Chiral Zn(II)-Bisamidine Complex as a Lewis-Brønsted Combined Acid Catalyst: Application to Asymmetric Mukaiyama Aldol Reactions of α-Ketoesters

Abstract
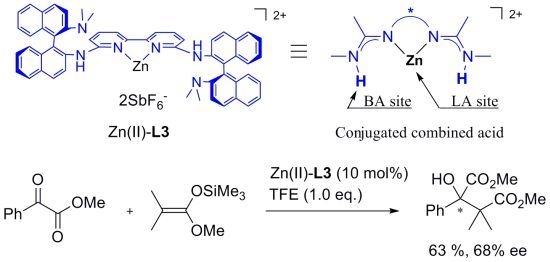
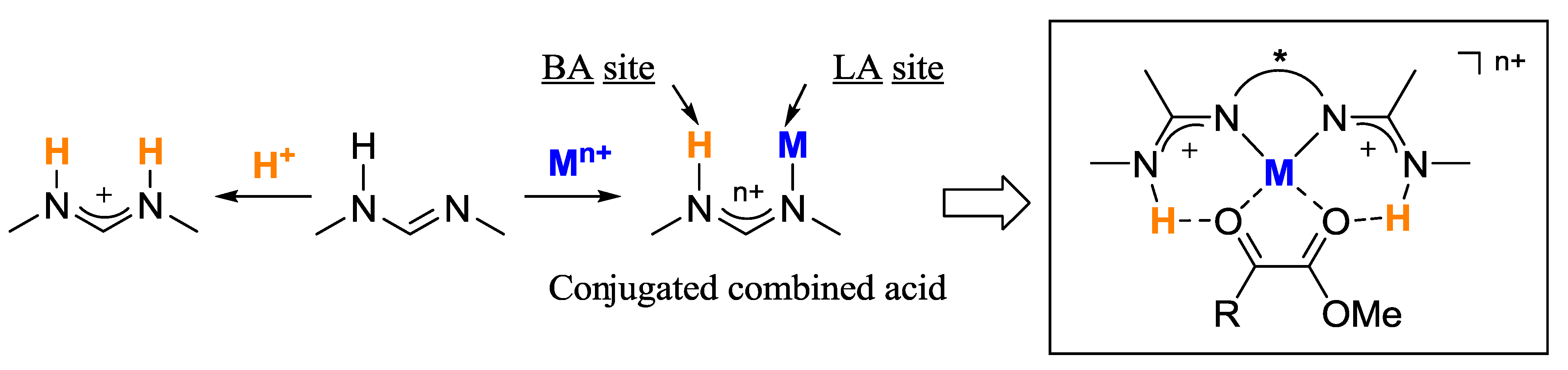
:
1. Introduction

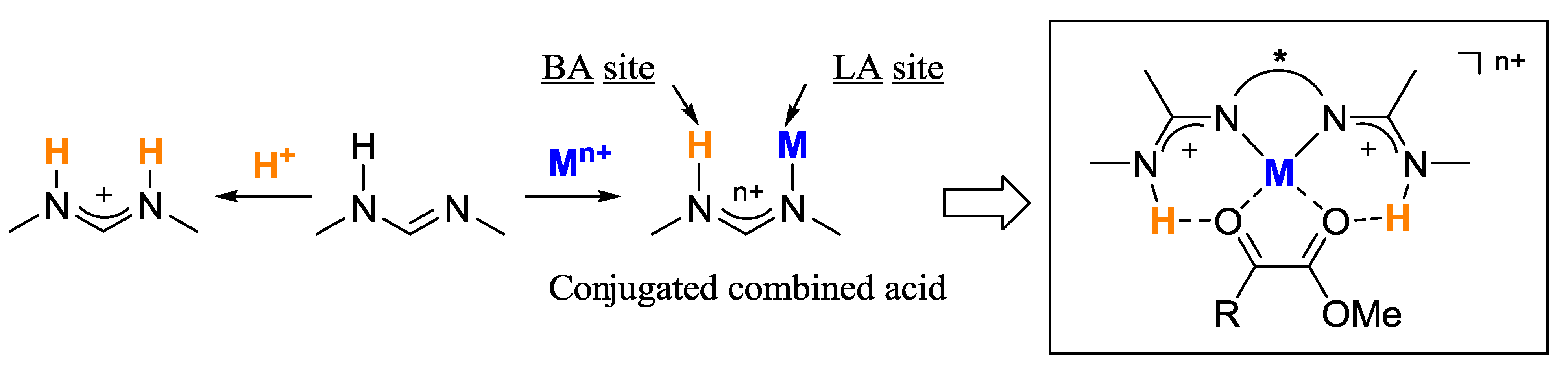
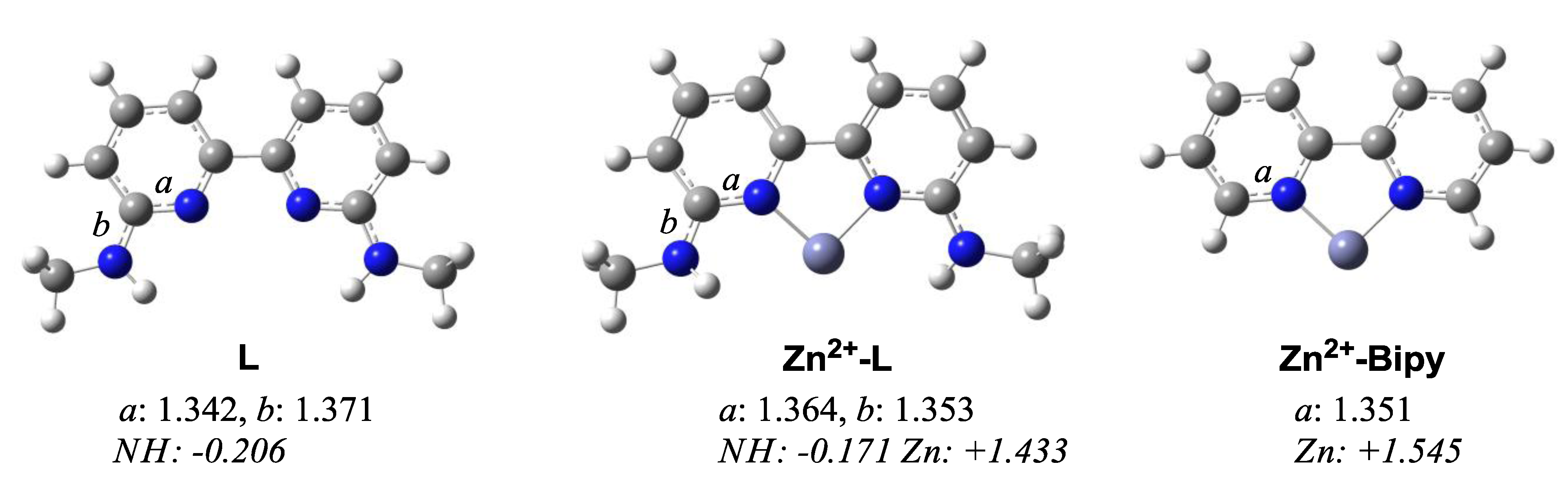
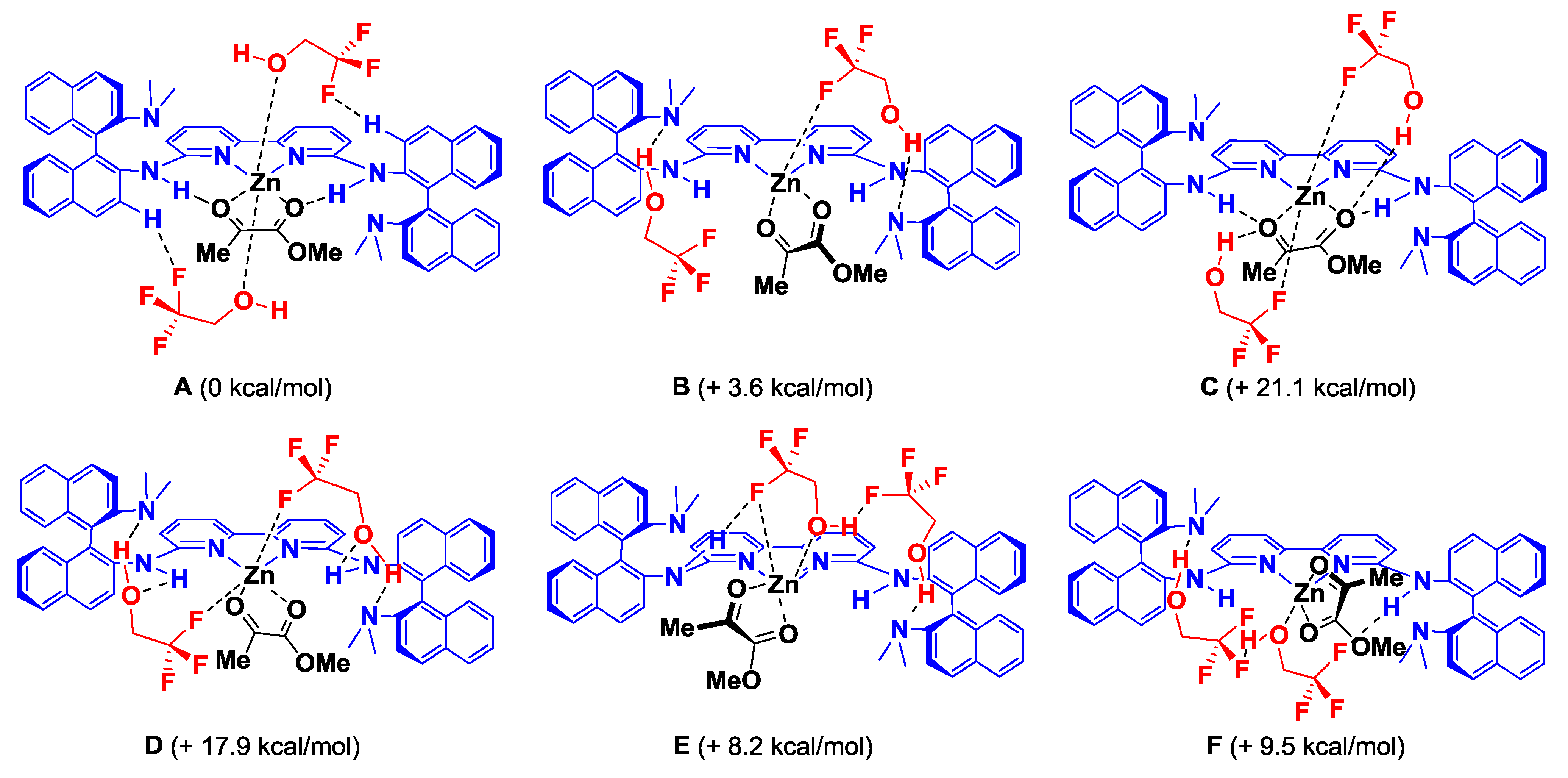
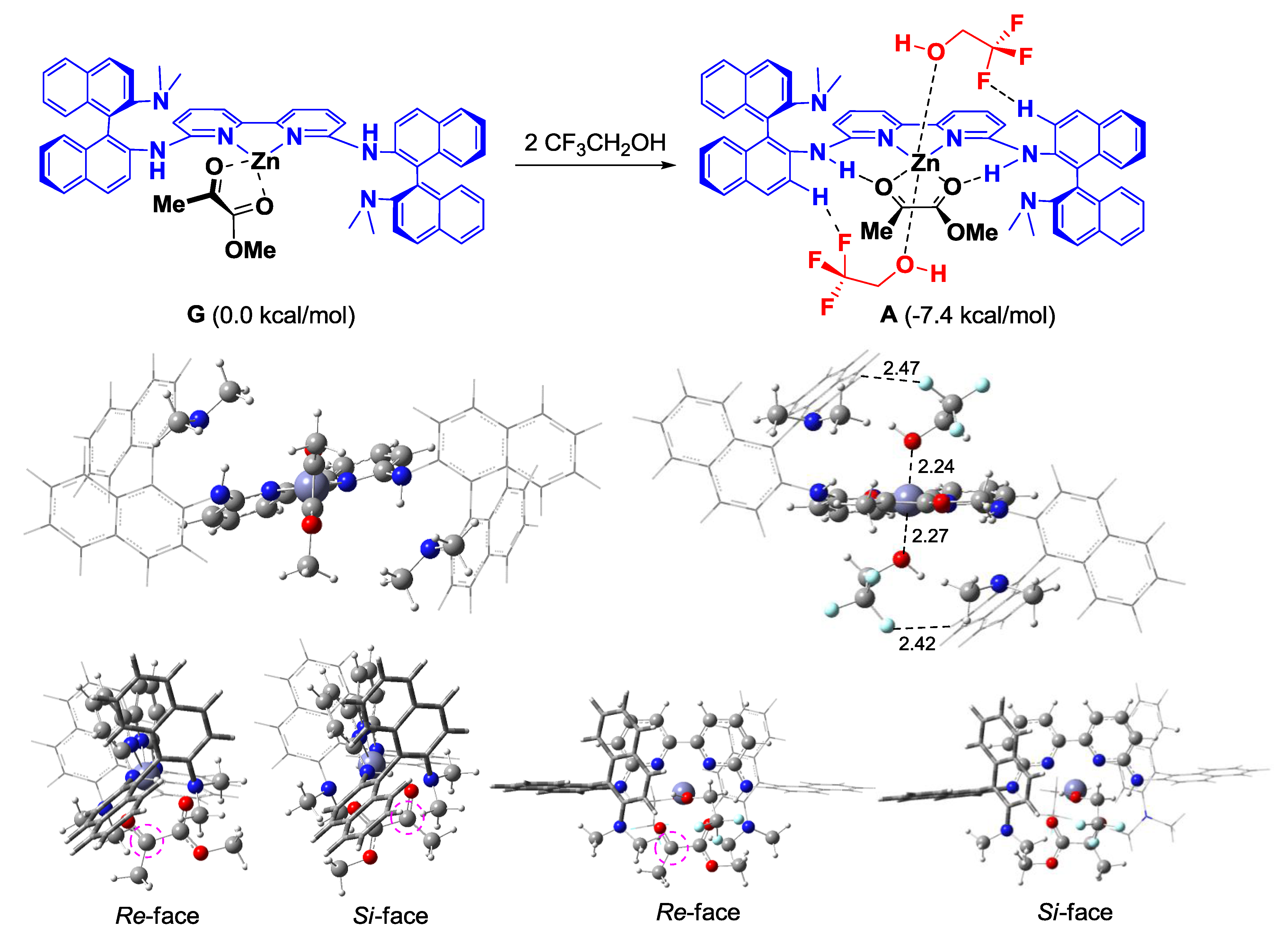
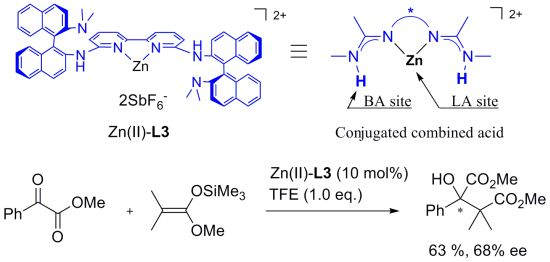
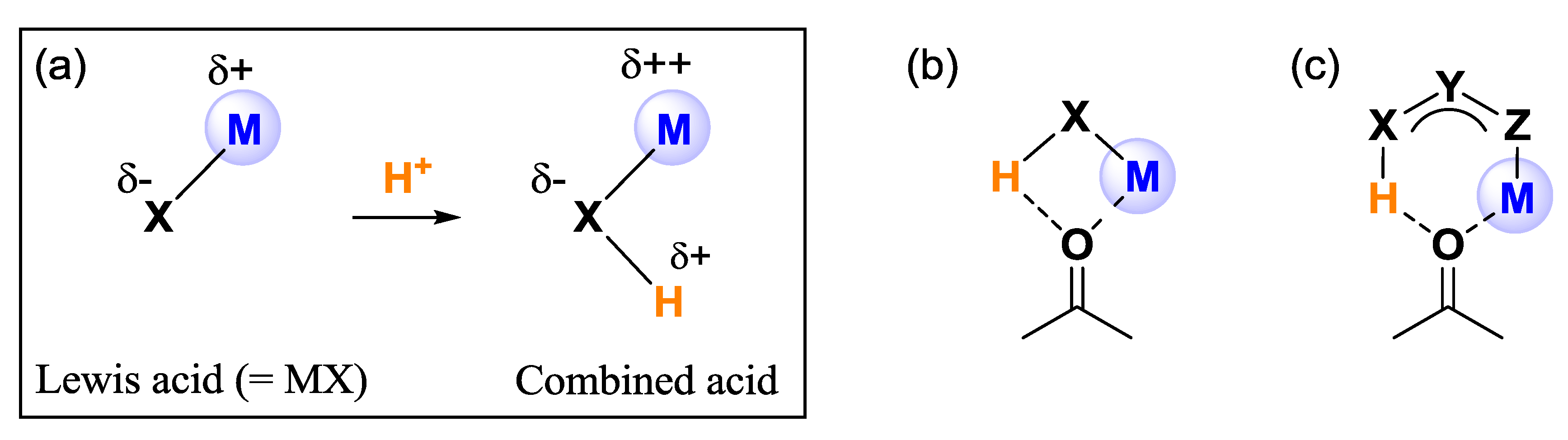
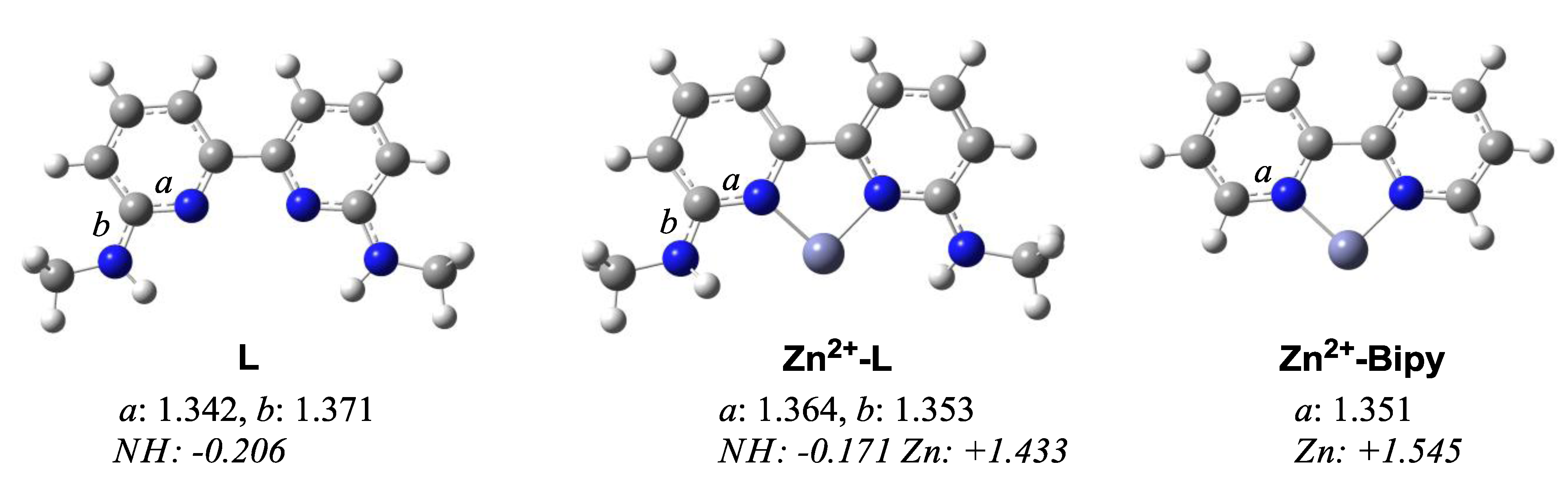
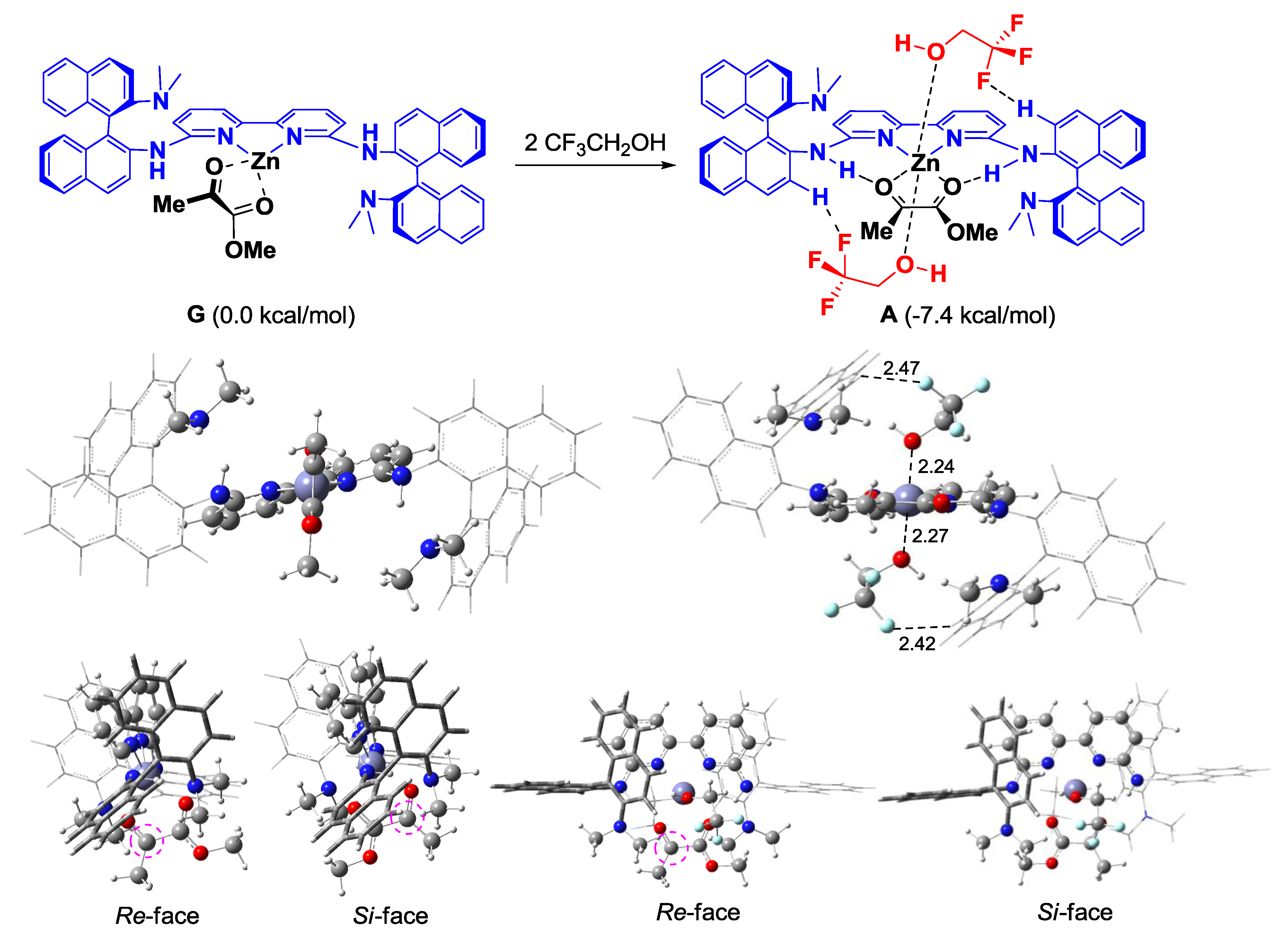
2. Results and Discussion

| Entry | MX2 | Ligand | Yield (%) a | Ligand | Yield (%) a |
|---|---|---|---|---|---|
| 1 | CuCl2 | L1 | 28 | L2 | 13 |
| 2 | ZnCl2 | L1 | 51 (89) b | L2 | 30 |
| 3 | FeCl2 | L1 | 48 | L2 | 15 |

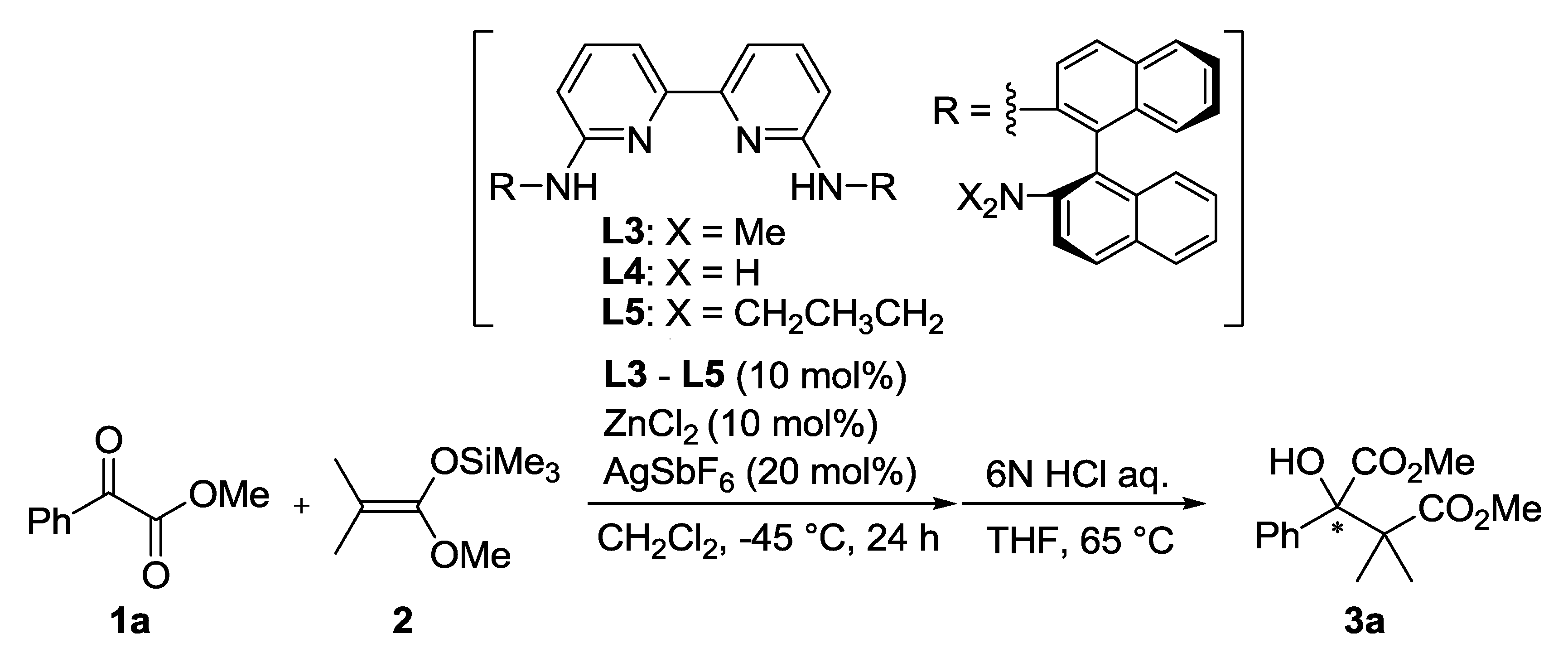
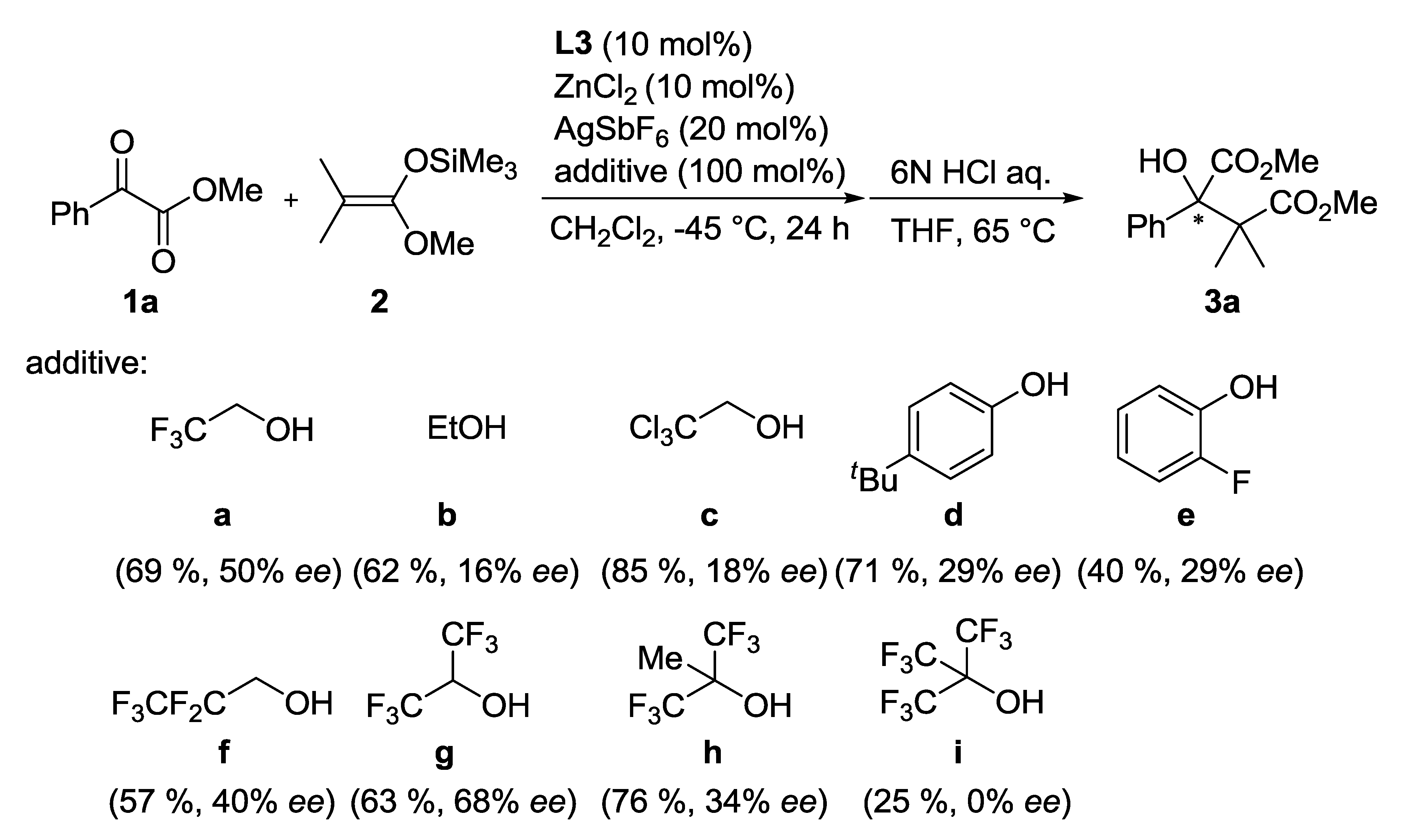
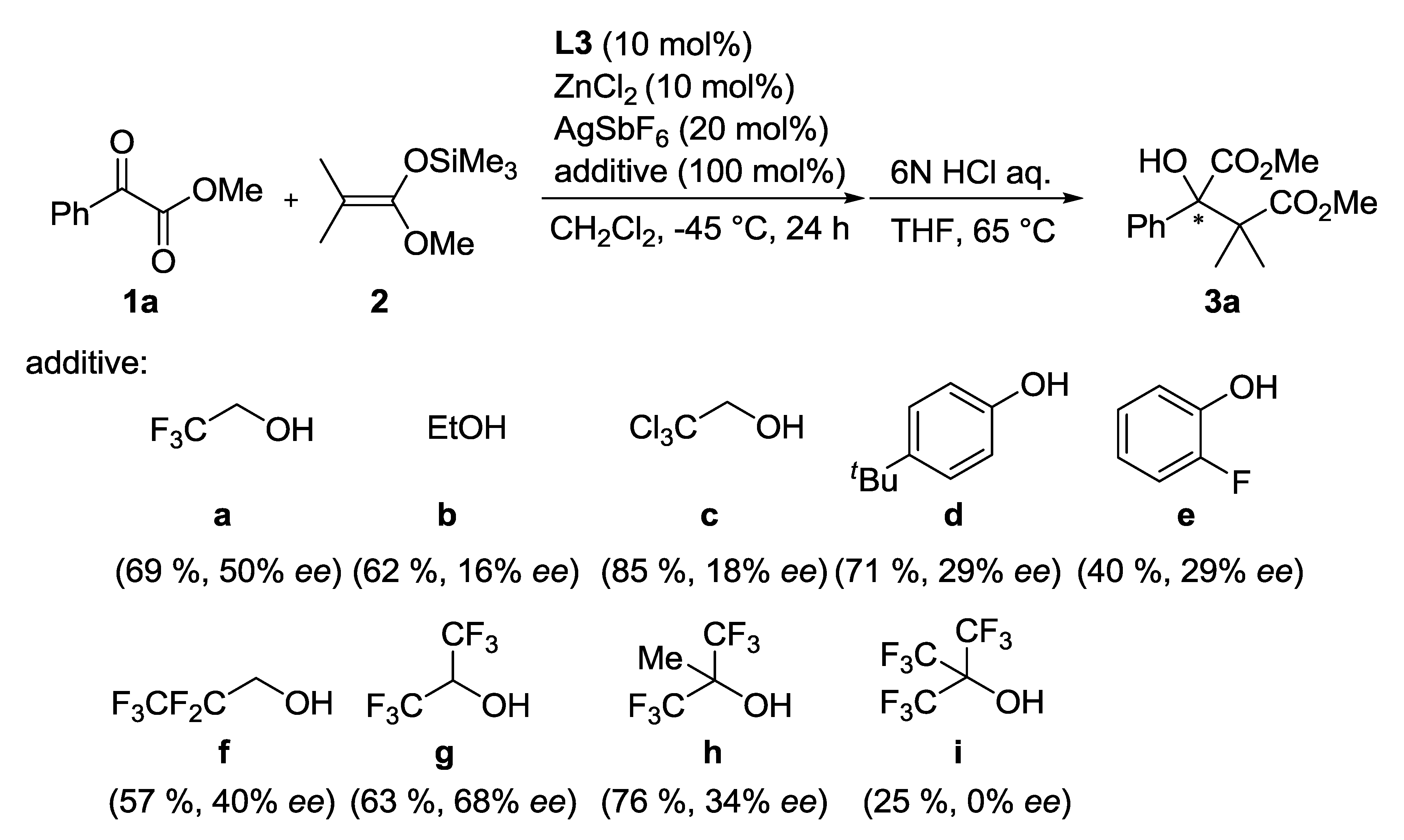
| Entry | Ligand | Y eq. of TFE | Yield (%) a | ee (%) b |
|---|---|---|---|---|
| 1 | L3 | - | 60 | 26 |
| 2 | L4 | - | 81 | 1 |
| 3 | L5 | - | 53 | 0 |
| 4 | L3 | 0.2 | 71 | 35 |
| 5 | L3 | 1.0 | 69 | 50 |
| 6 | L3 | 5.0 | 68 | 33 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
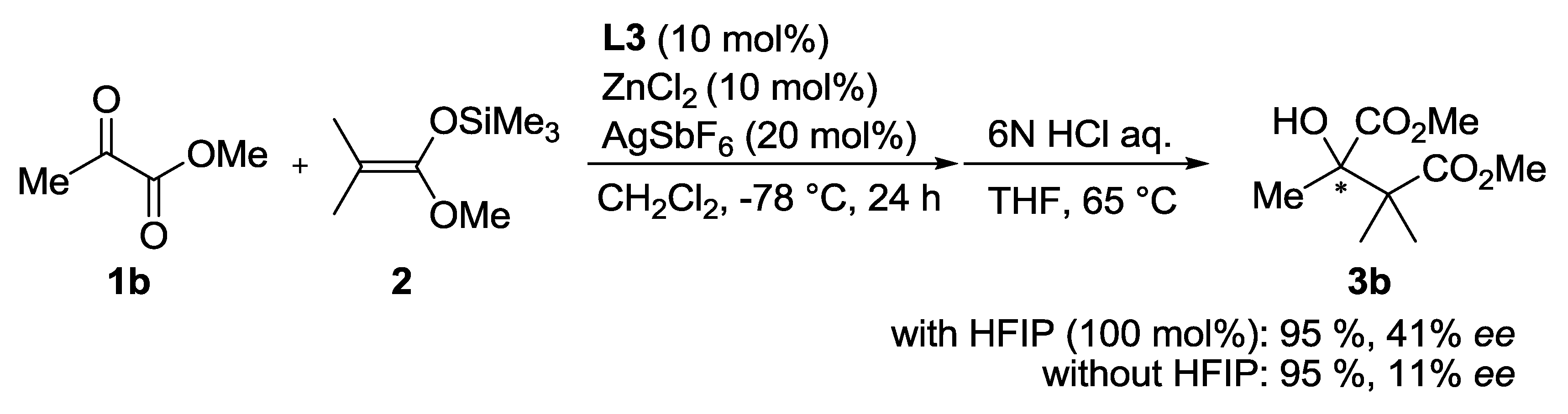{kind=link}
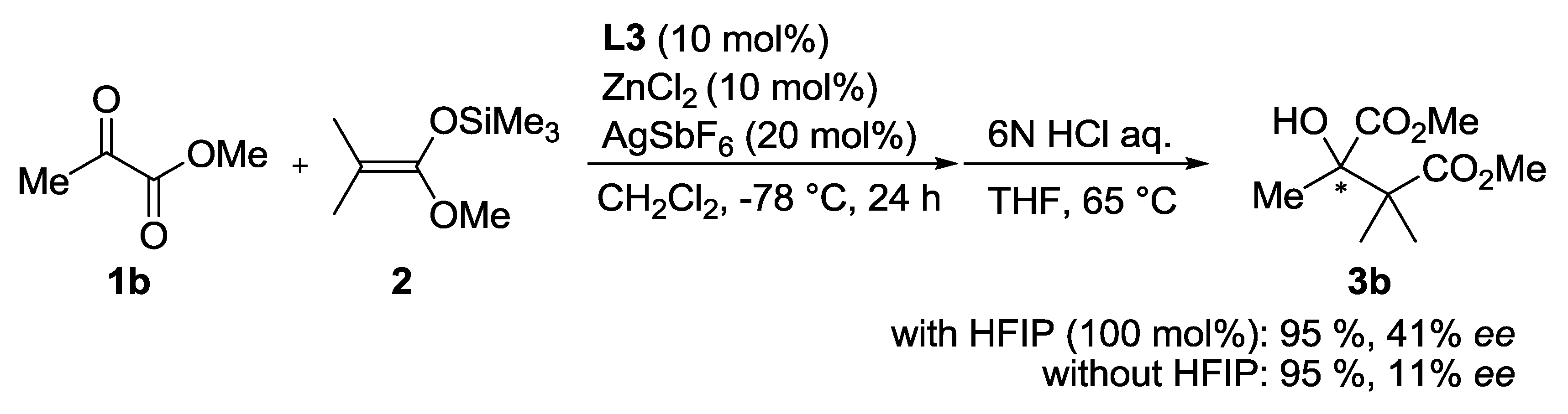
3. Experimental
3.1. General
