Simultaneous Quantification of Five Bioactive Components of Acanthopanax senticosus and Its Extract by Ultra Performance Liquid Chromatography with Electrospray Ionization Time-of-Flight Mass Spectrometry

Abstract
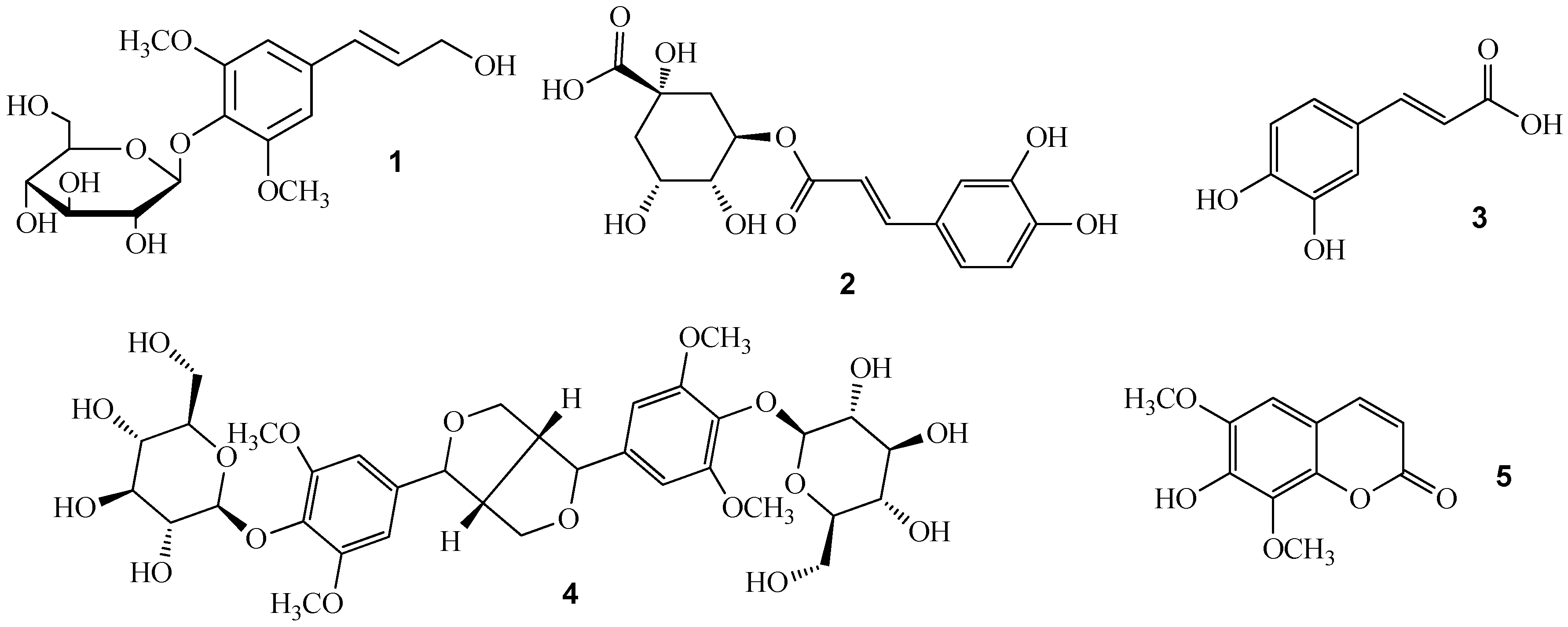
:1. Introduction

2. Results and Discussion
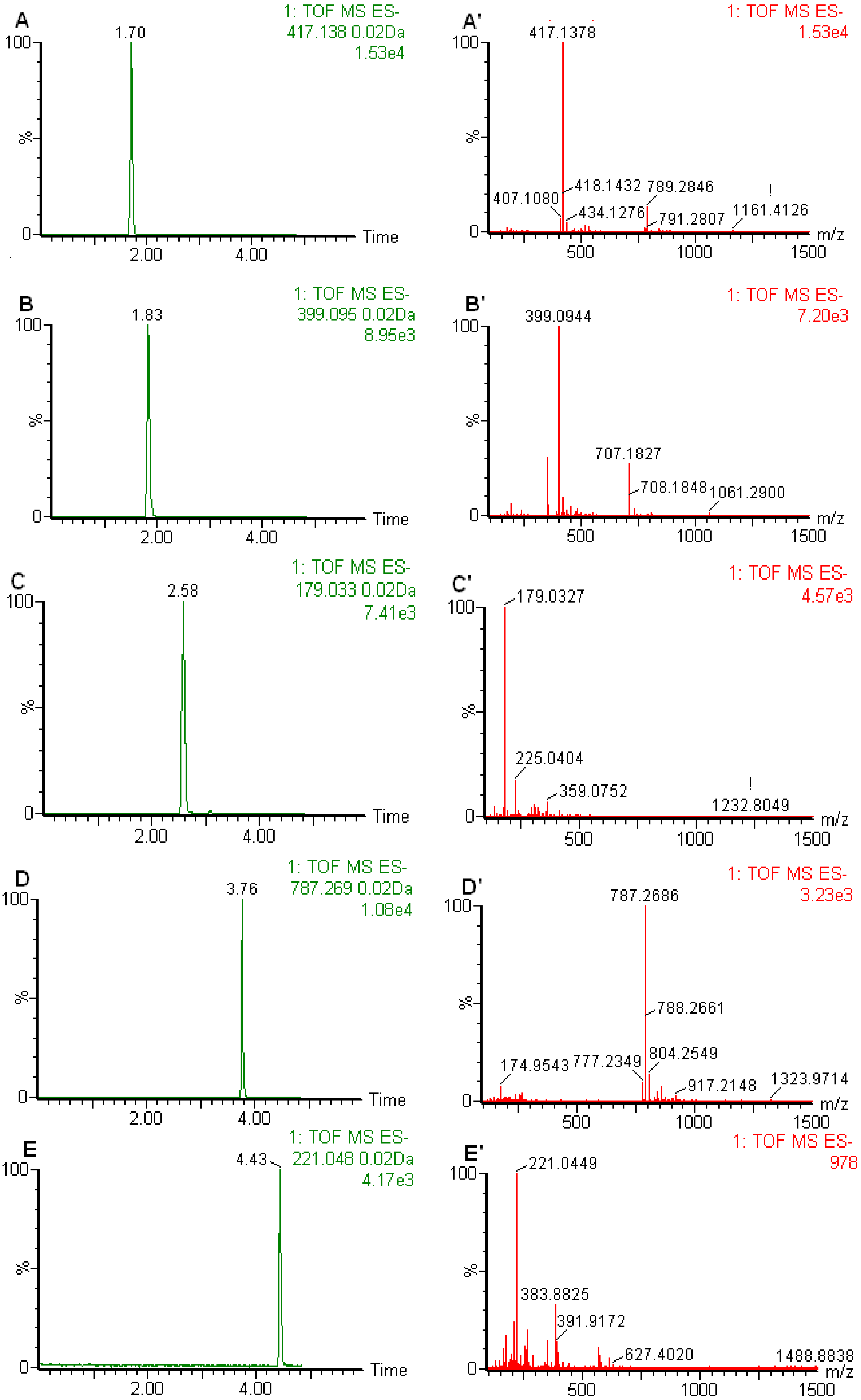
2.1. Optimization of the Chromatographic Conditions


2.2. Calibration curves, Limits of Detection and Quantification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Regression equation | Linear range (μg/mL) | r | LODs (μg/mL) | LOQs (μg/mL) |
|---|---|---|---|---|---|
| 1 | y = 24.49x + 91.44 | 0.2–1000 | 0.9994 | 0.008 | 0.03 |
| 2 | y = 14.70x + 75.46 | 0.2–1000 | 0.9992 | 0.02 | 0.06 |
| 3 | y = 14.86x + 78.13 | 1.0–1000 | 0.9991 | 0.04 | 0.12 |
| 4 | y = 11.15x + 49.04 | 0.2–1000 | 0.9994 | 0.008 | 0.03 |
| 5 | y = 5.49x + 29.14 | 1.0–1000 | 0.9991 | 0.20 | 0.60 |
2.3. Method Validation
| No. | Original (mg) | Spiked (mg) | Found (mg) | Mean recovery (%) | RSD (%) (n = 3) |
|---|---|---|---|---|---|
| 1 | 1.022 | 0.533 | 1.545 | 98.13 | 1.52 |
| 1.066 | 2.103 | 101.41 | 1.78 | ||
| 1.599 | 2.563 | 96.37 | 2.06 | ||
| 2 | 0.638 | 0.262 | 0.899 | 99.62 | 1.05 |
| 0.524 | 1.176 | 102.67 | 2.14 | ||
