Application of Paramagnetic NMR-Validated Molecular Dynamics Simulation to the Analysis of a Conformational Ensemble of a Branched Oligosaccharide

Abstract
:
1. Introduction
2. Results and Discussion
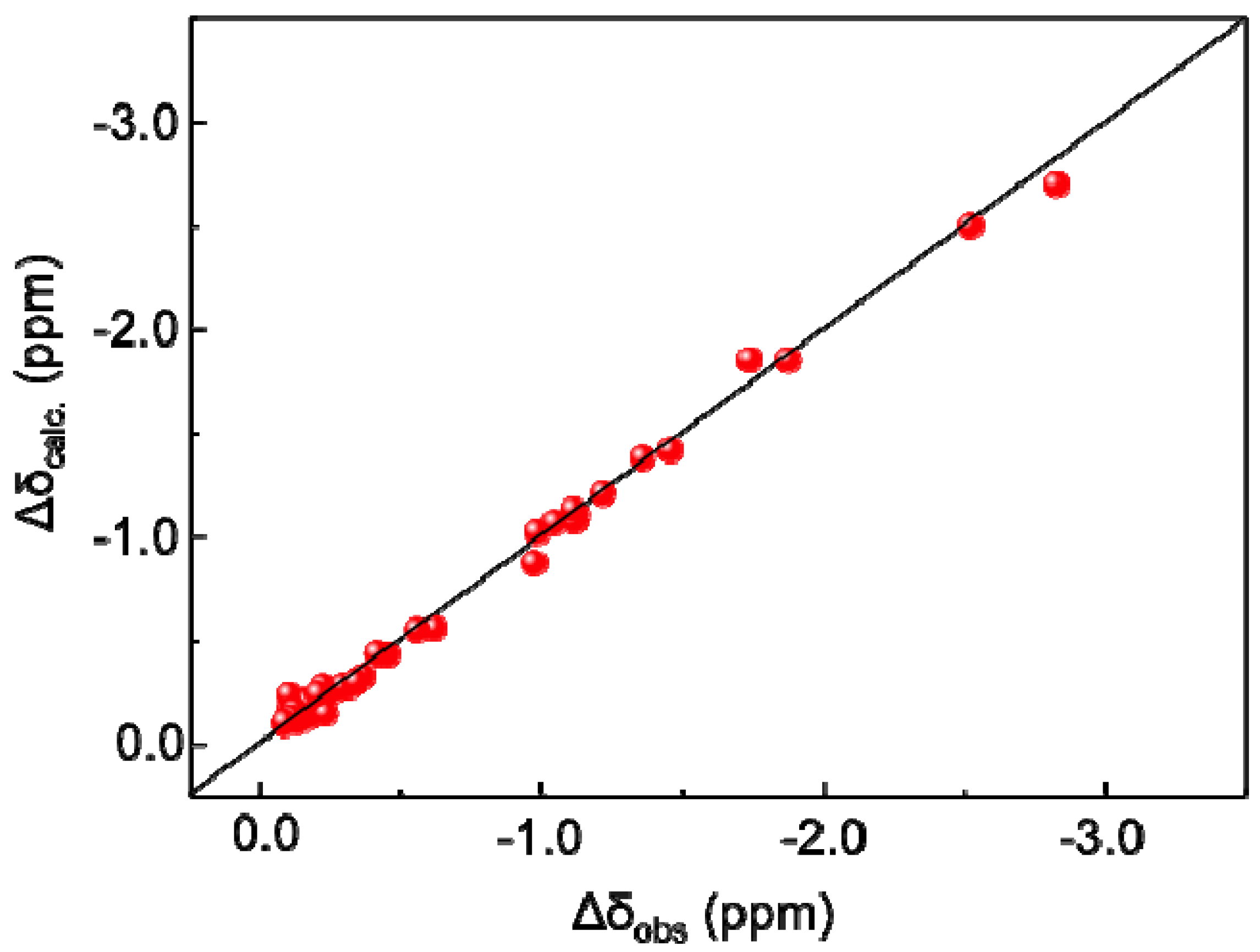
2.1. MD Simulations and PCS Analyses of the GM2 Tetrasaccharide





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| La3+ | Tm3+ | Tb3+ | ||||
|---|---|---|---|---|---|---|
| Δδ13C/ppm | Δδ1H/ppm | Δδ13C/ppm | Δδ1H/ppm | Δδ13C/ppm | Δδ1H/ppm | |
| Glc 1 | 79.95 | 5.132 | 77.12 | 2.620 | n.d. (a) | n.d. |
| 2 | 71.44 | 3.547 | 69.57 | 1.811 | n.d. | n.d. |
| 3 | 75.15 | 3.676 | 73.93 | 2.565 | n.d. | n.d. |
| 4 | 78.06 | 3.652 | 76.94 | 2.670 | 75.97 | 1.683 |
| 5 | 76.63 | 3.652 | 75.17 | 2.295 | n.d. | n.d. |
| 6 | 59.99 | 3.884 | 58.86 | 2.840 | 58.01 | 2.049 |
| 6 | 59.98 | 3.772 | 58.86 | 2.800 | 58.01 | 1.939 |
| Gal 1 | 102.7 | 4.480 | 102.1 | 3.922 | 101.6 | 3.325 |
| 2 | 70.10 | 3.305 | 69.65 | 2.890 | 69.20 | 2.416 |
| 3 | 74.43 | 4.074 | 74.09 | 3.778 | 73.75 | 3.419 |
| 4 | 77.31 | 4.046 | 77.00 | 3.823 | 76.63 | 3.514 |
| 5 | 74.12 | 3.698 | 73.75 | 3.360 | 73.34 | 2.932 |
