Diterpenoids from the Buds of Pinus banksiana Lamb.

Abstract
:1. Introduction
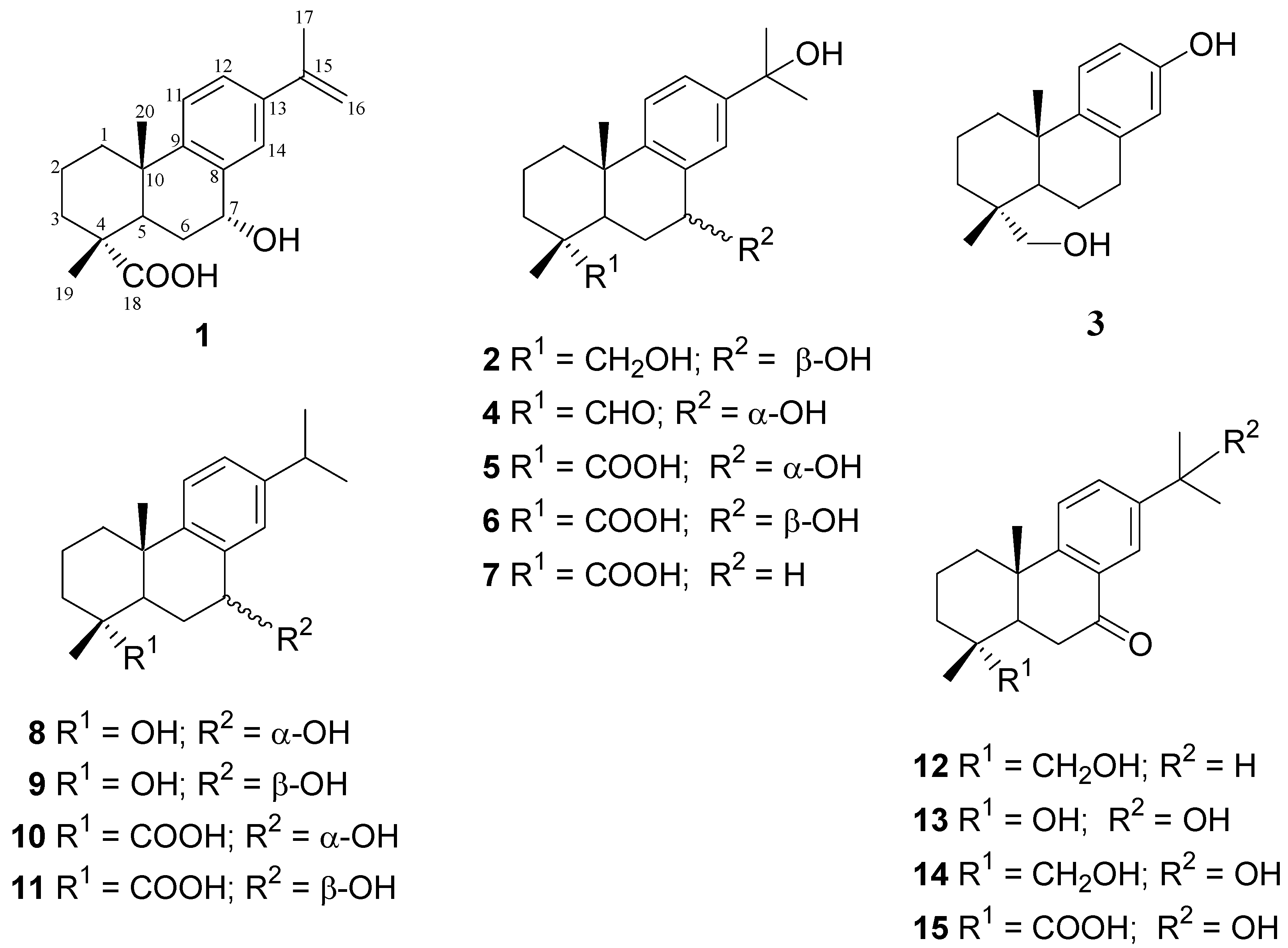
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (µg/mL) a,b | ||||
|---|---|---|---|---|---|
| A549 c | DLD-1 d | WS1 e | S. aureus f | E. colig | |
| Hexanes extract | 45 ± 4 | 44 ± 3 | 59 ± 6 | 29 ± 3 | >100 |
| CH2Cl2 extract | 26 ± 3 | 32 ± 3 | 42 ± 4 | 64 ± 5 | >100 |
| MeOH extract | >100 | >100 | >100 | >100 | >100 |
| Fraction F | 7 ± 2 | 12 ± 2 | 79 ± 3 | 51 ± 2 | >100 |
| Fraction G | 5 ± 1 | 14 ± 1 | 42 ± 3 | >100 | >100 |
| Fraction H | 6 ± 1 | 13 ± 1 | 39 ± 3 | >100 | >100 |
| Fraction K | 84 ± 1 | >100 | >100 | >100 | >100 |
| Etoposide h | 0.4 ± 0.2 | 1.7 ± 0.3 | 5.9 ± 0.6 | ||
| Chloramphenicol i | >1.6 | 0.12 ± 0.02 | |||
| Position | 1 a | 2 b | 3 a |
|---|---|---|---|
| 1 | 2.33 (m) | 2.27 (br d , 12.6) | 2.26 (m) |
