Synthesis and in Vitro Antiproliferative Activity of New Phenylaminoisoquinolinequinones against Cancer Cell Lines

Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry



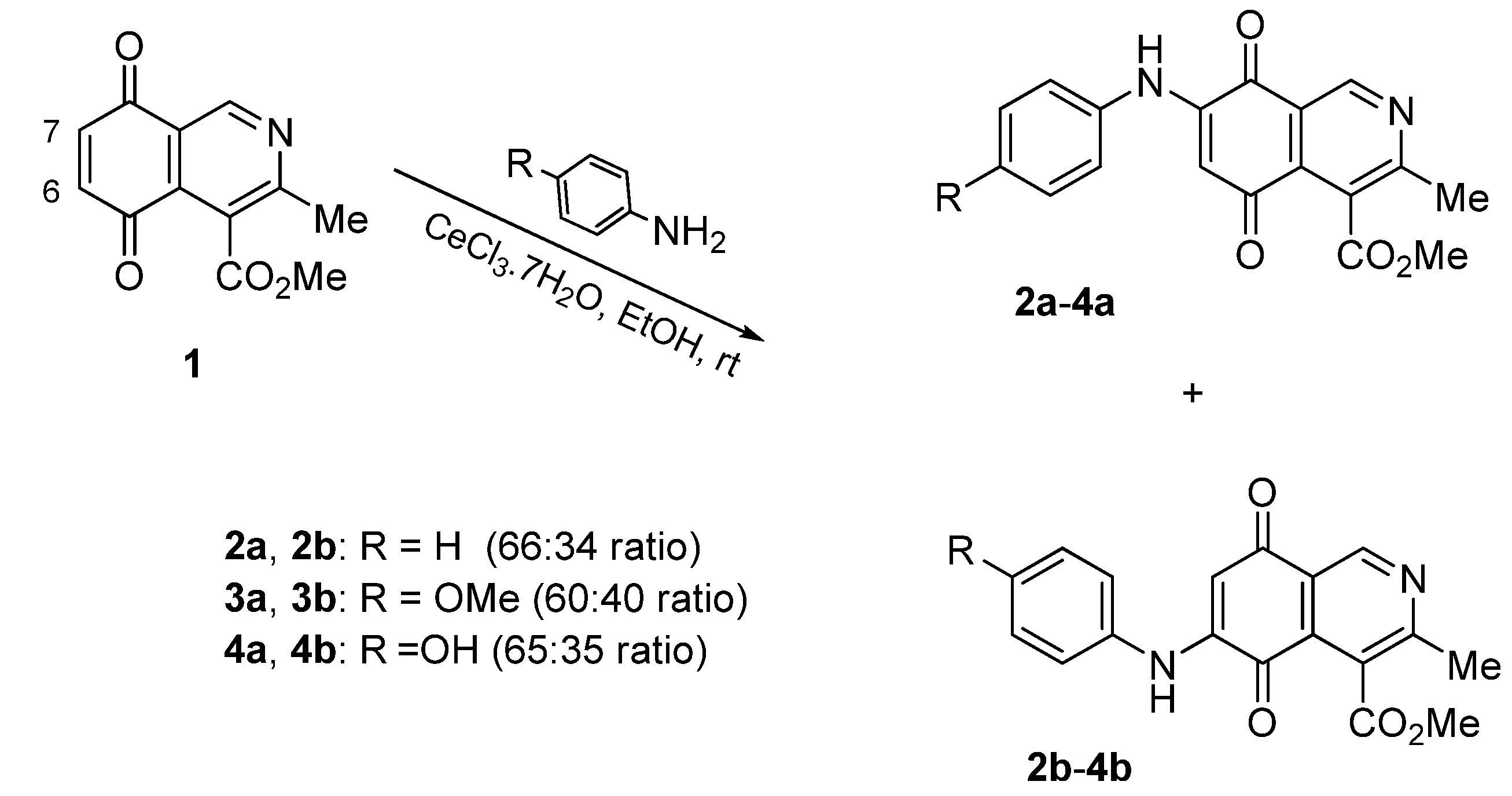{kind=link}
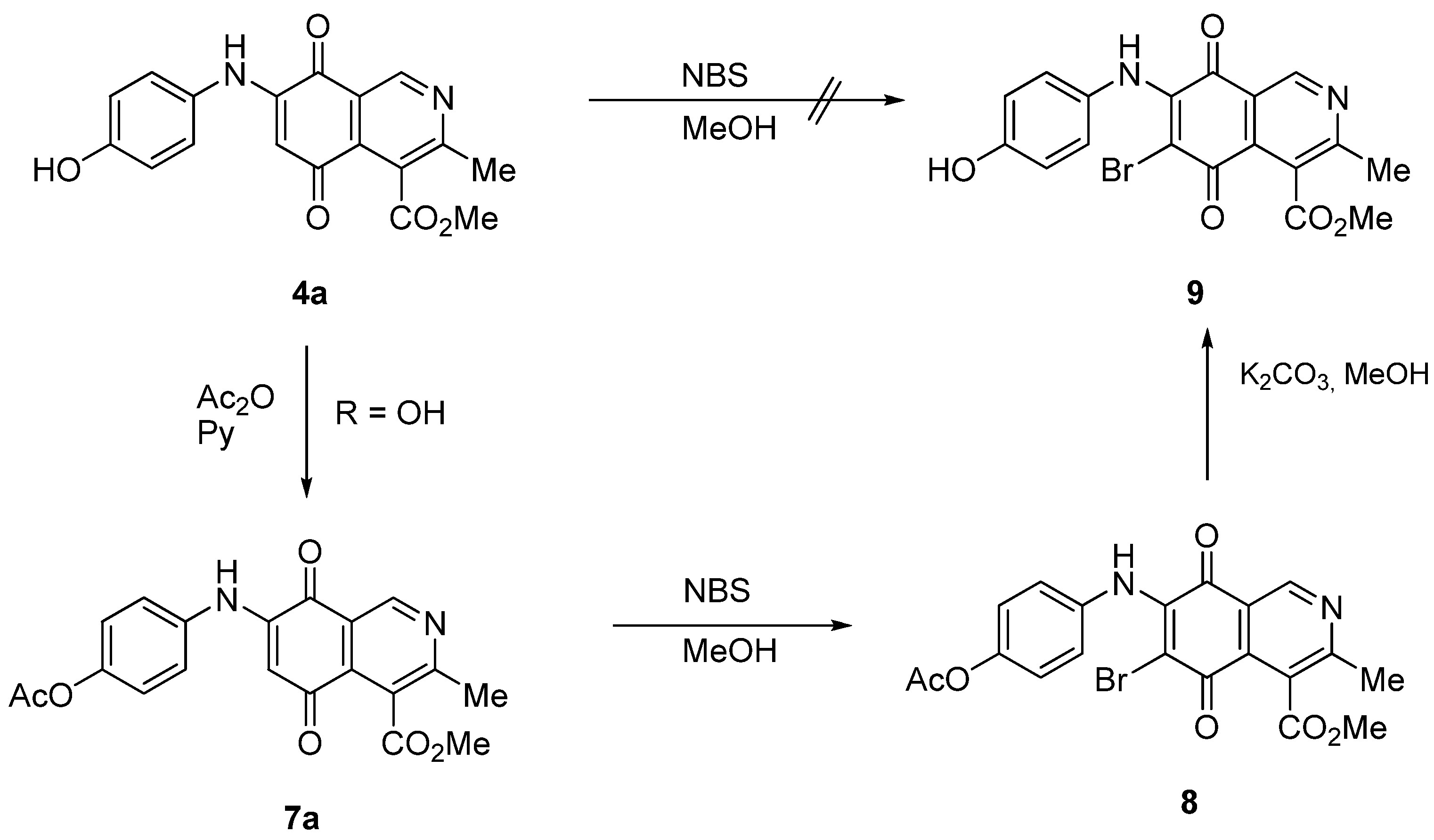{kind=link}
| Substrate | Bromine derivative N° (Yield %) a | Substrate | Bromine derivative N° (Yield) a |