| 6 | 60.67 | 3.751 | 60.39 | 3.530 | 60.00 | 3.142 |
| 6 | 60.66 | 3.702 | 60.39 | 3.480 | 60.00 | 2.985 |
| Neu5Ac 3 | 36.98 | 2.590 | 36.77 | 2.393 | 36.64 | 2.143 |
| 3 | 36.99 | 1.844 | 36.76 | 1.612 | 36.64 | 1.346 |
| 4 | 68.78 | 3.700 | 68.57 | 3.570 | 68.44 | 3.409 |
| 5 | 51.66 | 3.731 | 51.47 | 3.577 | 51.39 | 3.403 |
| 6 | 73.15 | 3.405 | 72.96 | 3.275 | 72.81 | 3.103 |
| 7 | 68.11 | 3.508 | 67.92 | 3.407 | 67.81 | 3.263 |
| 8 | 72.37 | 3.669 | 72.18 | 3.505 | 72.03 | 3.285 |
| 9 | 62.90 | 3.792 | 62.76 | 3.677 | 62.64 | 3.516 |
| 9 | 62.90 | 3.543 | 62.75 | 3.441 | 62.64 | 3.289 |
| GalNAc 1 | 102.8 | 4.680 | 102.6 | 4.480 | 102.3 | 4.227 |
| 2 | 52.40 | 3.845 | 52.17 | 3.641 | 51.97 | 3.324 |
| 3 | 71.38 | 3.594 | 71.20 | 3.470 | 71.02 | 3.278 |
| 4 | 67.88 | 3.844 | 67.71 | 3.747 | 67.54 | 3.564 |
| 5 | 74.82 | 3.637 | 74.64 | 3.524 | 74.45 | 3.315 |
| 6 | 61.23 | 3.693 | 61.10 | 3.609 | 60.93 | 3.405 |
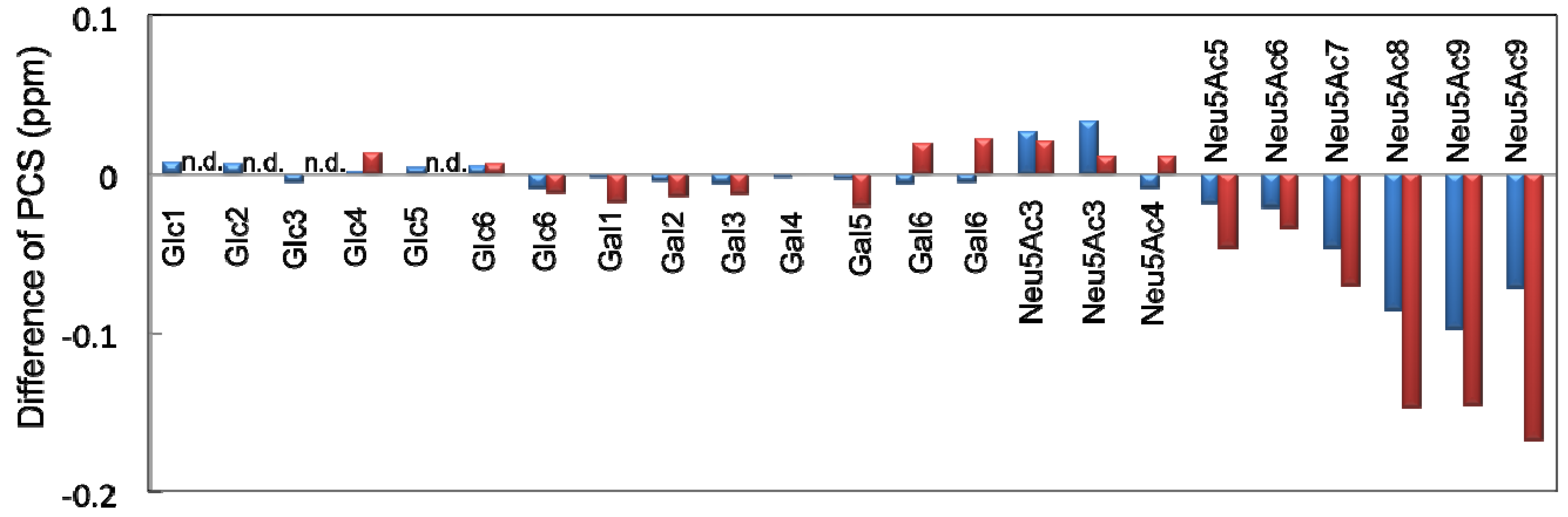
| Glc | Gal | Neu5Ac | GalNAc | |||||
|---|---|---|---|---|---|---|---|---|
| Δδ13C | Δδ1H | Δδ13C | Δδ1H | Δδ13C | Δδ1H | Δδ13C | Δδ1H | |
| 1 | −2.83 | −2.51 | −0.62 | −0.56 | −0.23 | −0.20 | ||