| 1.51 (m) | 1.35 | 1.30 (m) | |
| 2 | 1.79 (m) | 1.81 | 1.79 (m) |
| 1.69 | 1.64 (m) | ||
| 3 | 1.80 (m) | 1.48 | 1.51 (m) |
| 1.36 | 1.31 (m) | ||
| 5 | 2.51 (m) | 1.76 | 1.64 (m) |
| 6 | 2.14 (m) | 2.20 (dd, 11.0, 6.7) | 1.80 (m) |
| 1.72 (m) | 1.65 | ||
| 7 | 4.81 (d, 3.5) | 4.86 (t, 8.9) | 2.79 |
| 11 | 7.23 (d, 8.4) | 7.21 (d, 8.3) | 7.04 (d, 8.6) |
| 12 | 7.39 (dd, 8.4, 1.6) | 7.31 (dd, 8.3, 0.9) | 6.51 (dd, 8.6, 2.7) |
| 14 | 7.46 (d, 1.6) | 7.64 (d, 1.9) | 6.42 (d, 2.7) |
| 15 | 3.43 (d , 7.5) | ||
| 3.09 (d , 7.5) | |||
| 16 | 5.36 (s) | 1.57 (s) | 0.84 (s) |
| 5.06 (s) | |||
| 17 | 2.14 (s) | 1.57 (s) | 1.17 (s) |
| 18 | 3.50 (d , 10.9) | ||
| 3.20 (d , 10.9) | |||
| 19 | 1.30 (s) | 0.89 (s) | |
| 20 | 1.18 (s) | 1.29 (s) |
| Position | 1 a | 2 b | 3 a |
|---|---|---|---|
| 1 | 37.6 (t) | 38.3 (t) | 40.0 (t) |