3.2. Synthesis of the 2,2'-Bipyridyl Derived Bisamidine Ligands
3.2.1. Synthesis of 6,6'-Dibromo-2,2'-dipyridyl
3.2.2. Synthesis of the (R)-DABN Derivatives
3.2.3. Synthesis of L1, L3, L4, and L5
3.3. General Procedure for Asymmetric Mukaiyama Aldol Reaction of 1a and 2 (Scheme 1)
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Yamamoto, H.; Futatsugi, K. Designer acids”: Combined acid catalysis for asymmetric synthesis. Angew. Chem. Int. Ed. Engl. 2005, 44, 1924–1942. [Google Scholar] [CrossRef]
- Maruoka, K.; Sakurai, M.; Fujiwara, J.; Yamamoto, H. Asymmetric Diels-Alder reaction directed toward chiral anthracycline intermediates. Tetrahedron 1986, 27, 4895–4898. [Google Scholar] [CrossRef]
- Furuta, K.; Miwa, Y.; Iwanaga, K.; Yamamoto, H. Acyloxyborane: An activating device for carboxylic acids. J. Am. Chem. Soc. 1988, 110, 6254–6255. [Google Scholar] [CrossRef]
- Ishihara, K.; Miyata, M.; Hattori, K.; Tada, T.; Yamamoto, H.Y. A new chiral BLA promoter for asymmetric aza Diels-Alder and Aldol-type reactions of imines. J. Am. Chem. Soc. 1994, 116, 10520–10524. [Google Scholar]
- Ishihara, K.; Yamamoto, H. Brønsted acid assisted chiral Lewis acid (BLA) catalyst for asymmetric Diels-Alder reaction. J. Am. Chem. Soc. 1994, 116, 1561–1562. [Google Scholar] [CrossRef]