| 2 | −1.87 | −1.74 | −0.45 | −0.41 | −0.23 | −0.20 | ||
| 3 | −1.22 | −1.11 | −0.34 | −0.30 | −0.22 | −0.23/−0.20 | −0.18 | −0.12 |
| 4 | −1.12 | −0.98 | −0.31 | −0.22 | −0.22 | −0.13 | −0.16 | −0.10 |
| 5 | −1.46 | −1.36 | −0.36 | −0.34 | −0.20 | −0.15 | −0.17 | −0.11 |
| 6 | −1.12 | −1.04/−0.97 | −0.27 | −0.22 | −0.19 | −0.13 | −0.13 | −0.08 |
| 7 | −0.18 | −0.10 | ||||||
| 8 | −0.19 | −0.16 | ||||||
| 9 | −0.15 | −0.11/−0.10 | ||||||

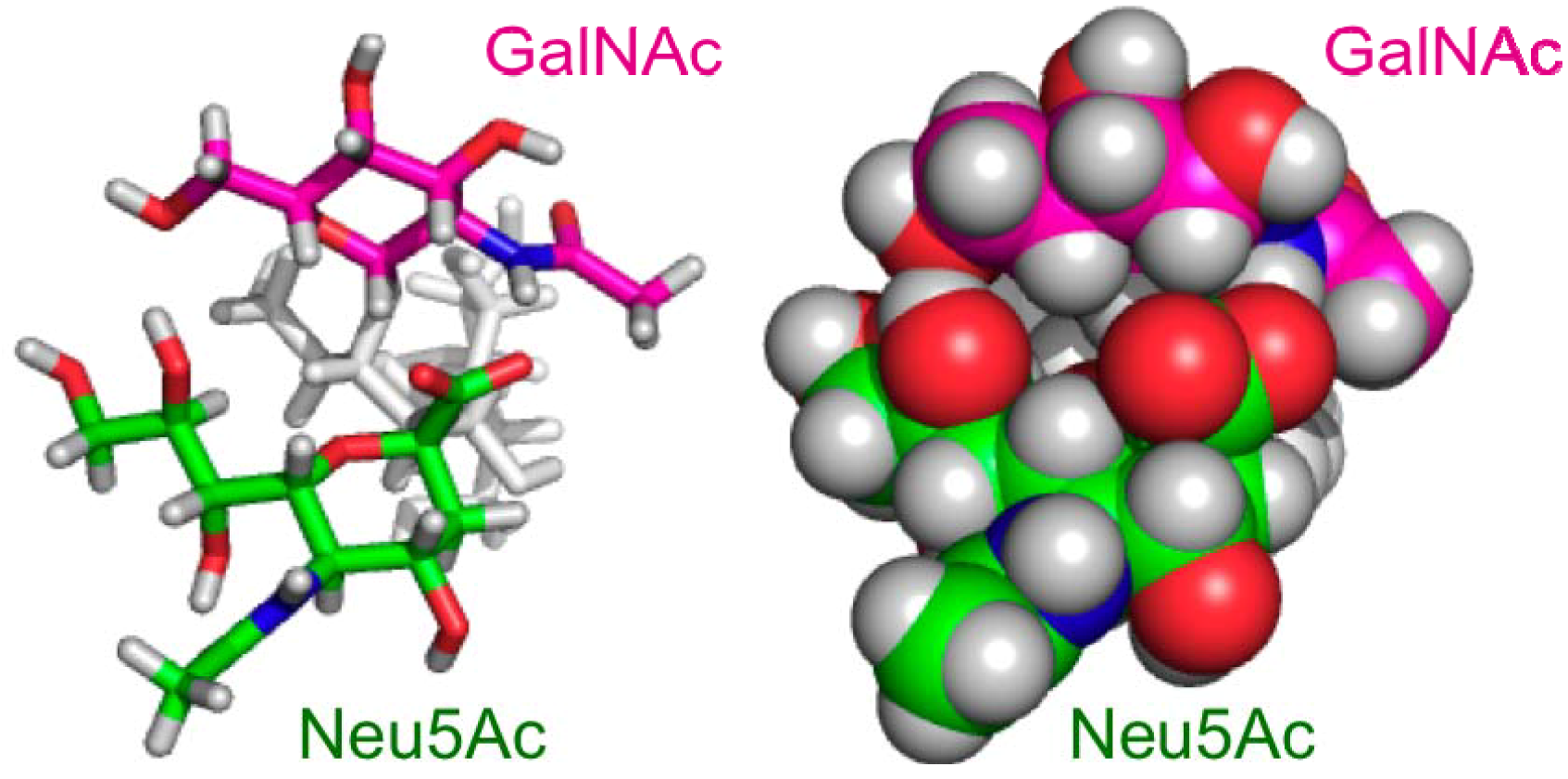
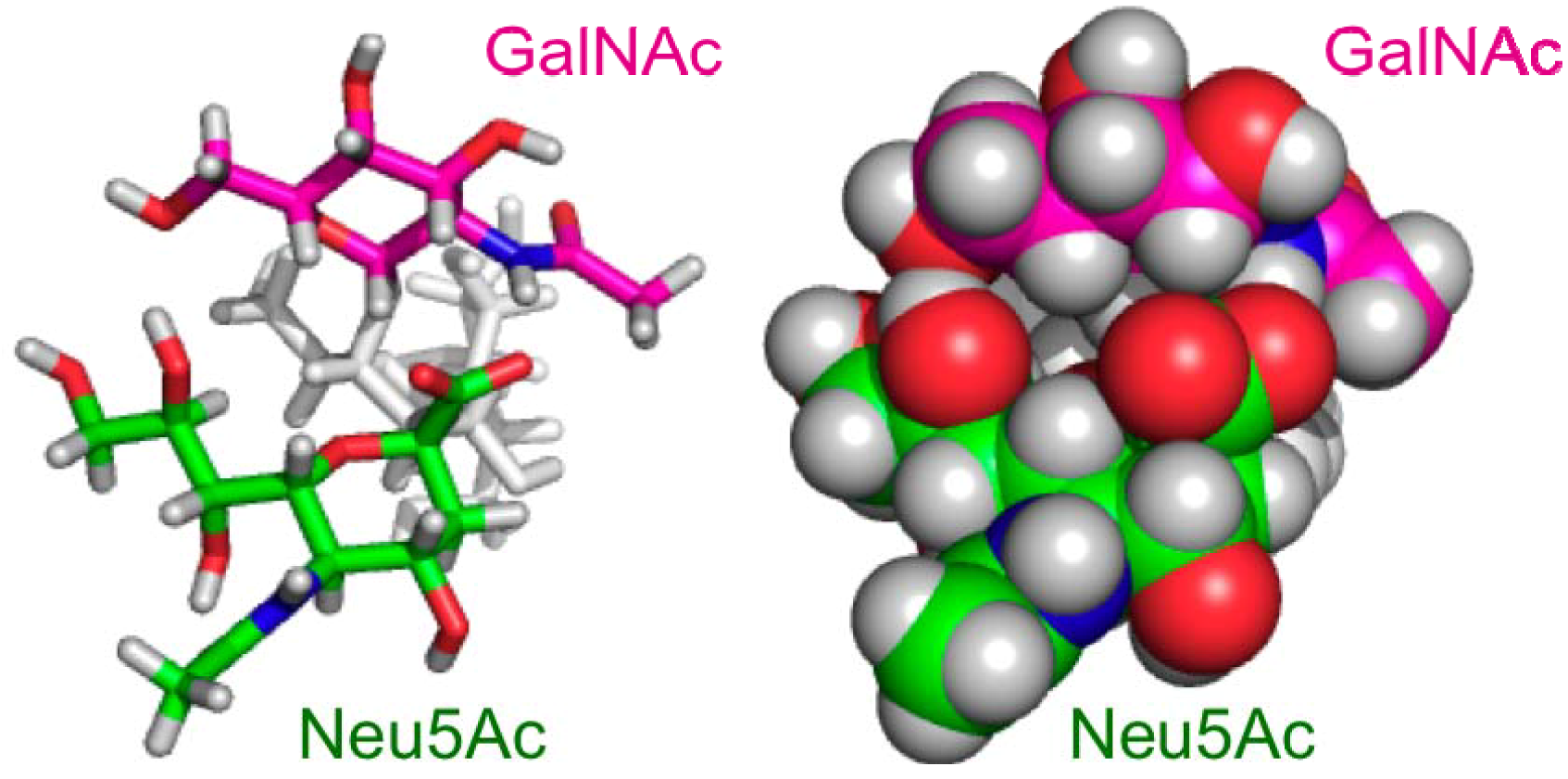
2.2. Conformational Comparison between the GM2 and GM3 Sugar Chains

3. Experimental
3.1. General
3.2. MD Simulations of the Sugar Moiety of GM2
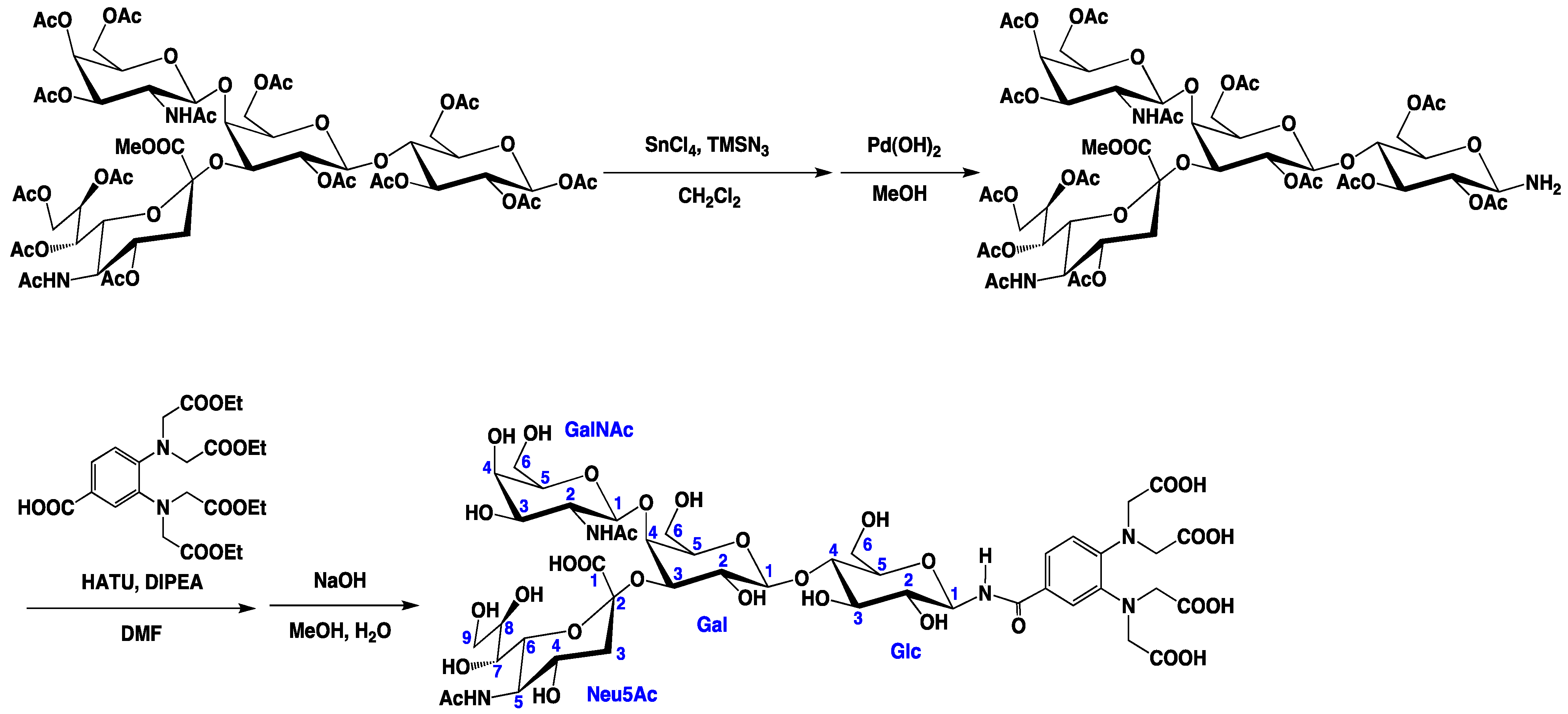
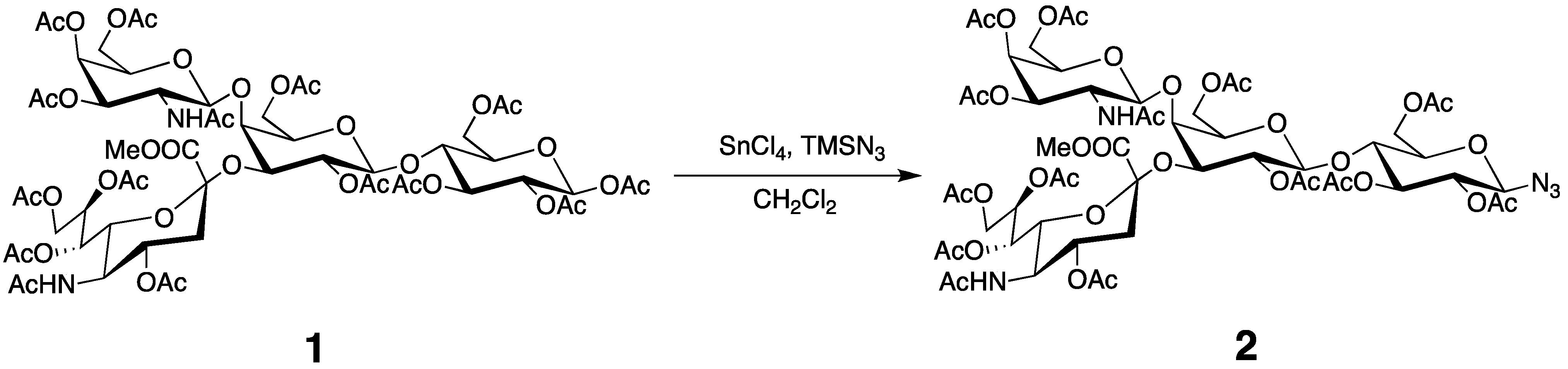
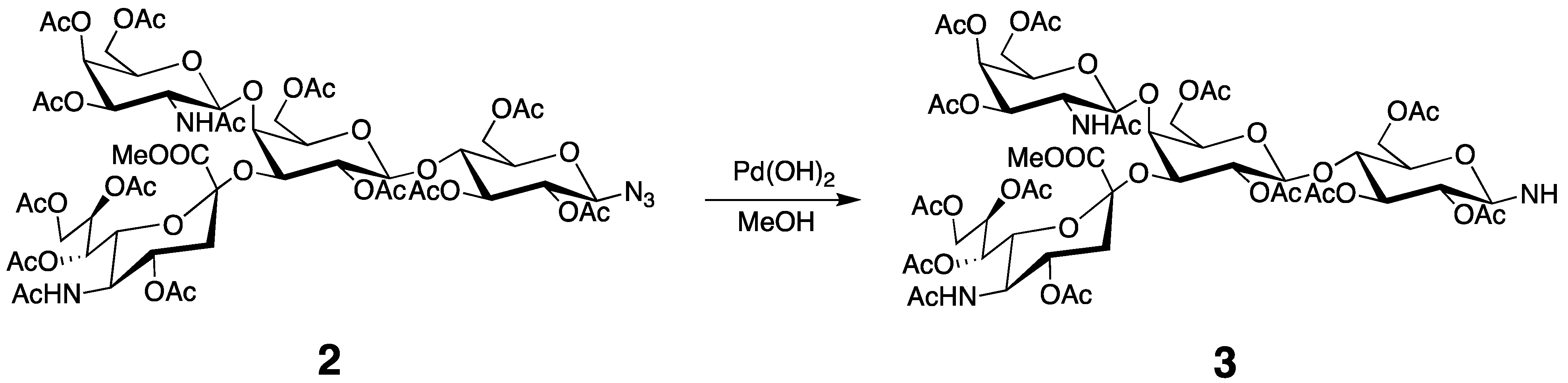
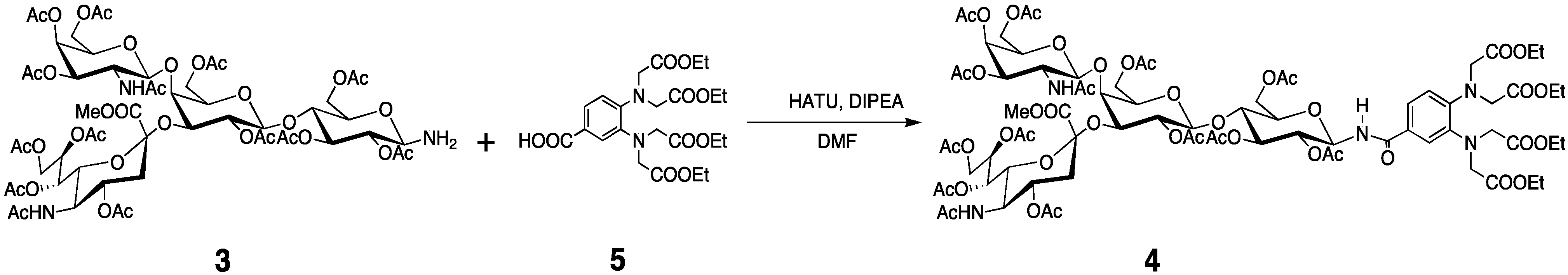
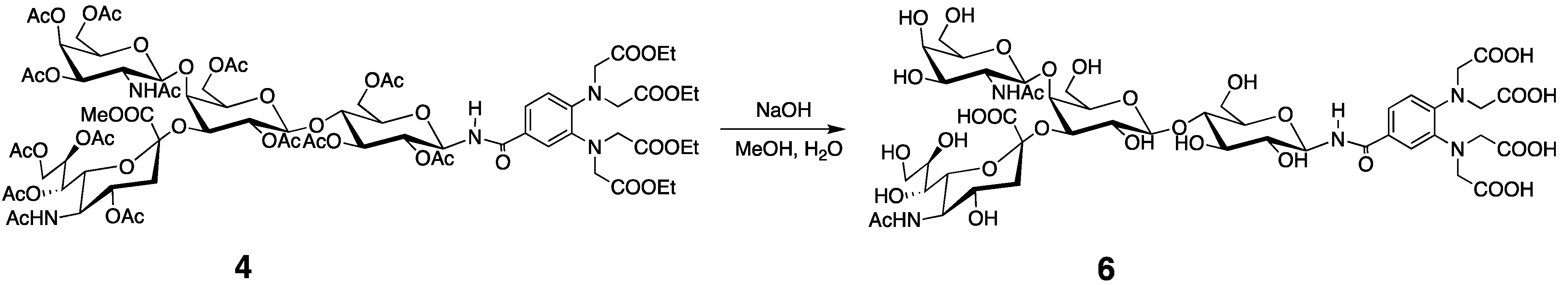
3.3. Preparation of the Tagged GM2 Tetrasaccharide




3.3. PCS Observation and Analyses of the GM2 Tetrasaccharide
4. Conclusions
Acknowledgments
- Sample Availability: Contact the authors.
References
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Sharon, N. Lectins: Carbohydrate-specific reagents and biological recognition molecules. J. Biol. Chem. 2007, 282, 2753–2764. [Google Scholar] [CrossRef]