| 0.786 | 1.405 | 97.58 | 1.25 | ||
| 3 | 0.044 | 0.033 | 0.078 | 103.03 | 0.96 |
| 0.066 | 0.109 | 98.48 | 1.49 | ||
| 0.099 | 0.145 | 102.02 | 2.82 | ||
| 4 | 1.172 | 0.608 | 1.773 | 98.85 | 1.76 |
| 1.216 | 2.380 | 99.34 | 1.03 | ||
| 1.824 | 2.994 | 99.89 | 2.89 | ||
| 5 | 0.050 | 0.027 | 0.076 | 96.30 | 0.89 |
| 0.054 | 0.106 | 103.70 | 1.81 | ||
| 0.081 | 0.131 | 98.77 | 1.67 |
2.4. Matrix Effects
2.5. Sample Analysis
| No. | 1 (syringin) | 2 (chlorogenic acid) | 3 (caffeic acid) | 4 (eleutheroside E) | 5 (isofraxidin) |
|---|---|---|---|---|---|
| 1 | 12.857 | 10.131 | Trace | 21.004 | 1.150 |
| 2 | 11.326 | 11.844 | Trace | 14.530 | 1.446 |
| 3 | 13.551 | 12.146 | Trace | 16.208 | 1.586 |
| 4 | 4.364 | 12.057 | Trace | 13.853 | 0.841 |
| 5 | 4.919 | 13.721 | Trace | 15.582 | 1.036 |
| 6 | 9.235 | 17.638 | Trace | 23.617 | 1.967 |
| 7 | 0.906 | 3.508 | Trace | 10.108 | 1.967 |
| 8 | 5.242 | 4.708 | 0.308 | 6.273 | 0.225 |
| 9 | 5.109 | 3.191 | 0.219 | 5.861 | 0.248 |
| 10 | 0.855 | 2.039 | Trace | 1.453 | 0.103 |
| 11 | 5.492 | 2.035 | 0.262 | 6.350 | 0.270 |
| 12 | 0.894 | 2.637 | Trace | 1.603 | 0.107 |
| 13 | 0.067 | 0.369 | Trace | 0.424 | Trace |
| 14 | 0.580 | 1.240 | 0.041 | 0.620 | 0.019 |
| 15 | 0.285 | 0.648 | 0.035 | 0.192 | 0.026 |