- Ishihara, K.; Kurihara, H.; Yamamoto, H. A new powerful and practical BLA catalyst for highly enantioselective Diels-Alder reaction: An extreme acceleration of reaction rate by Brønsted acid. J. Am. Chem. Soc. 1996, 118, 3049–3050. [Google Scholar] [CrossRef]
- Ishihara, K.; Kurihara, H.; Matsumoto, M.; Yamamoto, H. Design of Brønsted acid-assisted chiral Lewis acid (BLA) catalysts for highly enantioselective Diels-Alder reactions. J. Am. Chem. Soc. 1998, 120, 6920–6930. [Google Scholar] [CrossRef]
- Corey, E.J.; Shibata, T.; Lee, T.W. Asymmetric Diels-Alder reactions catalyzed by a triflic acid activated chiral oxazaborolidine. J. Am. Chem. Soc. 2002, 124, 3808–3809. [Google Scholar] [CrossRef]
- Ryu, D.H.; Lee, T.W.; Corey, E.J. Broad-spectrum enantioselective Diels-Alder catalysis by chiral, cationic oxazaborolidines. J. Am. Chem. Soc. 2002, 124, 9992–9993. [Google Scholar] [CrossRef]
- Ryu, D.H.; Corey, E.J. Triflimide activation of a chiral oxazaborolidine leads to a more general catalytic system for enantioselective Diels-Alder addition. J. Am. Chem. Soc. 2003, 125, 6388–6390. [Google Scholar] [CrossRef]
- Payette, J.N.; Yamamoto, H. Regioselective and asymmetric Diels-Alder reaction of 1- and 2-substituted cyclopentadienes catalyzed by a Brønsted acid activated chiral oxazaborolidine. J. Am. Chem. Soc. 2007, 129, 9536–9537. [Google Scholar] [CrossRef]