- Kato, K.; Kamiya, Y. Structural views of glycoprotein-fate determination in cells. Glycobiology 2007, 17, 1031–1044. [Google Scholar] [CrossRef]
- Wormald, M.R.; Petrescu, A.J.; Pao, Y.L.; Glithero, A.; Elliott, T.; Dwek, R.A. Conformational studies of oligosaccharides and glycopeptides: Complementarity of NMR, X-ray crystallography, and molecular modelling. Chem. Rev. 2002, 102, 371–386. [Google Scholar] [CrossRef]
- Kamiya, Y.; Yagi-Utsumi, M.; Yagi, H.; Kato, K. Structural and molecular basis of carbohydrate-protein interaction systems as potential therapeutic targets. Curr. Pharm. Des. 2011, 17, 1672–1684. [Google Scholar] [CrossRef]
- Kato, K.; Yamaguchi, Y. Glycoproteins and Antibodies: Solution NMR Studies. In Encyclopedia of Magnetic Resonance; Harris, R.K., Wasylishen, R.K., Eds.; John Wiley: Chichester, UK, 2011; pp. 1–12, doi:10.1002/9780470034590.emrstm1233. [Google Scholar]
- Kato, K.; Sasakawa, H.; Kamiya, Y.; Utsumi, M.; Nakano, M.; Takahashi, N.; Yamaguchi, Y. 920 MHz ultra-high field NMR approaches to structural glycobiology. Biochim. Biophys. Acta 2008, 1780, 619–625. [Google Scholar] [CrossRef]