| 2 | 18.5 (t) | 18.5 (t) | 19.9 (t) |
| 3 | 36.3 (t) | 34.7 (t) | 36.3 (t) |
| 4 | 46.9 (s) | 37.5 (s) | 38.9 (s) |
| 5 | 39.6 (d) | 42.3 (d) | 45.1 (d) |
| 6 | 31.4 (t) | 29.8 (t) | 19.9 (t) |
| 7 | 68.2 (d) | 70.9 (d) | 31.2 (t) |
| 8 | 135.8 (s) | 137.6 (s) | 137.5 (s) |
| 9 | 148.3(s) | 148.1 (s) | 142.7 (s) |
| 10 | 37.6 (s) | 38.1 (s) | 38.2 (s) |
| 11 | 124.2 (d) | 124.4 (d) | 126.4 (d) |
| 12 | 125.6 (d) | 123.9 (d) | 113.9 (d) |
| 13 | 139.1 (s) | 146.5 (s) | 155.5 (s) |
| 14 | 127.5 (d) | 123.1 (d) | 115.7 (d) |
| 15 | 142.6 (s) | 72.5 (s) | 72.0 (t) |
| 16 | 112.1 (t) | 31.7 (q) | 18.0 (q) |
| 17 | 21.8 (q) | 31.6 (q) | 26.0 (q) |
| 18 | 182.3 (s) | 71.6 (t) | |
| 19 | 16.3 (q) | 17.5 (q) | |
| 20 | 24.1 (q) | 25.7 (q) |
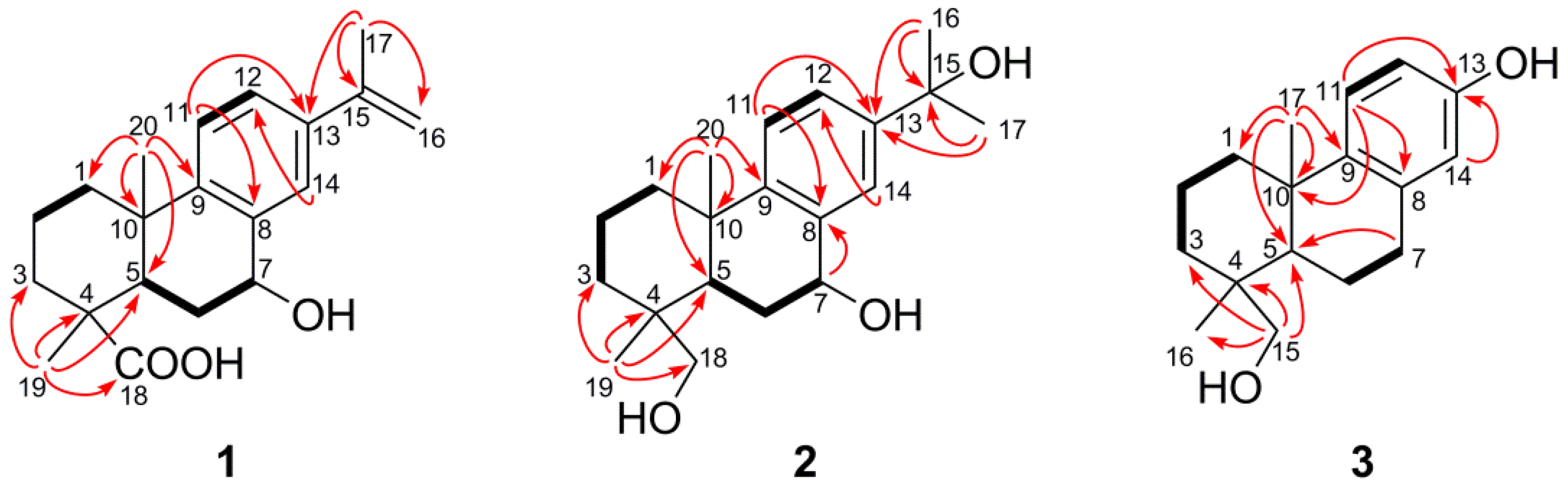
 C) correlations of compounds 1–3.
C) correlations of compounds 1–3.