|---|---|---|---|
 |  |  | 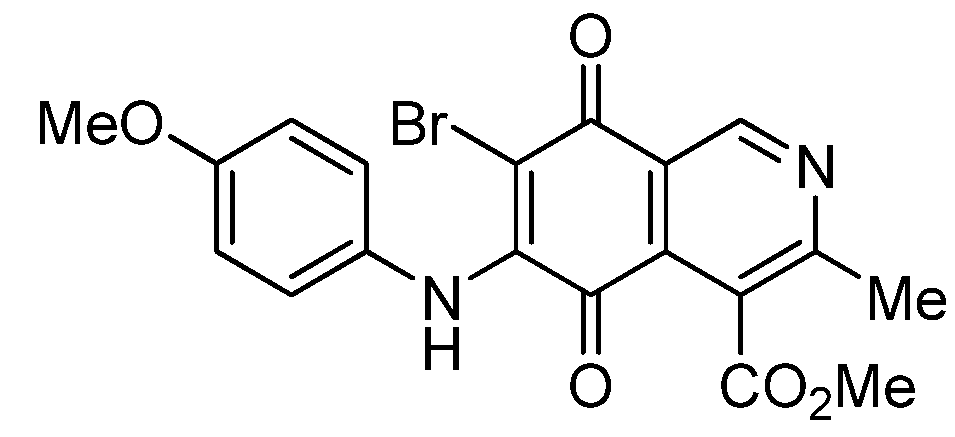 |
| 2a | 5a (87) | 3b | 6b (78) |
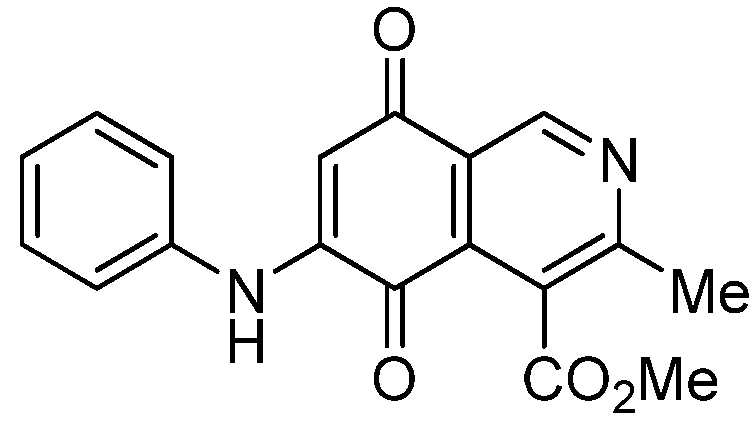 |  | 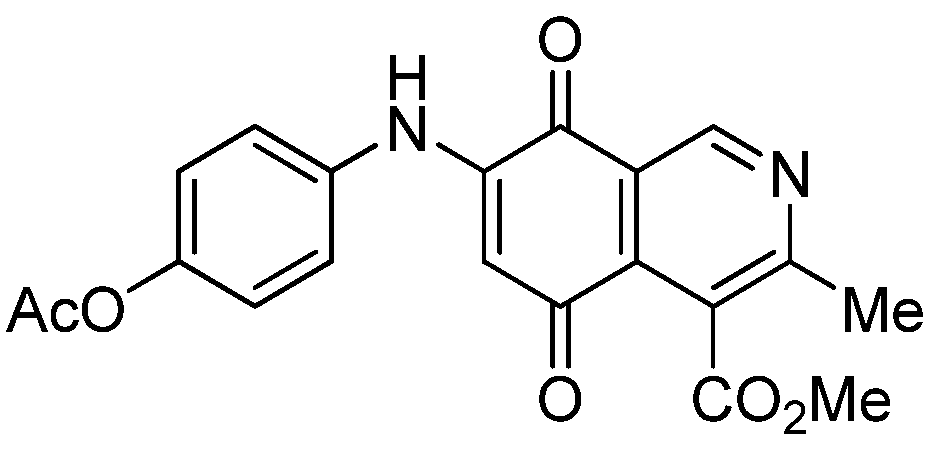 | 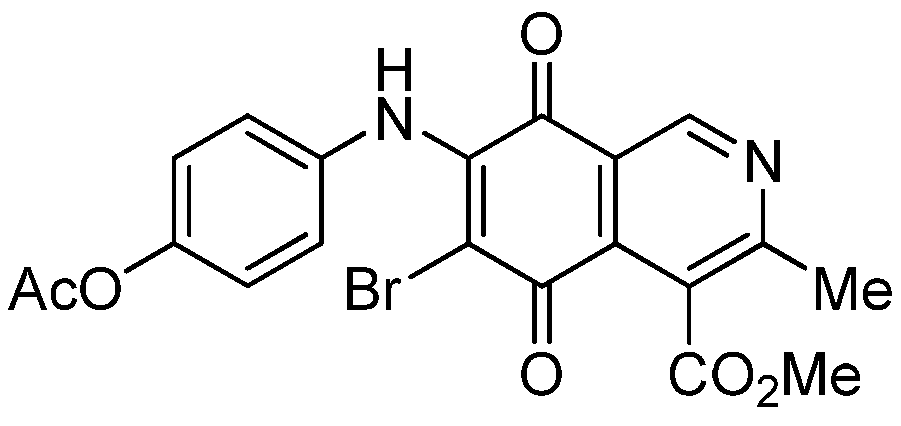 |
| 2b | 5b (74) | 7a | 8 (56) |
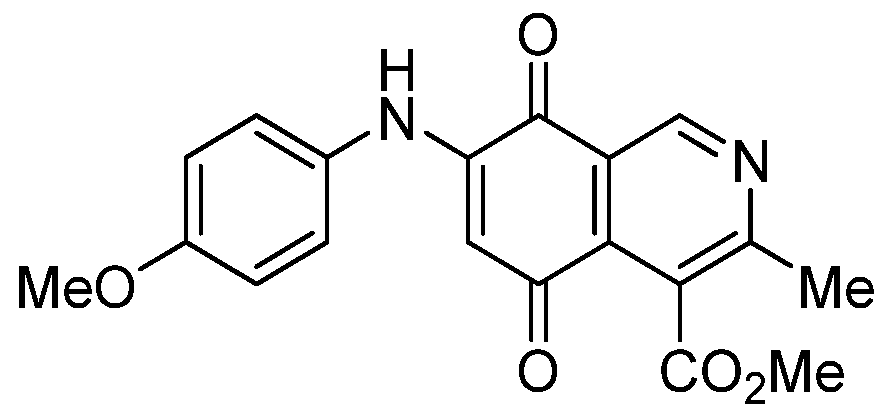 |  |  |  |
| 3a | 6a (80) | 8 | 9 (64) |