- Homans, S.W. Conformation and dynamics of oligosaccharides in solution. Glycobiology 1993, 3, 551–555. [Google Scholar] [CrossRef]
- Roldos, V.; Canada, E.J.; Jiménez-Barbero, J. Carbohydrate-protein interactions: A 3D view by NMR. Chembiochem 2011, 12, 990–1005. [Google Scholar] [CrossRef]
- Yamamoto, S.; Yamaguchi, T.; Erdélyi, M.; Griesinger, C.; Kato, K. Paramagnetic lanthanide tagging for NMR conformational analyses of N-Linked oligosaccharides. Chem. Eur. J. 2011, 17, 9280–9282. [Google Scholar] [CrossRef]
- Erdélyi, M.; d’Auvergne, E.; Navarro-Vázquez, A.; Leonov, A.; Griesinger, C. Dynamics of the glycosidic bond: Conformational space of lactose. Chem. Eur. J. 2011, 17, 9368–9376. [Google Scholar]
- DeMarco, M.L.; Woods, R.J.; Prestegard, J.H.; Tian, F. Presentation of membrane-anchored glycosphingolipids determined from molecular dynamics simulations and NMR paramagnetic relaxation rate enhancement. J. Am. Chem. Soc. 2010, 132, 1334–1338. [Google Scholar] [CrossRef]
