Schizandrin Protects Primary Rat Cortical Cell Cultures from Glutamate-Induced Apoptosis by Inhibiting Activation of the MAPK Family and the Mitochondria Dependent Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
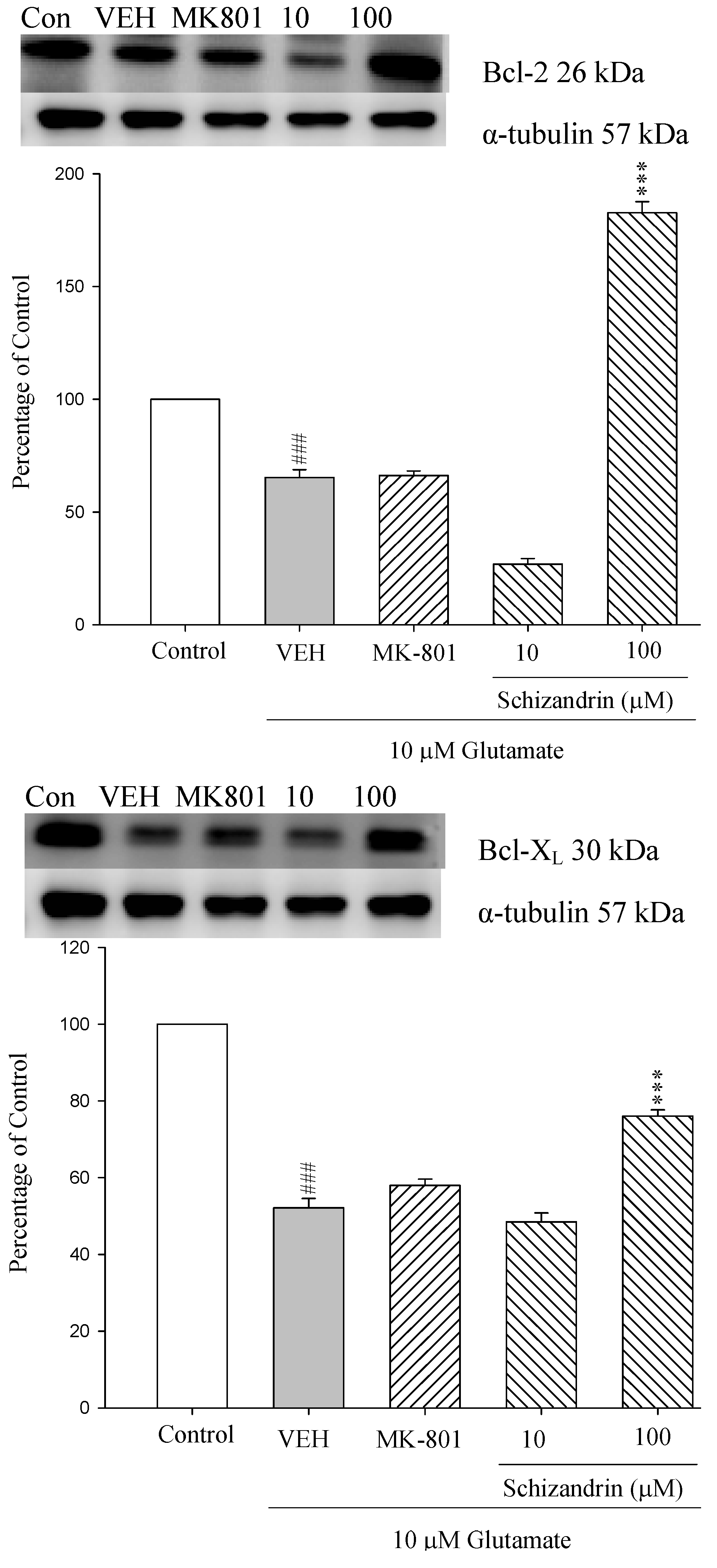
2.1. Western Blotting Analysis of the Effects of Schizandrin on Glu-Induced Protein Level Changes of Bcl-2 and Bcl-XL in Rat Cortical Cell Cultures
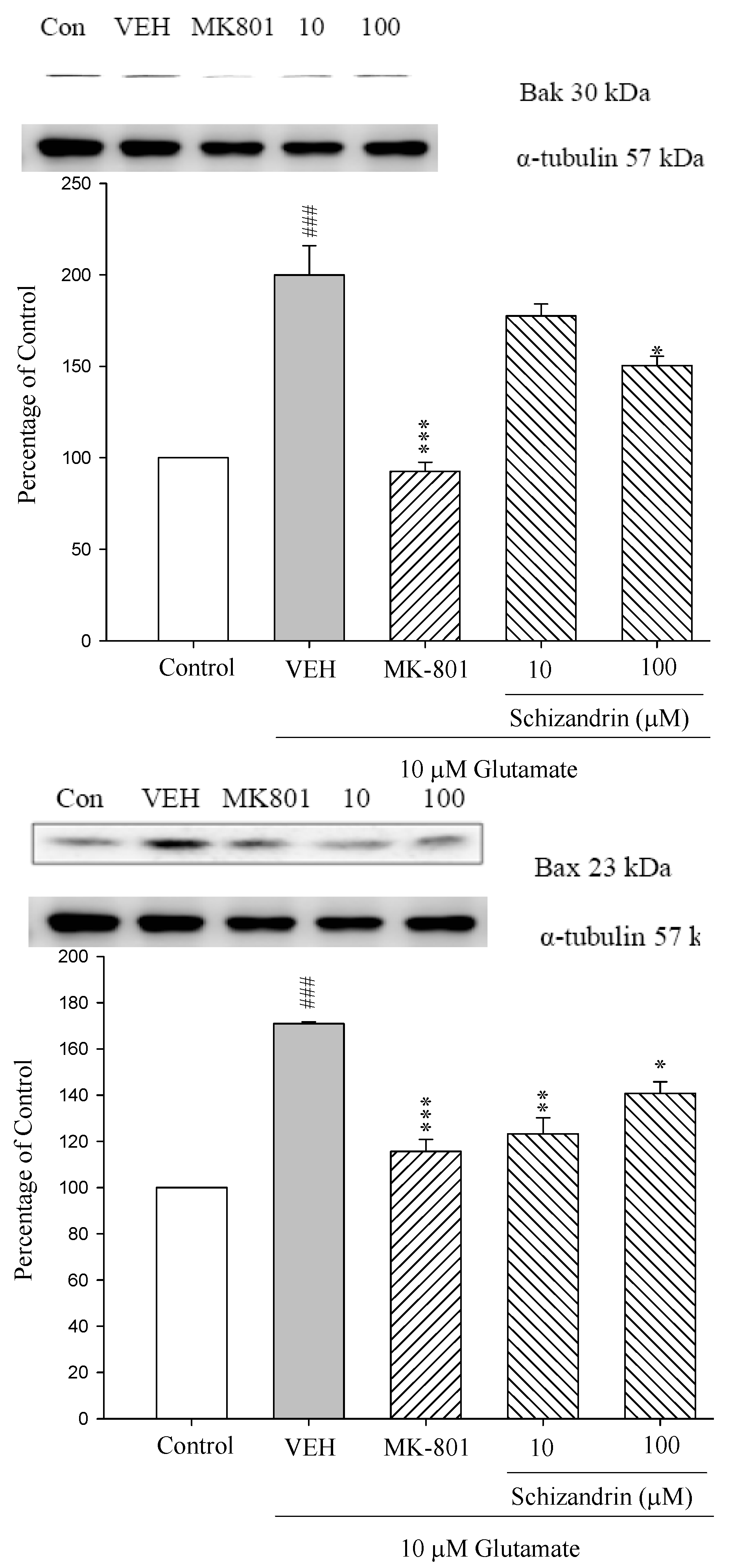
2.2. Western Blot Analysis of the Effects of Schizandrin on Glu-Induced Protein Level Changes of Bak and Bax in Rat Cortical Cell Cultures
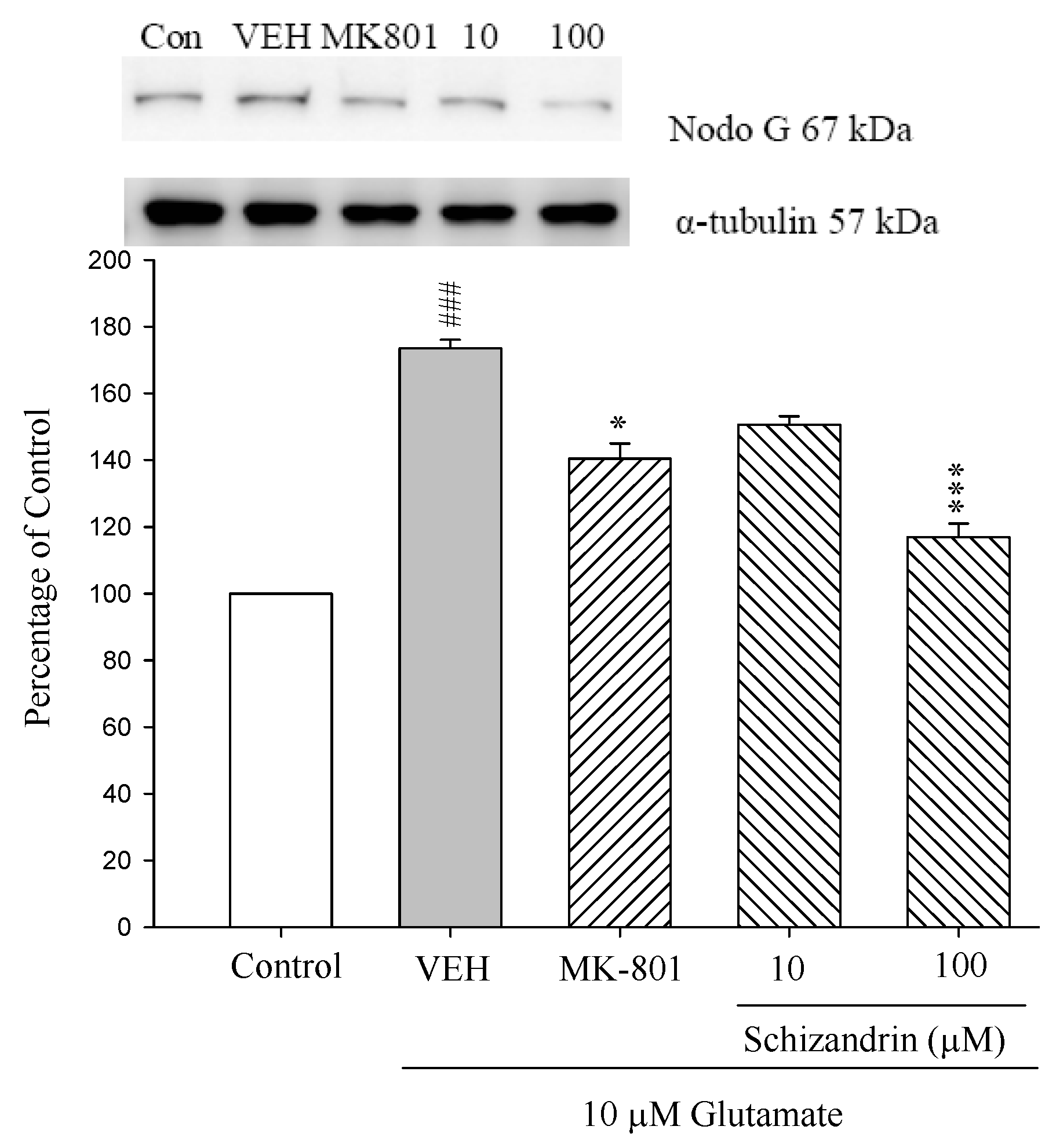
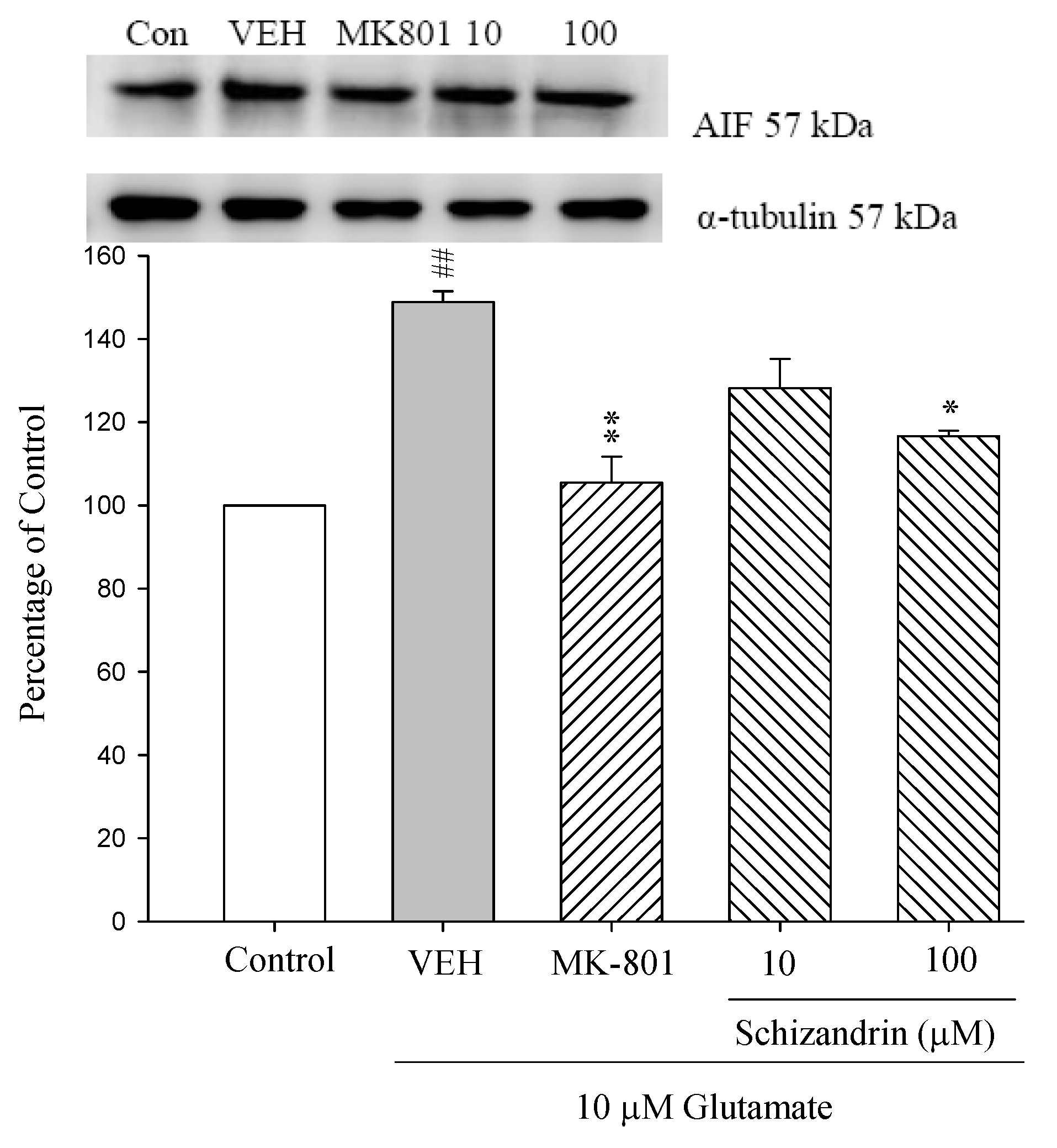
2.3. Western Blot Analysis of the Effects of Schizandrin on Glu-Induced Protein Level Changes of Nodo G and AIF in Rat Cortical Cell Cultures
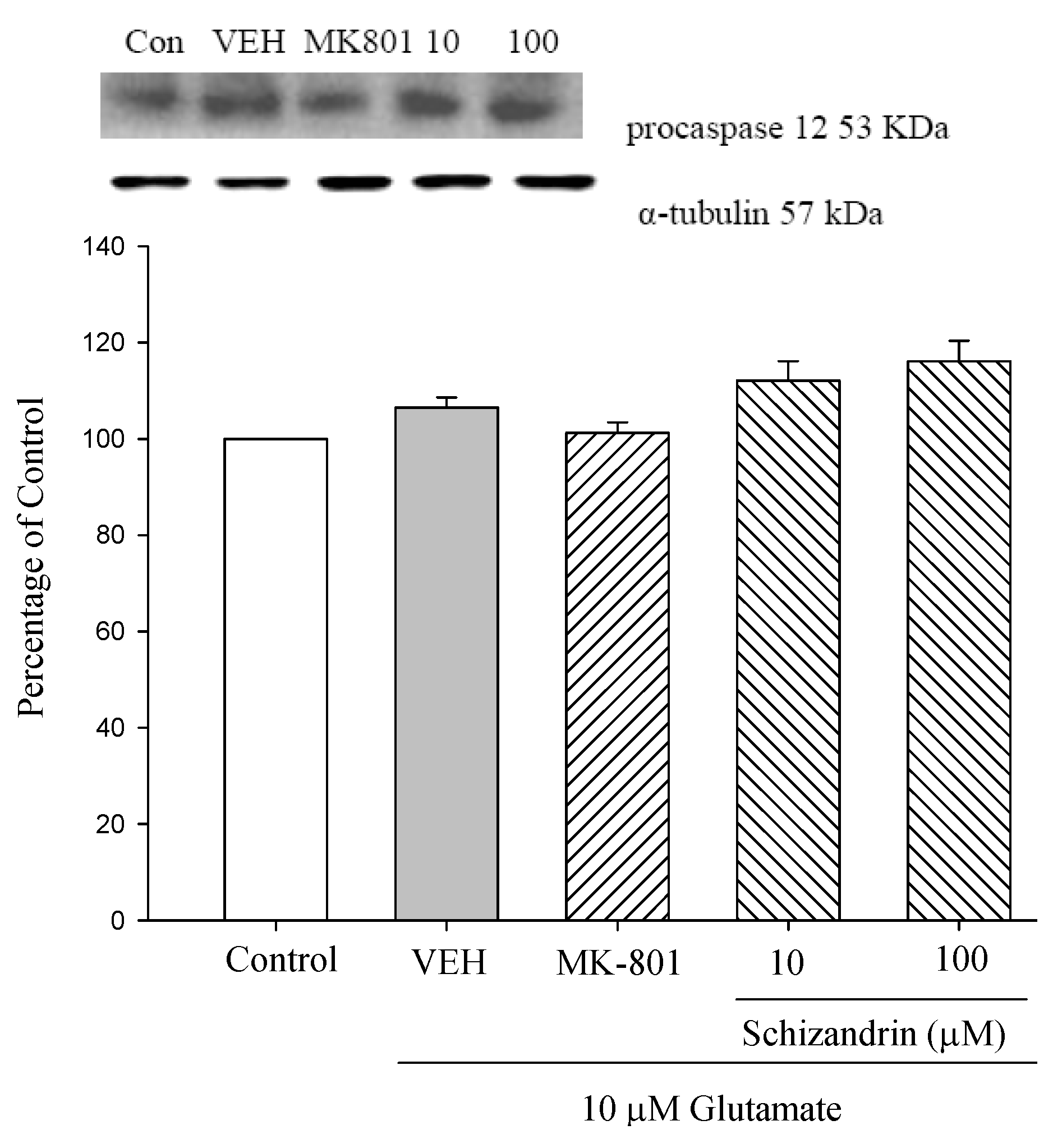
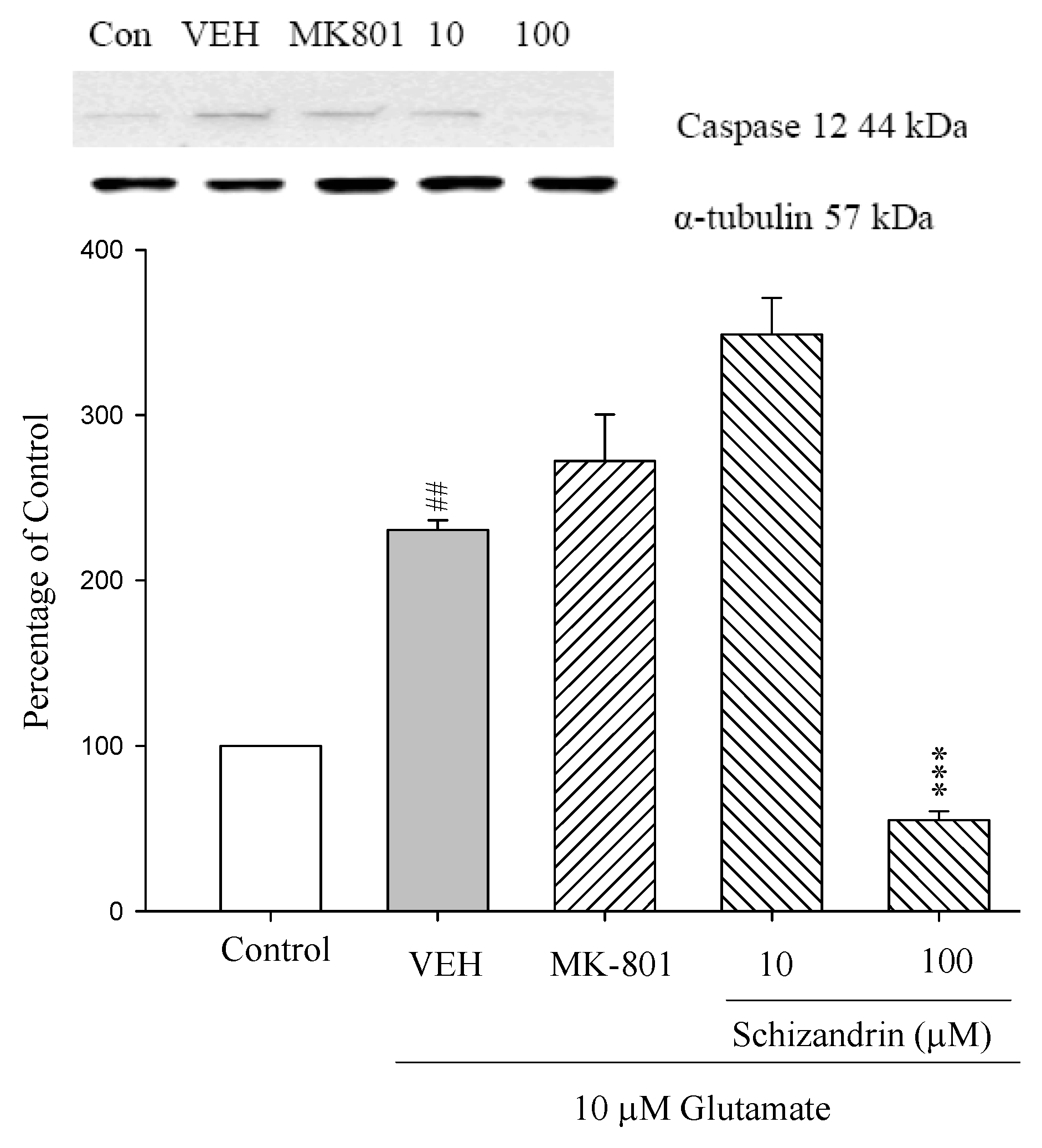
2.4. Western Blot Analysis of the Effects of Schizandrin on Glu-Induced Protein Level Changes of Procaspase 12 and Caspase 12 in Rat Cortical Cell Cultures
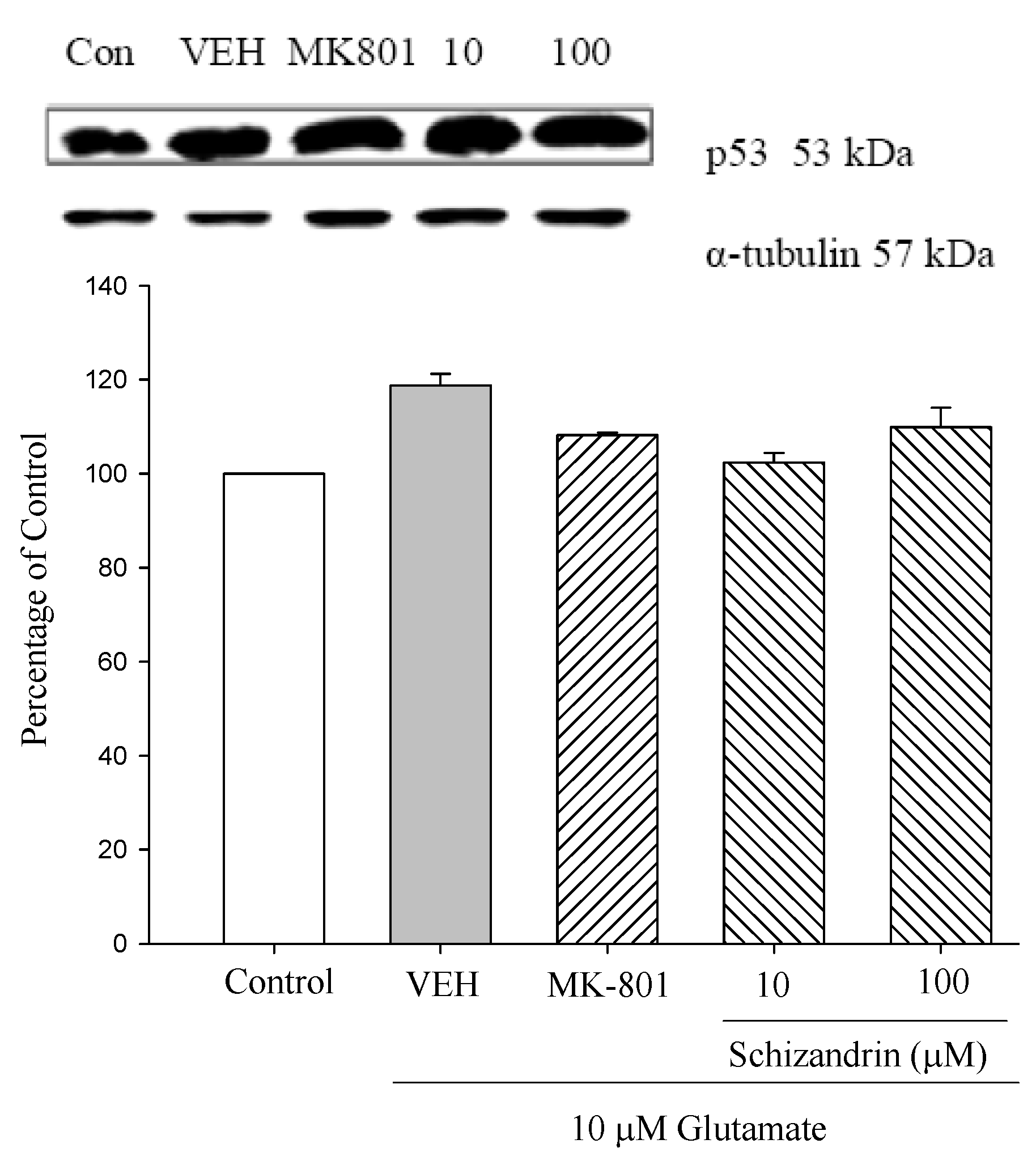
2.5. Western Blot Analysis of the Effects of Schizandrin on Glu-Induced Protein Level Changes of p53 in Rat Cortical Cell Cultures
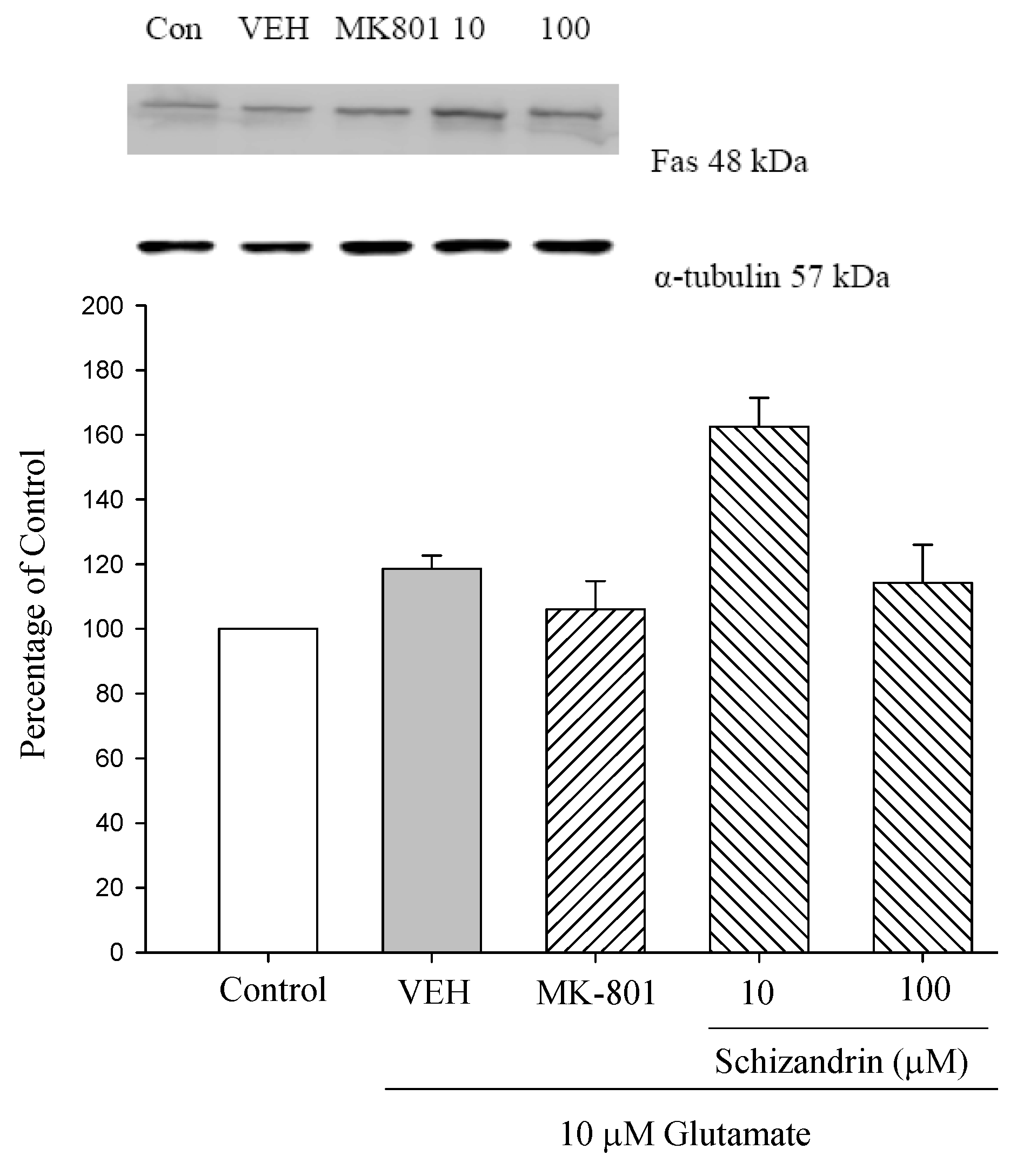
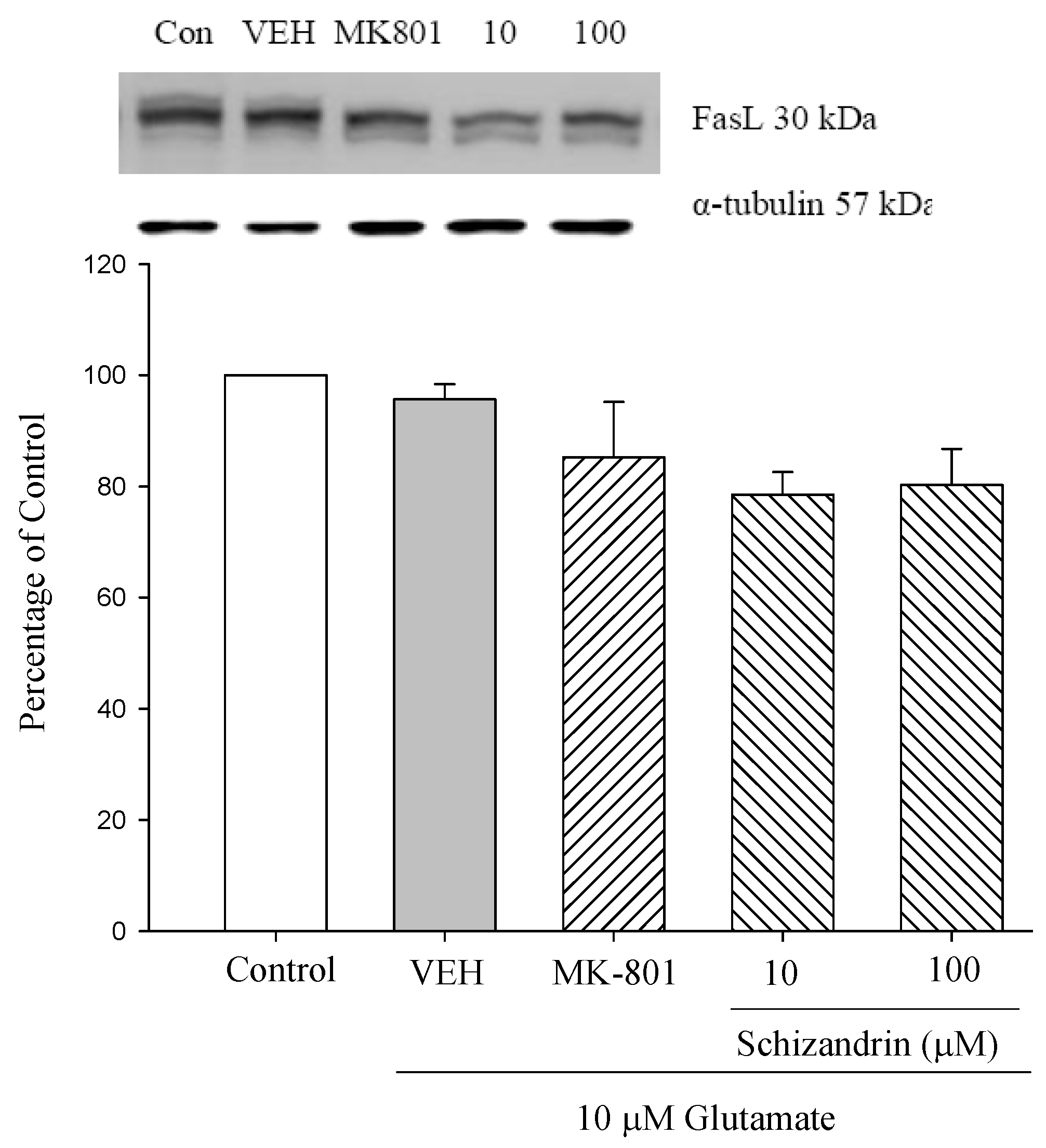