- Hu, G.; Huang, L.; Huang, R.H.; Wulff, W.D. Evidence for a boroxinate based Brønsted acid derivative of VAPOL as the active catalyst in the catalytic asymmetric aziridination reaction. J. Am. Chem. Soc. 2009, 131, 15615–15617. [Google Scholar]
- Rauniyar, V.; Hall, D.G. Rationally improved chiral Brønsted acid for catalytic enantioselective allylboration of aldehydes with an expanded reagent scope. J. Org. Chem. 2009, 74, 4236–4241. [Google Scholar] [CrossRef]
- Lv, J.; Li, X.; Zhong, L.; Luo, S.; Cheng, J.-P. Asymmetric binary-acid catalysis with chiral phosphoric acid and MgF2: catalytic enantioselective Friedel-Crafts reactions of β,γ-unsaturated α-ketoesters. Org. Lett. 2010, 12, 1096–1099. [Google Scholar]
- Nugent, B.M.; Yoder, R.A.; Johnston, J.N. Chiral proton catalysis: A catalytic enantioselective direct Aza-Henry reaction. J. Am. Chem. Soc. 2004, 126, 3418–3419. [Google Scholar] [CrossRef]
- Singh, A.; Yoder, R.A.; Shen, B.; Johnston, J.N. Chiral proton catalysis: Enantioselective Brønsted acid catalyzed additions of nitroacetic acid derivatives as glycine equivalents. J. Am. Chem. Soc. 2007, 129, 3466–3467. [Google Scholar]
- Davis, T.A.; Wilt, J.C.; Johnston, J.N. Bifunctional asymmetric catalysis: Amplification of Brønsted basicity can orthogonally increase the reactivity of a chiral Brønsted acid. J. Am. Chem. Soc. 2010, 132, 2880–2882. [Google Scholar] [CrossRef]