- Yamamoto, S.; Zhang, Y.; Yamaguchi, T.; Kameda, T.; Kato, K. Lanthanide-assisted NMR evaluation of a dynamic ensemble of oligosaccharide conformations. Chem. Commun. 2012, 48, 4752–4754. [Google Scholar]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; Gonzalez-Outeirino, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates.J. Comput. Chem. 2008, 29, 622–655. [Google Scholar]
- Li, Y.T.; LI, S.C.; Hasegawa, A.; Ishida, H.; Kiso, M.; Bernardi, A.; Brocca, P.; Raimondi, L.; Sonnino, S. Structural basis for the resistance of Tay-Sachs ganglioside GM2 to enzymatic degradation. J. Biol. Chem. 1999, 274, 10014–10018. [Google Scholar]
- Veluraja, K.; Rao, V.S.R. Theoretical studies on the conformation of monosialogangliosides and disialogangliosides. Carbohydr. Polym. 1983, 3, 175–192. [Google Scholar] [CrossRef]
- Sabesan, S.; Bock, K.; Lemieux, U. The conformational properties of the ganglioside GM2 and GM1 based on 1H and 13C nuclear magnetic resonance studies. Can. J. Chem. 1984, 62, 1034–1045. [Google Scholar] [CrossRef]
- Brocca, P.; Bernardi, A.; Raimondi, L.; Sonnino, S. Modeling ganglioside headgroups by conformational analysis and molecular dynamics. Glycoconjugate J. 2000, 17, 283–299. [Google Scholar] [CrossRef]