2.2. Electrochemical Results
| Compound N° | −EI1/2 (mV) | −EII1/2 (mV) | logP a | 6 or 7-H b |
|---|---|---|---|---|
| 1 | 352 | 1140 | 0.10 | 7.04 |
| 2a | 563 | 874 | 0.74 | 6.39 |
| 2b | 494 | 1035 | 0.74 | 6.38 |
| 3a | 551 | 1083 | 0.61 | 6.21 |
| 3b | 482 | 1040 | 0.61 | 6.20 |
| 4a | 555 | 869 | 0.35 | 6.20 |
| 4b | 525 | 881 | 0.35 | 6.18 |
| 5a | 373 | 932 | 1.05 | - |
| 5b | 518 | 906 | 1.05 | - |
| 6a | 426 | 795 | 0.92 | - |
| 6b | 373 | 953 | 0.92 | - |
| 7a | 521 | 1058 | 0.32 | 6.34 |
| 7b | 544 | 711 | 0.32 | 6.34 |
| 8 | 386 | 944 | 0.63 | - |
| 9 | 431 | 777 | 0.66 | - |
2.3. In Vitro Antiproliferative Activity of Phenylaminoisoquinolinequinones again Cancer Cell Lines
| IC50 ± SEM a (μM) | |||||
|---|---|---|---|---|---|