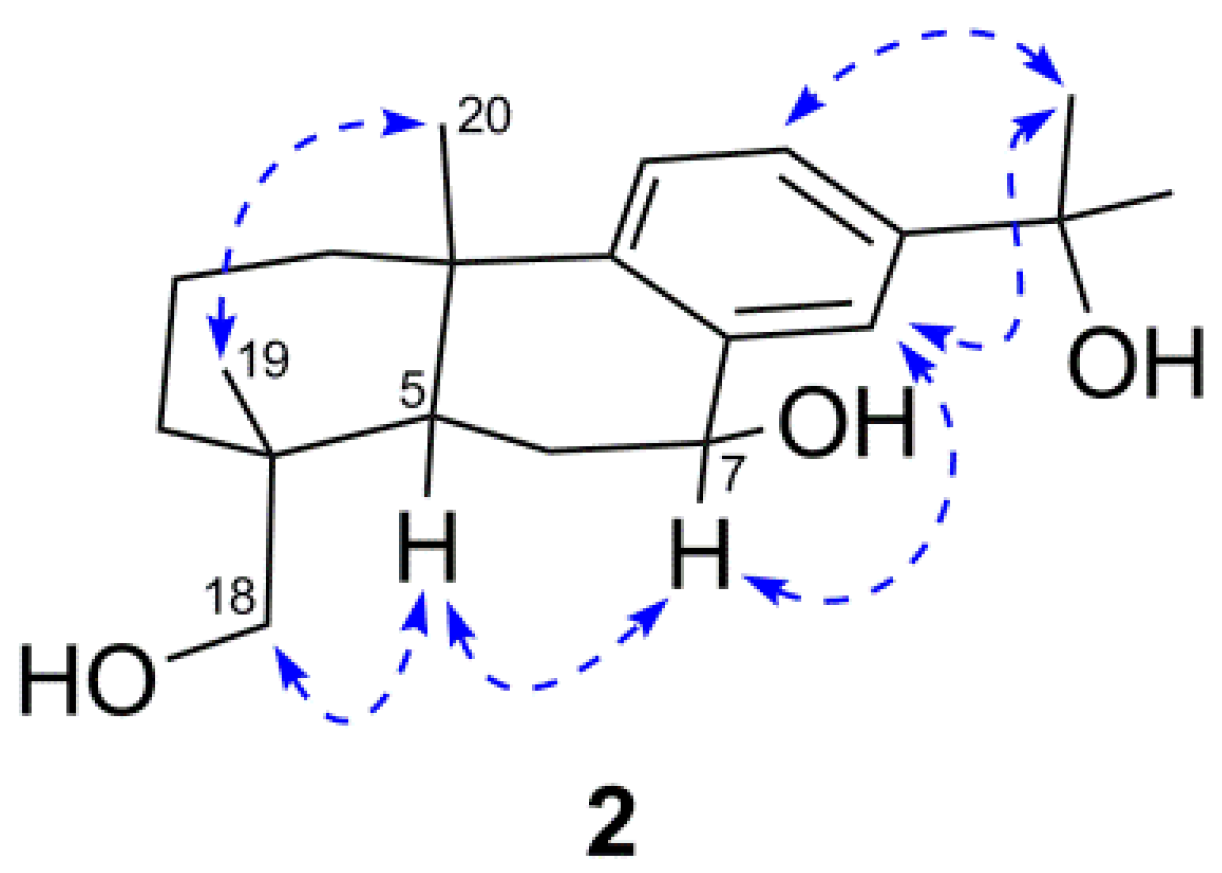
 ) correlations of compound 2.
) correlations of compound 2.
| Compounds | IC50 (µM) a,b | ||||
|---|---|---|---|---|---|
| A549 c | DLD-1 d | WS1 e | S. aureus f | E. coli g | |
| 1 | >100 | >100 | >100 | >100 | >100 |
| 2 | >100 | >100 | >100 | >100 | >100 |
| 3 | >100 | >100 | >100 | >100 | >100 |
| 4 | >100 | >100 | >100 | >100 | >100 |
| 5 | >100 | >100 | >100 | >100 | >100 |
| 6 | >100 | >100 | >100 | >100 | >100 |
| 7 | 98 ± 2 | >100 | >100 | >100 | >100 |
| 8 | 63 ± 7 | 91 ± 9 | >100 | >100 | >100 |
| 9 | >100 | >100 | >100 | >100 | >100 |
| 10 | >100 | >100 | >100 | >100 | >100 |
| 11 | >100 | >100 | >100 | >100 | >100 |
| 12 | 34 ± 4 | 47 ± 9 | >100 | >100 | >100 |
| 13 | 46 ± 3 | >100 | >100 | >100 | >100 |
| 14 | >100 | >100 | >100 | >100 | >100 |
| 15 | >100 | >100 | >100 | >100 | >100 |
| Etoposide h | 0.7 ± 0.3 | 2.9 ± 0.5 | 10 ±2 | ||
| Chloramphenicoli | >5 | 0.37 ± 0.06 | |||
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Compound Characterization
3.5. Cell Lines and Culture Conditions
3.6. Cytotoxicity Assay
3.7. Antibacterial Assay
4. Conclusions
Acknowledgments
References
- Little, E.L. Atlas of United States Trees. Conifers and Important Hardwoods; USDA Forest Service Miscellaneous Publication 1146: Washington, DC, USA, 1971; Volume 1. [Google Scholar]
- Critchfield, W.B. The late Quaternary history of lodgepole and jack pines. Can. J. For. Res. 1985, 15, 749–772. [Google Scholar] [CrossRef]
- Rudolph, T.D.; Laidly, P.R. Silvic of North America; U.S.D.A. Forest Service, Agriculture Handbook 654: Washington, DC, USA, 1990; Volume 1. [Google Scholar]
- Kuhlein, H.V.; Turner, N.J. Traditional Plant Foods of Canadian Indigenous Peoples. Nutrition, Botany and Use; Gordon and Breach Publishers: Philadelphia, PA, USA, 1991. [Google Scholar]
- Leighton, A.L. Wild Plant Use by the Woods Cree (Nihithawak) of East-Central Saskatchewan; National Museums of Canada: Ottawa, ON, Canada, 1985. [Google Scholar]
- Moerman, D. Native American Ethnobotany; Timber press: Portland, OR, USA, 1998. [Google Scholar]
- Marles, R.; Clavelle, C.; Monteleone, L.; Tays, N.; Burns, D. Aboriginal Plant Uses in Canada’s Northwest Boreal Forest; UBC Press: Vancouver, BC, Canada, 2000. [Google Scholar]
- Pichette, A.; Garneau, F.X.; Jean, F.I.; Riedl, B.; Girard, M. Chemical differences between the wood extract of jake pine, black spruce and balsam fir from eastern Canada. J. Wood Chem. 1998, 18, 427–438. [Google Scholar] [CrossRef]