- Veluraja, K.; Selvin, J.F.A.; Venkateshwari, S.; Priyadarzini, T.R.K. 3DSDSCAR-a three dimensional structural database for sialic acid-containing carbohydrates through molecular dynamics simulation. Carbohydr. Res. 2010, 345, 2030–2037. [Google Scholar] [CrossRef]
- Levery, S.B. 1H-NMR study of GM2 ganglioside: Evidence that an interresidue amide-carboxyl hydrogen bond contributes to stabilization of a preferred conformation. Glycoconjugate J. 1991, 8, 484–92. [Google Scholar] [CrossRef]
- Koerner, T.A.W., Jr.; Prestegard, J.H.; Demou, P.C.; Yu, R.K. High-resolution proton NMR studies of gangliosides. 1. Use of homonuclear two-dimensional spin-echo J-correlated spectroscopy for determination of residue composition and anomeric configurations. Biochemistry 1983, 22, 2676–2687. [Google Scholar]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. Amber11; University of California, San Francisco, CA, USA, 2010. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.V.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3691. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar]
- Goddard, T.D.; Kneller, D.G. SPARKY 3; University of California, San Francisco, CA, USA, 2008. [Google Scholar]
- Sánchez-Pedregal, V.M.; Santamaría-Fernández, R.; Navarro-Vázquez, A. Residual dipolar couplings of freely rotating groups in small molecules. stereochemical assignment and side-chain conformation of 8-phenylmenthol. Org. Lett. 2009, 11, 1471–1474. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Y.; Yamamoto, S.; Yamaguchi, T.; Kato, K. Application of Paramagnetic NMR-Validated Molecular Dynamics Simulation to the Analysis of a Conformational Ensemble of a Branched Oligosaccharide. Molecules 2012, 17, 6658-6671. https://doi.org/10.3390/molecules17066658
Zhang Y, Yamamoto S, Yamaguchi T, Kato K. Application of Paramagnetic NMR-Validated Molecular Dynamics Simulation to the Analysis of a Conformational Ensemble of a Branched Oligosaccharide. Molecules. 2012; 17(6):6658-6671. https://doi.org/10.3390/molecules17066658
Chicago/Turabian StyleZhang, Ying, Sayoko Yamamoto, Takumi Yamaguchi, and Koichi Kato. 2012. "Application of Paramagnetic NMR-Validated Molecular Dynamics Simulation to the Analysis of a Conformational Ensemble of a Branched Oligosaccharide" Molecules 17, no. 6: 6658-6671. https://doi.org/10.3390/molecules17066658