2.6. Western Blot Analysis of the Effects of Schizandrin on Glu-Induced Protein Level Changes of Fas and FasL in Rat Cortical Cell Cultures
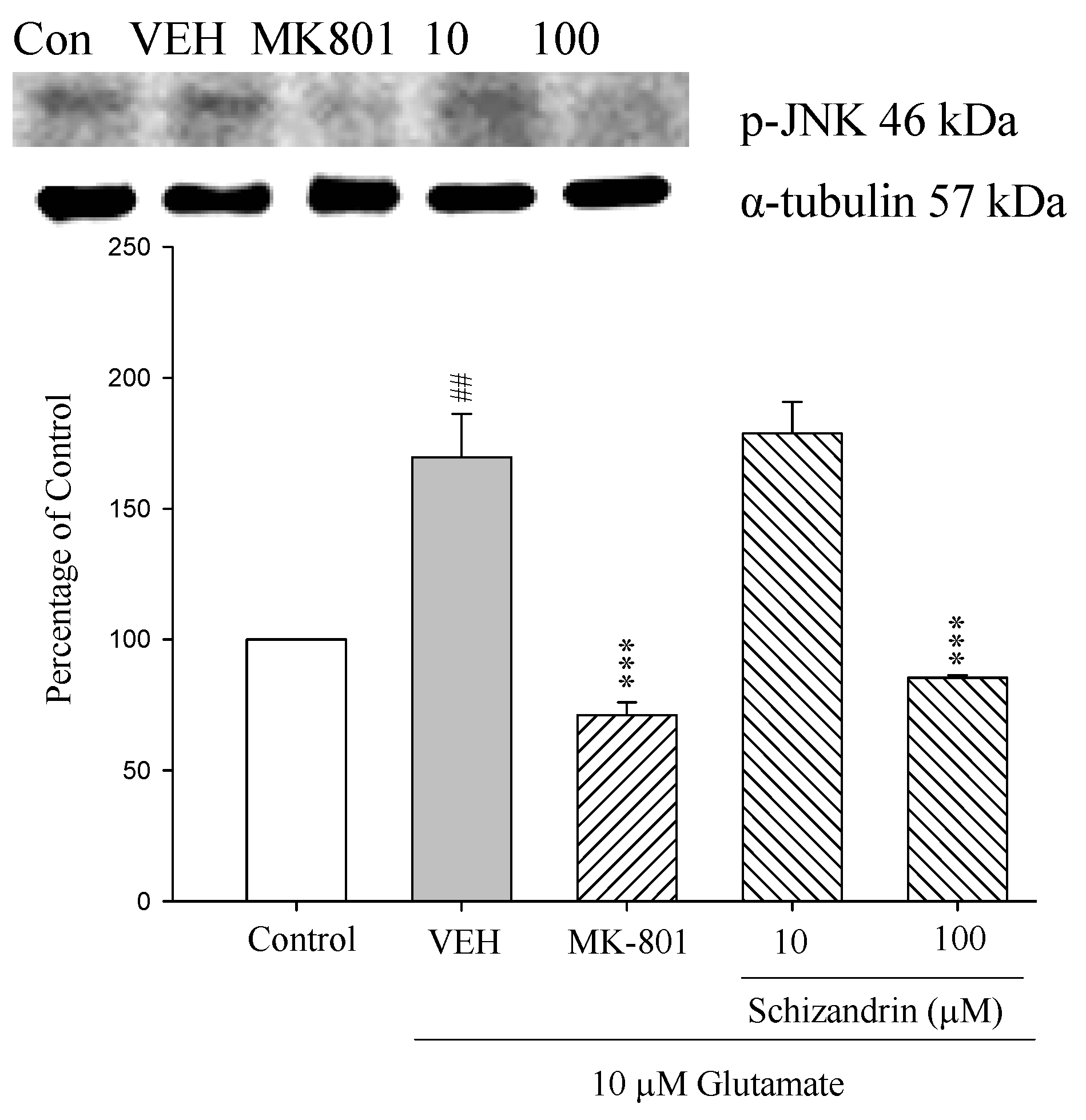
2.7. Western Blot Analysis of the Effects of Schizandrin on Glu-Induced Protein Level Changes of Phosphor-JNK and α-Tubulin in Rat Cortical Cell Cultures
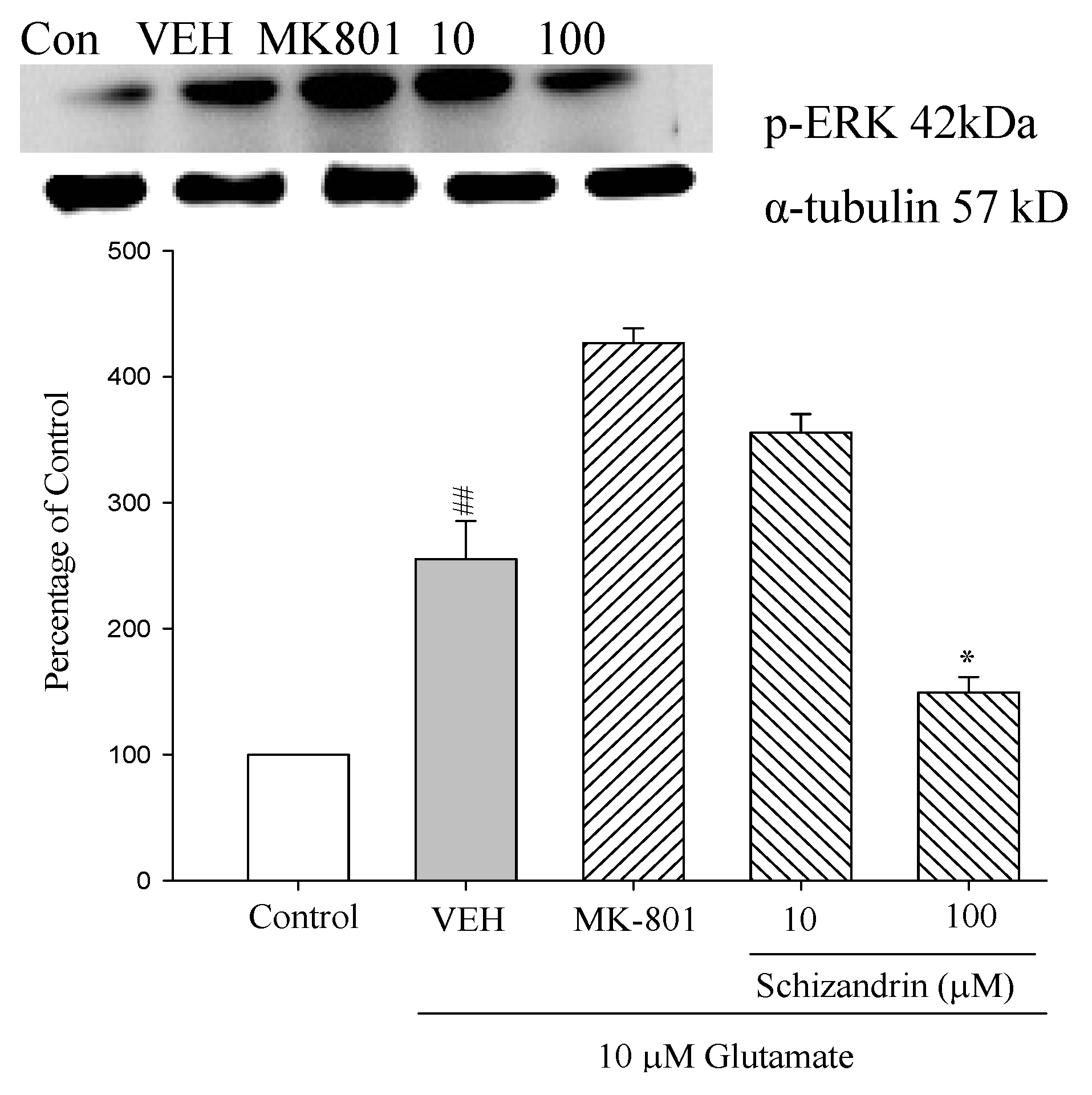
2.8. Western Blot Analysis of the Effects of Schizandrin on Glu-Induced Protein Level Changes of Phosphor-ERK in Rat Cortical Cell Cultures
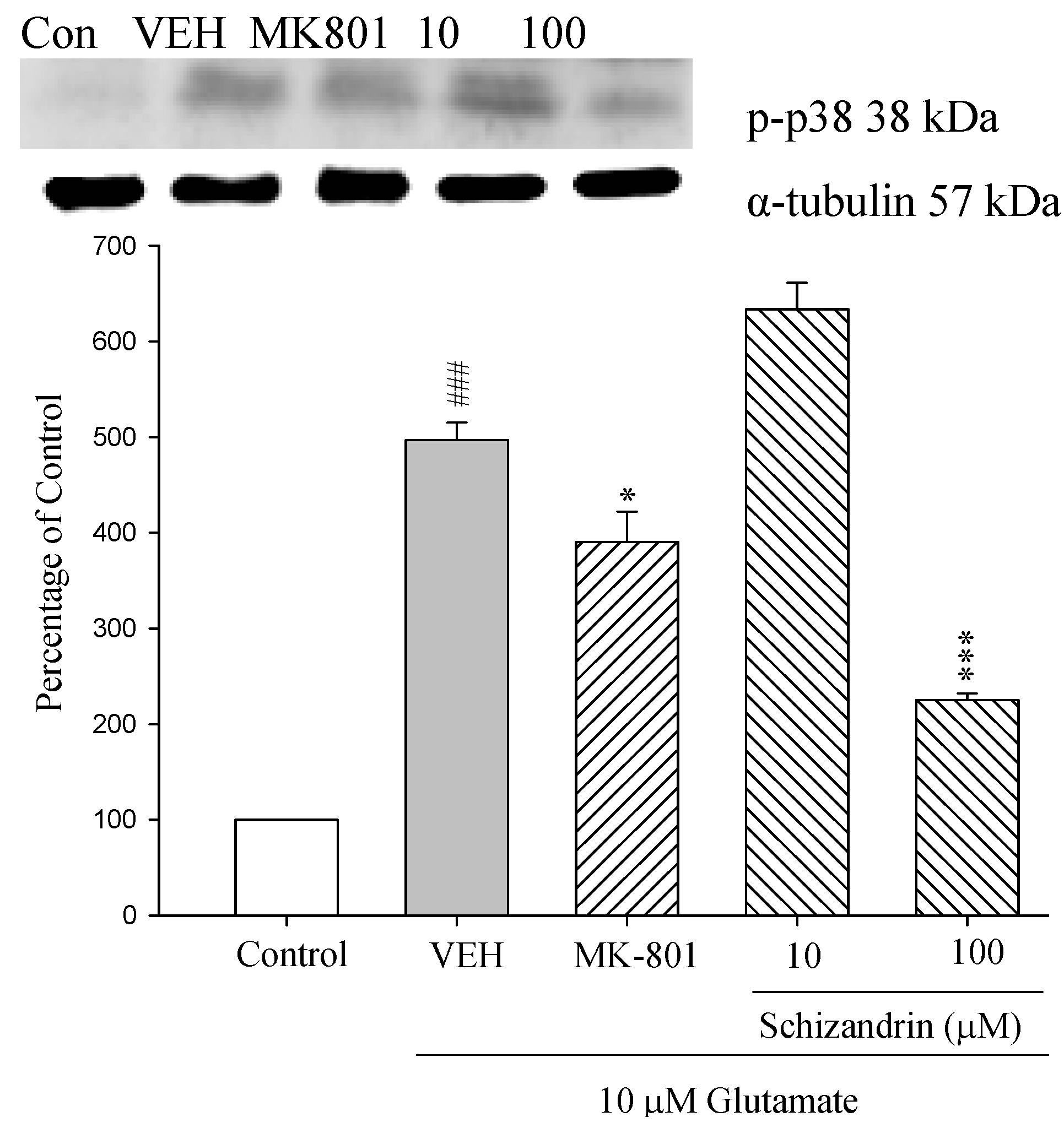
2.9. Western Blot Analysis of the Effects of Schizandrin on Glu-Induced Protein Level Changes of Phosphorylated-p38 in Rat cOrtical Cell Cultures
2.10. Discussion
3. Experimental
3.1. Materials and Reagents
3.2. Primary Cultures of Rat Cortical Cell
3.3. Excitotoxicity Induction and Drug Treatment
3.4. Protein Extraction and Western Blot Analysis
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Meldrum, B.; Garthwaite, J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. Sci. 1990, 9, 379–387. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 8, 623–634. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Green, D.R. Protease activation during apoptosis: Death by a thousand cuts? Cell 1995, 82, 349–352. [Google Scholar] [CrossRef]
- Rao, R.V.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 4, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 4, 239–257. [Google Scholar] [CrossRef]