- Wallin, K.F.; Raffa, K.F. Association of within-tree jack pine budworm feeding patterns with canopy level and within-needle variation of water, nutrient, and monoterpene concentrations. Can. J. For. Res. 1998, 28, 228–233. [Google Scholar] [CrossRef]
- Von Ruddlof, E.; Sato, A. The heartwood extractives of Pinus banksiana lamb. Can. J. Chem. 1963, 41, 2165–2174. [Google Scholar]
- Bower, C.; Rowe, J.W. Extractives of jack pine bark: Occurence of (+)-13-epimanoyl oxide and related labdane diterpenes. Phytochemistry 1967, 6, 151–153. [Google Scholar] [CrossRef]
- Rowe, J.W.; Nagasampagi, B. Derivatives of nordehydroabietane from pine bark. Phytochemistry 1971, 10, 1647–1651. [Google Scholar]
- Savidge, R.A. Coniferin, a biochemical indicator of commitment to tracheid differentiation in conifers. Can. J. Bot. 1989, 67, 2663–2668. [Google Scholar] [CrossRef]
- Sinclair, G.D.; Dymond, D.K. The distribution and composition of extractives in jack pine trees. Can. J. For. Res. 1973, 3, 516–521. [Google Scholar] [CrossRef]
- Ohtsu, H.; Tanaka, R.; Matsunaga, S. Abietane diterpenoids from the cones of Larix kaempferi. J. Nat. Prod. 1998, 61, 1307–1309. [Google Scholar]
- Prinz, S.; Müllner, U.; Heilmann, J.; Winkelmann, K.; Sticher, O.; Haslinger, E.; Hüfner, A. Oxidation products of abietic acid and its methyl ester. J. Nat. Prod. 2002, 65, 1530–1534. [Google Scholar]
- Miguel Del Corral, J.M.; Gordaliza, M.; Salinero, M.A.; San Feliciano, A. 13C-NMR data for abieta-8,11,13-triene diterpenoids. Magn. Reson. Chem. 1994, 32, 774–781. [Google Scholar]
- Ohmoto, T.; Kanatani, K.; Yamaguchi, K. Constituent of Pollen. XIII. Constituents of Cedrus deodava Loud. Chem. Pharm. Bull. 1987, 35, 229–234. [Google Scholar] [CrossRef]
- Cheng, Y.S.; Lu, S.B. The chemical constituents of the wood of Keteleeria davidiana Beissner. J. Chin. Chem. Soc. 1978, 25, 47–53. [Google Scholar]
- Ohtsu, H.; Tanaka, R.; Matsunaga, S. 18-nor-abietatrienes from the cones of Larix kaempferi. J. Nat. Prod. 1998, 61, 406–408. [Google Scholar] [CrossRef]
- Barrero, A.F.; Alvarez-Manzaneda, E.J.; Alvarez-Manzaneda, R.; Chahboun, R.; Mencses, R.; Marta Aparicio, B. Ring A functionalization of terpenoids by the unusual Baeyer-Villiger rearrangement of aliphatic aldehydes. Synlett 1999, 6, 713–716. [Google Scholar]
- Ayer, W.A.; Migaj, B.S. Acids from blue-stain diseased lodgepole pine. Can. J. Bot. 1989, 67, 1426–1428. [Google Scholar] [CrossRef]
