–3825. [Google Scholar] [CrossRef]
- Kisanga, P.B.; Verkade, J.G. pKa Measurements of P(RNCH2CH3)3N. J. Org. Chem. 2000, 65, 5431–5432. [Google Scholar] [CrossRef]
- Kisanga, P.B.; Ilankumaran, P.; Fetterly, B.M.; Verkade, J.G. P(RNCH2CH2)3N: Efficient 1,4-addition catalysts. J. Org. Chem. 2002, 67, 3555–3560. [Google Scholar] [CrossRef]
- Verkade, J.G.; Kisanga, P.B. Recent applications of proazaphosphatranes in organic synthesis. Aldrichimica Acta 2004, 1, 3–14. [Google Scholar]
- Sriramurthy, V.; Kwon, O. University of California, Los Angeles, CA, USA. Chiral 4-(N,N-dialkyl)aminomethyl-5-diphenylphosphinomethyl-1,3-dioxolane. 2012. [Google Scholar]
- Blinn, D.A.; Button, R.S.; Farazi, V.; Neeb, M.K.; Tapley, C.L.; Treheame, T.E.; West, S.D.; Kruger, T.L.; Storhoff, B.N. Addition of diphenylphosphine to Michael-type olefins: The preparation of phosphine-nitrile and phosphine-ester ligands. J. Organomet. Chem. 1990, 393, 143–152. [Google Scholar]
- Shi, M.; Chen, L.-H.; Li, C.-Q. Chiral phosphine Lewis bases catalyzed asymmetric aza-Baylis-Hillman reaction of N-sulfonated imines with activated olefins. J. Am. Chem. Soc. 2005, 127, 3790–3800. [Google Scholar]
- Jiang, Y.-Q.; Shi, Y.-L.; Shi, M. Chiral phosphine-catalyzed enantioselective construction of γ-butenolides through substitution of Morita-Baylis-Hillman acetates with 2-trimethylsilyloxy furan. J. Am. Chem. Soc. 2008, 130, 7202–7203. [Google Scholar] [CrossRef]
- Cowen, B.J.; Miller, S.J. Enantioselective [3+2]-cycloadditions catalyzed by a protected, multifunctional phosphine-containing α-amino acid. J. Am. Chem. Soc. 2007, 129, 10988–10989. [Google Scholar] [CrossRef]
- Xiao, H.; Chai, Z.; Zheng, C.-W.; Yang, Y.-Q.; Liu, W.; Zhang, J.-K.; Zhao, G. Asymmetric [3+2] cycloadditions of allenoates and dual activated olefins catalyzed by simple bifunctional N-acyl aminophosphines. Angew. Chem. Int. Ed. Engl. 2010, 49, 4467–4470. [Google Scholar]
- Cao, D.-D.; Liu, W.; Lu, Y.-P.; Yang, Y.-Q.; Zhao, G. Bifunctional N-acyl-aminophosphine-catalyzed asymmetric [4+2] cycloadditions of allenoates and imines. Chem. Eur. J. 2011, 17, 10562–10565. [Google Scholar] [CrossRef]
- List, B. Proline-catalyzed asymmetric reactions. Tetrahedron 2002, 58, 5573–5590. [Google Scholar] [CrossRef]
- List, B. Enamine catalysis is a powerful strategy for the catalytic generation and use of carbanion equivalents. Acc. Chem. Res. 2004, 37, 548–557. [Google Scholar] [CrossRef]
- Whitesell, J.K. C2 Symmetry and asymmetric induction. Chem. Rev. 1989, 89, 1581–1590. [Google Scholar] [CrossRef]
- Vogl, E.M.; Matsunaga, S.; Kanai, M.; Iida, T.; Shibasaki, M. Linking BINOL: C2-Symmetric ligands for investigations on asymmetric catalysis. Tetrahedron Lett. 1998, 39, 7917–7920. [Google Scholar] [CrossRef]
- Noyori, R.; Takaya, H. BINAP: An efficient chiral element for asymmetric catalysis. Acc. Chem. Res. 1990, 23, 345–350. [Google Scholar] [CrossRef]
- Noyori, R. Asymmetric catalysis: Science and opportunities. Angew. Chem. Int. Ed. Engl. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Chen, Y.; Yekta, S.; Yudin, A.K. Modified BINOL ligands in asymmetric catalysis. Chem. Rev. 2003, 103, 3155–3212. [Google Scholar] [CrossRef]
- Ooi, T.; Kameda, M.; Maruoka, K. Design of N-spiro C2-symmetric chiral quaternary ammonium bromides as novel chiral phase-transfer catalysts: Synthesis and application to practical asymmetric synthesis of α-amino acids. J. Am. Chem. Soc. 2003, 125, 5139–5151. [Google Scholar] [CrossRef]
- Demuth, M.; Mikhail, G. A convenient in situ preparation of trimethylsilyl trifluoromethanesulfonate. Synthesis 1982, 10, 827. [Google Scholar] [CrossRef]
- Demuth, M.; Mikhail, G. Electrophile-initiated selective ring transformations of cyclopropyl ketones. Tetrahedron 1983, 39, 991–997. [Google Scholar] [CrossRef]