- Dreyer, E.B.; Zhang, D.; Lipton, S.A. Transcriptional or translational inhibition blocks low dose NMDA-mediated cell death. Neuroreport 1995, 6, 942–944. [Google Scholar] [CrossRef] [PubMed]
- Hockenbery, D.; Nunez, G.; Milliman, C.R.; Schreiber, D.; Korsmeyer, S.J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, C.; Chai, J.; Shi, Y.; Xue, D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 2002, 298, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, X.; Bhalla, K.C.; Kim, N.; Ibrado, A.M.; Cai, J.; Peng, T.I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Mielke, K.; Herdegen, T. JNK and p38 stresskinases—Degenerative effectors of signal-transduction-cascades in the nervous system. Prog. Neurobiol. 2000, 1, 45–60. [Google Scholar] [CrossRef]
- Zhu, X.; Lee, H.G.; Raina, A.K.; Perry, G.; Smith, M.A. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 2002, 5, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Gu, Z.; Zhang, G.; Jing, G. Diphosphorylation and involvement of extracellular signal-regulated kinases (ERK1/2) in glutamate-induced apoptotic-like death in cultured rat cortical neurons. Brain Res. 2000, 857, 71–77. [Google Scholar] [CrossRef]
- Segura Torres, J.E.; Chaparro-Huerta, V.; Rivera Cervantres, M.C.; Montes-Gonzalez, R.; Flores Soto, M.E.; Beas-Zarate, C. Neuronal cell death due to glutamate excitotocity is mediated by p38 activation in the rat cerebral cortex. Neurosci. Lett. 2006, 403, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Z. An Illustrated Chinese Material Medica in Hong Kong; Chung Hwa Book: Hong Kong, China, 2004. [Google Scholar]
- Hsieh, M.T.; Tsai, M.L.; Peng, W.H.; Wu, C.R. Effects of Fructus schizandrae on cycloheximide-induced amnesia in rats. Phytother. Res. 1999, 3, 256–257. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Hsieh, M.T.; Wu, C.R.; Tsai, F.H.; Lu, T.C.; Hsieh, C.C.; Li, W.C.; Lin, Y.T.; Peng, W.H. Schizandrin protects primary cultures of rat cortical cells from glutamate-induced excitotoxicity. J. Pharmacol. Sci. 2008, 1, 21–31. [Google Scholar] [CrossRef]
- Ramoa, A.S.; Alkondon, M.; Aracava, Y.; Irons, J.; Lunt, G.G.; Deshpande, S.S.; Wonnacott, S.; Aronstam, R.S.; Albuquerque, E.X. The anticonvulsant MK-801 interacts with peripheral and central nicotinic acetylcholine receptor ion channels. J. Pharmacol. Exp. Ther. 1990, 1, 71–82. [Google Scholar]
- Drian, M.J.; Kamenka, J.M.; Pirat, J.L.; Privat, A. Non-competitive antagonists of N-methyl-d-aspartate prevent spontaneous neuronal death in primary cultures of embryonic rat cortex. J. Neurosci. Res. 1991, 29, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Mackey, M.R.; Perkins, G.; Ellisman, M.H.; Latterich, M.; Schneiter, R.; Green, D.R; Newmeyer, D.D. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002, 111, 331–342. [Google Scholar] [CrossRef]
- Saito, M.S.; Korsmeyer, J.; Schlesinger, P.H. BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat. Cell Biol. 2000, 8, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Korsmeyer, S.J.; Wei, M.C.; Saito, M.; Weiler, S.; Oh, K.J.; Schlesinger, P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 12, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, G.; Saelens, X.; van Gurp, M.; MacFarlane, M.; Martin, S.J.; Vandenabeele, P. The role of mitochondrial factors in apoptosis: A Russian roulette with more than one bullet. Cell Death Differ. 2002, 10, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Beyaert, R.; van Loo, G.; Heyninck, K.; Vandenabeele, P. Signaling to gene activation and cell death by tumor necrosis factor receptors and Fas. Int. Rev. Cytol. 2002, 2149, 225–272. [Google Scholar]
- Jiang, Q.; Gu, Z.; Zhang, G. Activation, involvement and nuclear translocation of c-Jun N-terminal protein kinase 1 and 2 in glutamate-induced apoptosis in cultured rat cortical neurons. Brain Res. 2002, 956, 194–201. [Google Scholar] [CrossRef]
- Kurino, M.; Fukunaga, K.; Ushio, Y.; Miyamoto, E. Activation of mitogen-activated protein kinase in cultured rat hippocampal neurons by stimulation of glutamate receptors. J Neurochem. 1995, 3, 1282–1289. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Grossmann, A.; Zedlick, D.; Bruckner, M.K. Increased expression and subcellular translocation of the mitogen activated protein kinase kinase and mitogen-activated protein kinase in Alzheimer’s disease. Neuroscience 1995, 1, 5–18. [Google Scholar] [CrossRef]
- Olney, J.W. Inciting excitotoxic cytocide among central neurons. Adv. Exp. Med. Biol. 1986, 203, 631–645. [Google Scholar] [PubMed]
- Raoul, C.; Pettmann, B.; Henderson, C.E. Active killing of neurons during development and following stress: A role for p75(NTR) and Fas? Curr. Opin. Neurobiol. 2000, 1, 111–117. [Google Scholar] [CrossRef]
- Blatt, N.B.; Glick, G.D. Signaling pathways and effector mechanisms pre-programmed cell death. Bioorg. Med. Chem. 2001, 6, 1371–1384. [Google Scholar] [CrossRef]
- Zimmermann, K.C.; Bonzon, C.; Green, D.R. The machinery of programmed cell death. Pharmacol. Ther. 2001, 1, 57–70. [Google Scholar] [CrossRef]
- Behl, C.; Hovey, L., III; Krajewski, S.; Schubert, D.; Reed, J.C. BCL-2 prevents killing of neuronal cells by glutamate but not by amyloid beta protein. Biochem. Biophys. Res. Commun. 1993, 197, 949–956. [Google Scholar] [CrossRef]