- Akalay, D.; Dürner, G.; Bats, J.W.; Bolte, M.; Göbel, M.W. Synthesis of C2-Symmetric Bisamidines: A New Type of Chiral Metal-Free Lewis Acid Analogue Interacting with Carbonyl Groups. J. Org. Chem. 2007, 72, 5618–5624. [Google Scholar] [CrossRef]
- Akalay, D.; Dürner, G.; Bats, J. W.; Göbel, M.W. C2-symmetric bisamidines: Chiral Brønsted bases catalysing the Diels-Alder reaction of anthrones. Beilstein J. Org. Chem. 2008, 4. [Google Scholar] [CrossRef]
- Miyata, K.; Kutsuna, H.; Kawakami, S.; Kitamura, M. A Chiral Bidentate sp2-N Ligand, Naph-diPIM: Application to CpRu-Catalyzed Asymmetric Dehydrative C-, N-, and O-Allylation. Angew. Chem. Int. Ed. Engl. 2011, 50, 4649–4653. [Google Scholar] [CrossRef]
- Catalytic enantioselective aldol additions to ketones, see: Adachi, S.; Harada, T. Catalytic Enantioselective Aldol additions to ketones. Eur. J. Org. Chem. 2009, 3661–3671. [Google Scholar] [CrossRef]
- Evans, D.A.; Kozlowski, M.C.; Burgey, C.S.; MacMillan, D.W.C. C2-symmetric copper(II) complexes as chiral Lewis acids. Catalytic enantioselective aldol additions of enolsilanes to pyruvate esters. J. Am. Chem. Soc. 1997, 119, 7893–7894. [Google Scholar] [CrossRef]
- Evans, D.A.; MacMillan, D.W.C.; Campos, K.R. C2-Symmetric tin(II) complexes as chiral Lewis acids. Catalytic enantioselective anti aldol additions of enolsilanes to glyoxylate and pyruvate esters. J. Am. Chem. Soc. 1997, 119, 10859–10860. [Google Scholar]