| N° | Structure | AGS b | SK-MES1 c | J82 d | −EI1/2 (eV) |
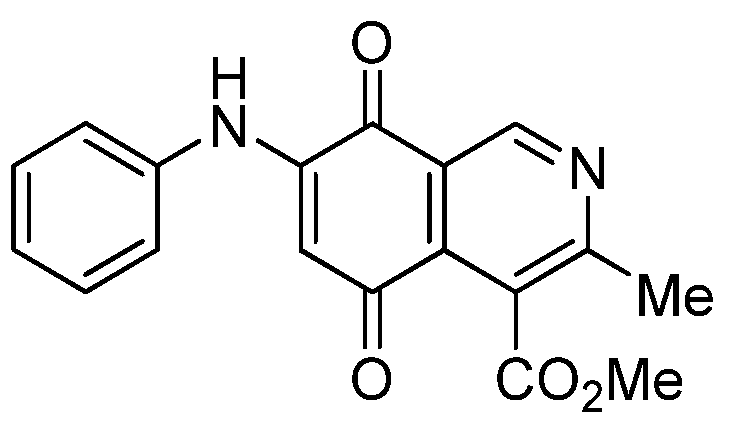
| 1 | 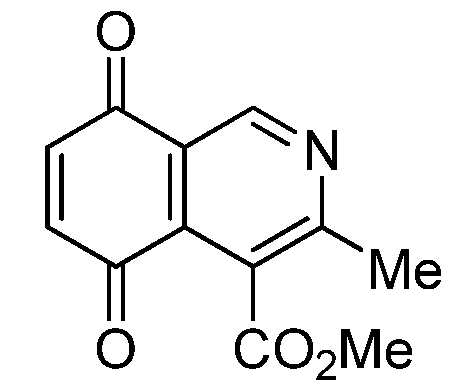 | 17.34 ± 1.64 | 25.90 ± 1.60 | 14.81 ± 0.74 | 352 |
| 2a |  | 1.10 ± 0.03 | 4.40 ± 0.09 | 1.50 ± 0.70 | 563 |
| 2b | 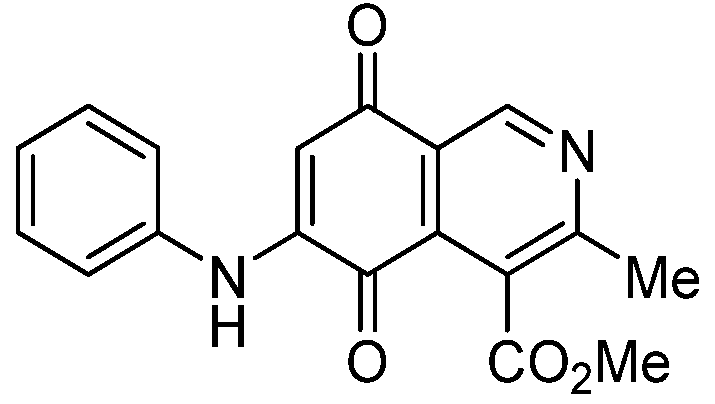 | 0.57 ± 0.20 | 2.60 ± 0.30 | 1.60 ± 0.70 | 494 |