- Schelman, W.R.; Andres, R.D.; Sipe, K.J.; Kang, E.; Weyhenmeyer, J.A. Glutamate mediates cell death and increases the Bax to Bcl-2 ratio in a differentiated neuronal cell line. Brain Res. 2004, 128, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.I.; Marzo, B.; Snow, E.G.; Brothers, M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 128, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Daugas, E.; Susin, S.A.; Zamzami, N.; Ferri, K.F.; Irinopoulou, T.; Larochette, N.; Prevost, M.C.; Leber, B.; Andrews, D.; Penninger, J.; et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. Fed. Proc. 2000, 5, 729–739. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [PubMed]
- Thomenius, M.J.; Distelhorst, C.W. Bcl-2 on the endoplasmic reticulum: Protecting the mitochondria from a distance. J. Cell Sci. 2003, 116, 4493–4499. [Google Scholar] [CrossRef] [PubMed]
- Owada, K.; Sanjo, N.; Kobayashi, T.; Mizusawa, H.; Muramatsu, H.; Muramatsu, T.; Michikawa, M. Midkine inhibits caspase-dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. J. Neurochem. 1999, 5, 2084–2092. [Google Scholar]
- Choi, D.W. Calcium-mediated neurotoxicity: Relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988, 10, 465–469. [Google Scholar] [CrossRef]