- Evans, D.A.; Burgey, C.S.; Kozlowski, M.C.; Tregay, S.W. C2-Symmetric copper(II) complexes as chiral Lewis acids. Scope and mechanism of the catalytic enantioselective aldol additions of enolsilanes to pyruvate esters. J. Am. Chem. Soc. 1999, 121, 686–699. [Google Scholar] [CrossRef]
- Langner, M.; Bolm, C. C1-Symmetric sulfoximines as ligands in copper-catalyzed asymmetric Mukaiyama-type aldol reactions. Angew. Chem. Int. Ed. Engl. 2004, 43, 5984–5987. [Google Scholar] [CrossRef]
- Sedelmeier, J.; Hammerer, T.; Bolm, C. C1-Symmetric oxazolinyl sulfoximines as ligands in copper-catalyzed asymmetric Mukaiyama aldol reactions. Org. Lett. 2008, 10, 917–920. [Google Scholar] [CrossRef]
- Akullian, L.C.; Snapper, M.L.; Hoveyda, A.H. A chiral Ag-based catalyst for practical, efficient, and highly enantioselective additions of enolsilanes to α-ketoesters. J. Am. Chem. Soc. 2006, 128, 6532–6533. [Google Scholar] [CrossRef]
- Engers, J.L.; Pagenkopf, B.L. A general asymmetric aldol reaction of silyl ketene acetals derived from simple esters to aryl α-keto esters. Eur. J. Org. Chem. 2009, 6109–6111. [Google Scholar]
- Ohkouchi, M.; Masui, D.; Yamaguchi, M.; Yamagishi, T. Efficient Mukaiyama aldol reaction by silver(I) carboxylate-bis(phosphine) catalysts. Nippon Kagaku Kaishi 2002, 223–229. [Google Scholar]