- Eastham, G.R.; Waugh, M.; Pringle, P.; Fanjul Solares, T. Process for the Carbonylation of Ethylenically Unsaturated Compounds, Novel Carbonylation Ligands and Catalyst Systems Incorporating such Ligands. WO/2010/001174, 2010. [Google Scholar]
- Gulyás, H.; Benet-Buchholz, J.; Escudero-Adan, E.C.; Freixa, Z.; van Leeuwen, P.W.N.M. Ionic interaction as a powerful driving force for the formation of heterobidentate assembly ligands. Chem. Eur. J. 2007, 13, 3424–3430. [Google Scholar] [CrossRef]
- Adams, H.; Bawa, R.A.; McMillan, K.G.; Jones, S. Asymmetric control in Diels-Alder cycloadditions of chiral 9-aminoanthracenes by relay of stereochemical information. Tetrahedron: Asymmetry 2007, 18, 1003–1012. [Google Scholar] [CrossRef]
- Galbo, F.L.; Occhiato, E.G.; Guarna, A.; Faggi, C. Preparation and cycloaddition reactions of enantiopure 2-(N-acylamino)-1,3-dienes for the synthesis of octahydroquinoline derivatives. J. Org. Chem. 2003, 68, 6360–6368. [Google Scholar] [CrossRef]
- Wallén, E.A.A.; Christiaans, J.A.M.; Saario, S.M.; Forsberg, M.M.; Venäläinen, J.I.; Paso, H.M.; Männistö, P.T.; Gynthera, J. 4-Phenylbutanoyl-2(S)-acylpyrrolidines and 4-phenylbutanoyl-L-prolyl-2(S)-acylpyrrolidines as prolyl oligopeptidase inhibitors. Bioorg. Med. Chem. 2002, 10, 2199–2206. [Google Scholar] [CrossRef]
- Alza, E.; Cambeiro, X.C.; Jimeno, C.; Pericàs, M.A. Highly enantioselective Michael additions in water catalyzed by a PS-supported pyrrolidine. Org. Lett. 2007, 9, 3717–3720. [Google Scholar]
- Havran, L.M.; Chong, D.C.; Childers, W.E.; Dollings, P.J.; Dietrich, A.; Harrison, B.L.; Marathias, V.; Tawa, G.; Aulabaugh, A.; Cowling, R.; et al. 3,4-Dihydropyrimido(1,2-α)indol-10(2H)-ones as potent non-peptidic inhibitors of caspase-3. Bioorg. Med. Chem. 2009, 17, 7755–7768. [Google Scholar] [CrossRef]
- Dokuzovič, Z.; Roberts, N.K.; Sawyer, J.F.; Whelan, J.; Bosnich, B. Asymmetric synthesis. Metal complex mediated synthesis of chiral glycine by enantioselective proton exchange. J. Am. Chem. Soc. 1986, 108, 2034–2039. [Google Scholar]
- Kanai, M.; Nakagawa, Y.; Tomioka, K. An asymmetric conjugate addition reaction of lithium organocopper reagent controlled by a chiral amidophosphine. Tetrahedron 1999, 55, 3831–3842. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Dalpozzo, R.; Giuliani, A.; Marcantoni, E.; Mecozzi, T.; Sambri, L.; Torregiani, E. An efficient procedure for the preparation of (E)-a-alkylidenecycloalkanones mediated by a CeCl3. 7H2O–NaI system. Novel methodology for the synthesis of (S)-(−)-pulegone. J. Org. Chem. 2002, 67, 9111–9114. [Google Scholar] [CrossRef]
- Hayashi, T.; Konishi, M.; Fukushima, M.; Kanehira, K.; Hioki, T.; Kumada, M. Chiral (β-aminoalkyl)phosphines. Highly efficient phosphine ligands for catalytic asymmetric Grignard cross-coupling. J. Org. Chem. 1983, 48, 2195–2202. [Google Scholar] [CrossRef]
- Gautier, F.-M.; Jones, S.; Martin, S. Asymmetric reduction of ketimines with trichlorosilane employing an imidazole derived organocatalyst. J. Org. Biomol. Chem. 2009, 7, 229–231. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kinoshita, T.; Uehara, H.; Sudo, T.; Ryu, I. Organocatalytic enantioselective synthesis of nitrogen-substituted dihydropyran-2-ones, a key synthetic intermediate of 1β-methylcarbapenems. Org. Lett. 2009, 11, 3934–3937. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Khong, S.N.; Kwon, O. Chiral Aminophosphines as Catalysts for Enantioselective Double-Michael Indoline Syntheses. Molecules 2012, 17, 5626-5650. https://doi.org/10.3390/molecules17055626
Khong SN, Kwon O. Chiral Aminophosphines as Catalysts for Enantioselective Double-Michael Indoline Syntheses. Molecules. 2012; 17(5):5626-5650. https://doi.org/10.3390/molecules17055626
Chicago/Turabian StyleKhong, San N., and Ohyun Kwon. 2012. "Chiral Aminophosphines as Catalysts for Enantioselective Double-Michael Indoline Syntheses" Molecules 17, no. 5: 5626-5650. https://doi.org/10.3390/molecules17055626