2.6. Quality Assessment of A. senticosus Samples by Principal Component Analysis

3. Experimental
3.1. Chemicals and Reagents
| Samples | Plant origins | Collection time | Factories | Properties |
|---|---|---|---|---|
| 1–3 | Wuchang, Heilongjiang | March 2011 | Renhuang | Extract |
| 4–7 | Wuchang, Heilongjiang | January 2011 | Wusulijiang | Extract |
| 8–9 | Shangzhi, Heilongjiang | December 2010 | Wusulijiang | Extract |
| 10 | Harbin, Heilongjiang | April 2011 | Wusulijiang | Extract |
| 11 | Shangzhi, Heilongjiang | March 2011 | Wusulijiang | eExtract |
| 12 | Harbin, Heilongjiang | May 2011 | Wusulijiang | Extract |
| 13 | Shangzhi, Heilongjiang | November 2010 | - | Medical materials |
| 14 | Wuchang, Heilongjiang | September 2010 | - | Medical materials |
| 15 | Anhui | February 2011 | - | Medical materials |
3.2. Sample Preparation of Acanthopanax Extract
3.3. Analytical Sample Preparation
3.4. UPLC-MS Conditions
3.5. Standard Preparation and Calibration Curves
4. Conclusions
Acknowledgments
References
- Missouri Botanical Garden. Tropicos. Available online: http://www.tropicos.org/NameSearch.aspx (accessed on 13 May 2012).
- National Committee of Chinese Pharmacopoeia, The Pharmacopoeia of the People’s Republic of China; Chemical Industry Press: Beijing, China, 2010.