- Synthesis of α-hydroxyester having sequential quarternary carbons by asymmetric Mukaiyama aldol reaction of methyl pyruvate and cyclopropyl silylketenehemithioacetal, see: Siegel, D.S.; Piizzi, G.; Piersanti, G.; Movassaghi, M. Enantioselective total synthesis of (−)-Acylfulvene and (−)-Irofulven. J. Org. Chem. 2009, 74, 9292–9304. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Vogl, E.M.; Gröger, H.; Shibasaki, M. Towards perfect asymmetric catalysis: Additives and cocatalysts. Angew. Chem. Int. Ed. Engl. 1999, 38, 1570–1577. [Google Scholar] [CrossRef]
- Evans, D.A.; Rovis, T.; Kozlowski, M.C.; Tedrow, J.S. C2-Symmetric Cu(II) complexes as chiral Lewis acids. Catalytic enantioselective Michael addition of silylketene acetals to alkylidene malonates. J. Am. Chem. Soc. 1999, 121, 1994–1995. [Google Scholar] [CrossRef]
- Evans, D.A.; Willis, M.C.; Johnston, J.N. Catalytic enantioselective Michael additions to unsaturated ester derivatives using chiral copper(II) Lewis acid complexes. Org. Lett. 1999, 1, 865–868. [Google Scholar] [CrossRef]
- Shuklov, I.A.; Dubrovina, N.V.; Börner, A. Fluorinated alcohols as solvents, cosolvents and additives in homogeneous catalysis. Synthesis 2007, 2925–2943. [Google Scholar]
- Wang, J.; Li, H.; Duan, W.; Zu, L.; Wang, W. Organocatalytic asymmetric Michael addition of 2,4-pentandione to nitroolefins. Org. Lett. 2005, 21, 4713–4716. [Google Scholar]