- Chiu, P.Y.; Leung, H.Y.; Siu, A.H.; Poon, M.K.; Ko, K.M. Schisandrin B decreases the sensitivity of mitochondria to calcium ion-induced permeability transition and protects against ischemia-reperfusion injury in rat hearts. Acta Pharmacol. Sin. 2007, 10, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |












© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, M.-S.; Chao, J.; Yen, J.-C.; Lin, L.-W.; Tsai, F.-S.; Hsieh, M.-T.; Peng, W.-H.; Cheng, H.-Y. Schizandrin Protects Primary Rat Cortical Cell Cultures from Glutamate-Induced Apoptosis by Inhibiting Activation of the MAPK Family and the Mitochondria Dependent Pathway. Molecules 2013, 18, 354-372. https://doi.org/10.3390/molecules18010354
Lee M-S, Chao J, Yen J-C, Lin L-W, Tsai F-S, Hsieh M-T, Peng W-H, Cheng H-Y. Schizandrin Protects Primary Rat Cortical Cell Cultures from Glutamate-Induced Apoptosis by Inhibiting Activation of the MAPK Family and the Mitochondria Dependent Pathway. Molecules. 2013; 18(1):354-372. https://doi.org/10.3390/molecules18010354
Chicago/Turabian StyleLee, Meng-Shiou, Jung Chao, Jiin-Cherng Yen, Li-Wei Lin, Fan-Shiu Tsai, Ming-Tsuen Hsieh, Wen-Huang Peng, and Hao-Yuan Cheng. 2013. "Schizandrin Protects Primary Rat Cortical Cell Cultures from Glutamate-Induced Apoptosis by Inhibiting Activation of the MAPK Family and the Mitochondria Dependent Pathway" Molecules 18, no. 1: 354-372. https://doi.org/10.3390/molecules18010354