- Lee, S.; Shen, D.S.; Oh, K.B.; Shin, K.H. Antibacterial compounds from the leaves of Acanthopanax senticosus. Arch. Pharm. Res. 2006, 26, 40–42. [Google Scholar]
- Zhang, X.L.; Ren, F.; Huang, W.; Ding, R.T.; Zhou, Q.S.; Liu, X.W. Anti-fatigue activity of extracts of stem bark from Acanthopanax senticosus. Molecules 2010, 16, 28–37. [Google Scholar] [CrossRef]
- Wang, X.; Hai, C.X.; Liang, X.; Yu, S.X.; Zhang, W.; Li, Y.L. The protective effects of Acanthopanax senticosus Harms aqueous extracts against oxidative stress: Role of Nrf2 and antioxidant enzymes. J. Ethnopharmacol. 2010, 127, 424–432. [Google Scholar] [CrossRef]
- Park, S.H.; Nhiem, N.X.; Kiem, P.V.; Choi, E.M.; Kim, J.A.; Kim, Y.H. A new norlupane triterpene from the leaves of Acanthopanax koreanum increases the differentiation of osteoblastic MC3T3-e1 cells. Arch. Pharm. Res. 2010, 33, 75–80. [Google Scholar] [CrossRef]
- Li, Q.; Jia, Y.; Xu, L.; Wang, X.; Shen, Z.; Liu, Y.; Bi, K. Simultaneous determination of protocatechuic acid, syringin, chlorogenic acid, caffeic acid, liriodendrin and isofraxidin in Acanthopanax senticosus Harms by HPLC-DAD. Biol. Pharm. Bull. 2006, 29, 532–534. [Google Scholar] [CrossRef]
- Lanza, A.; Martinez, M; Matellano, L.; Carretero, C.; Castillo, L.; Sen, A.; Benito, P. Lignan and phenylpropanoid glycosides from Phillyrea latifolia and their in vitro anti-inflammatory activity. Planta Med. 2001, 67, 219–223. [Google Scholar] [CrossRef]
- Tohda, C.; Ichimura, M.; Bai, Y.; Tanaka, K.; Zhu, S.; Komatsu, K. Inhibitory effects of Eleutherococcus senticosus extracts onamyloid beta (25–35)-induced neuritic atrophy and synaptic loss. J. Pharmacol. Sci. 2008, 107, 329–339. [Google Scholar] [CrossRef]
- Sun, H.; Lv, H.; Zhang, Y.; Wang, X.; Bi, K.; Cao, H. A rapid and sensitive UPLC-ESIMS method for analysis of isofraxidin, a natural antistress compound, and its metabolites in rat plasma. J. Sep. Sci. 2007, 30, 3202–3206. [Google Scholar] [CrossRef]
- Meng, L.; Lozano, Y.F.; Gaydou, E.M.; Li, B. Antioxidant activities of polyphenols extracted from Perilla frutescens Varieties. Molecules 2009, 14, 133–140. [Google Scholar]
- Sample Availability: Samples are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, S.-P.; An, J.-T.; Wang, R.; Li, Q. Simultaneous Quantification of Five Bioactive Components of Acanthopanax senticosus and Its Extract by Ultra Performance Liquid Chromatography with Electrospray Ionization Time-of-Flight Mass Spectrometry. Molecules 2012, 17, 7903-7913. https://doi.org/10.3390/molecules17077903
Liu S-P, An J-T, Wang R, Li Q. Simultaneous Quantification of Five Bioactive Components of Acanthopanax senticosus and Its Extract by Ultra Performance Liquid Chromatography with Electrospray Ionization Time-of-Flight Mass Spectrometry. Molecules. 2012; 17(7):7903-7913. https://doi.org/10.3390/molecules17077903
Chicago/Turabian StyleLiu, Shi-Ping, Jing-Tao An, Rui Wang, and Qiang Li. 2012. "Simultaneous Quantification of Five Bioactive Components of Acanthopanax senticosus and Its Extract by Ultra Performance Liquid Chromatography with Electrospray Ionization Time-of-Flight Mass Spectrometry" Molecules 17, no. 7: 7903-7913. https://doi.org/10.3390/molecules17077903