- Wolfe, J.P.; Wagaw, S.; Buchwald, S.L. An Improved Catalyst System for Aromatic Carbon-Nitrogen Bond Formation: The Possible Involvement of Bis(Phosphine) Palladium Complexes as Key Intermediates. J. Am. Chem. Soc. 1996, 118, 7215–7216. [Google Scholar] [CrossRef]
- Doucet, H.; Parrain, J.-L.; Santelli, M. Ruthenium(II) and palladium(II) complexes mediated addition of ketene silyl ketal to aldehydes and ketones: Remarkable influence of the nature of the ligand. Synlett 2000, 871–873. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gotoh, R.; Yamanaka, M. Chiral Zn(II)-Bisamidine Complex as a Lewis-Brønsted Combined Acid Catalyst: Application to Asymmetric Mukaiyama Aldol Reactions of α-Ketoesters. Molecules 2012, 17, 9010-9022. https://doi.org/10.3390/molecules17089010
Gotoh R, Yamanaka M. Chiral Zn(II)-Bisamidine Complex as a Lewis-Brønsted Combined Acid Catalyst: Application to Asymmetric Mukaiyama Aldol Reactions of α-Ketoesters. Molecules. 2012; 17(8):9010-9022. https://doi.org/10.3390/molecules17089010
Chicago/Turabian StyleGotoh, Ryo, and Masahiro Yamanaka. 2012. "Chiral Zn(II)-Bisamidine Complex as a Lewis-Brønsted Combined Acid Catalyst: Application to Asymmetric Mukaiyama Aldol Reactions of α-Ketoesters" Molecules 17, no. 8: 9010-9022. https://doi.org/10.3390/molecules17089010