- Tanaka, R.; Ohtsu, H.; Matsunaga, S. Abietane diterpene acids and other constituents from the leaves of Larix Kaempferi. Phytochemistry 1997, 46, 1051–1057. [Google Scholar] [CrossRef]
- Kuo, Y.H.; Yeh, M.H. Norditerpenes from the heartwood of Picea morrisonicola. Phytochemistry 1998, 49, 2453–2455. [Google Scholar]
- Matsumoto, T.; Imai, S.; Sunaoka, Y.; Yoshinari, T. The conversion of (+)-dehydroabietic acid into steroidal hormones. Bull. Chem. Soc. Jpn. 1988, 61, 723–727. [Google Scholar]
- Rago, R.; Mitchen, J.; Wilding, G. DNA fluorometric assay in 96-well tissue culture plates using Hoechst 33 258 after cell lysis by freezing in distilled water. Anal. Biochem. 1990, 191, 31–34. [Google Scholar]
- Barrero, A.F.; Quílez Del Moral, J.F.; Mar Herrador, M.; Arteaga, J.F.; Akssira, M.; Benharref, A.; Dakir, M. Abietane diterpenes from the cone of Cedrus atlantica. Phytochemistry 2005, 66, 105–111. [Google Scholar] [CrossRef]
- Sultan, Z.; Jeon, Y.M.; Moon, S.S. Labdane type diterpenes active against acne from pine cones. Planta Med. 2008, 74, 449–452. [Google Scholar] [CrossRef]
- Gouiric, S.C.; Feresin, G.E.; Tapia, A.A.; Rossomando, P.C.; Schmeda-Hirschmann, G.; Bustos, D.A. 1β,7β-dihydroxydehydroabietic acid, a new biotransformation product of dehydroabietic acid by Aspergillus niger. World J. Microb. Biot. 2004, 20, 281–284. [Google Scholar] [CrossRef]
- Xue, J.J.; Fan, C.Q.; Dong, L.; Yang, S.P.; Yue, J.M. Novel antibacterial diterpenoids from Larix chinensis Beissn. Chem. Biodivers. 2004, 1, 1702–1707. [Google Scholar] [CrossRef]
- Banfi, E.; Scialino, G.; Monti-Bragadin, C. Development of a microdilution method to evaluate Mycobacterium tuberculosis drug susceptibility. J. Antimicrob. Chemother. 2003, 52, 796–800. [Google Scholar]
- Sample Availability: Samples of the compounds 1–3 are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Georges, P.; Legault, J.; Lavoie, S.; Grenon, C.; Pichette, A. Diterpenoids from the Buds of Pinus banksiana Lamb. Molecules 2012, 17, 9716-9727. https://doi.org/10.3390/molecules17089716
Georges P, Legault J, Lavoie S, Grenon C, Pichette A. Diterpenoids from the Buds of Pinus banksiana Lamb. Molecules. 2012; 17(8):9716-9727. https://doi.org/10.3390/molecules17089716
Chicago/Turabian StyleGeorges, Patricia, Jean Legault, Serge Lavoie, Carole Grenon, and André Pichette. 2012. "Diterpenoids from the Buds of Pinus banksiana Lamb." Molecules 17, no. 8: 9716-9727. https://doi.org/10.3390/molecules17089716