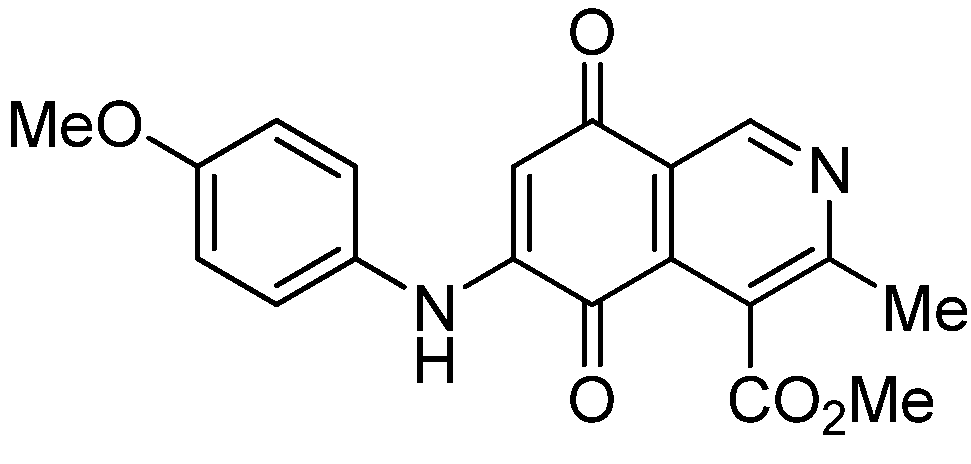
| 3a | 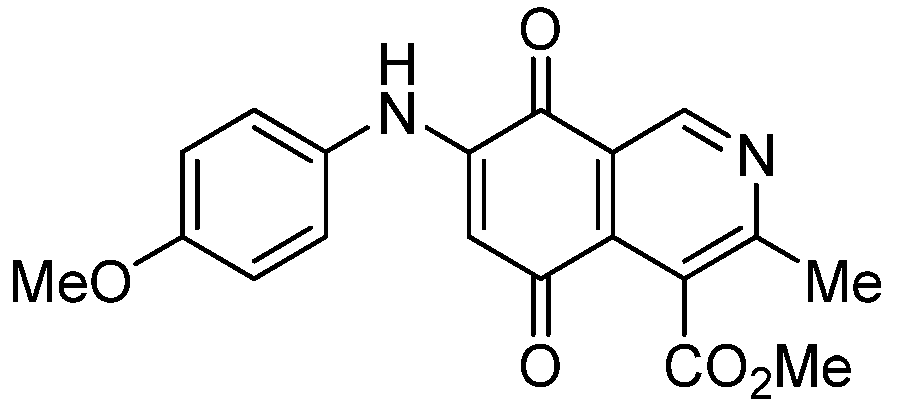 | 1.10 ± 0.10 | 3.40 ± 0.60 | 3.80 ± 0.70 | 551 |
| 3b | 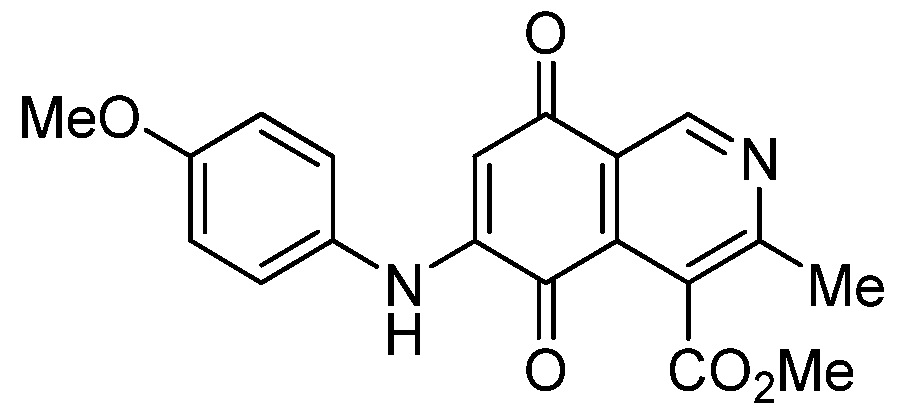 | 0.32 ± 0.04 | 1.10 ± 0.09 | 1.90 ± 0.2 0 | 482 |
| 4a |  | 0.72 ± 0.19 | 2.40 ± 0.12 | 2.06 ± 0.10 | 555 |
| 4b |  | 0.24 ± 0.01 | 0.89 ± 0.05 | 0.76 ± 0.04 | 525 |
| 7a | 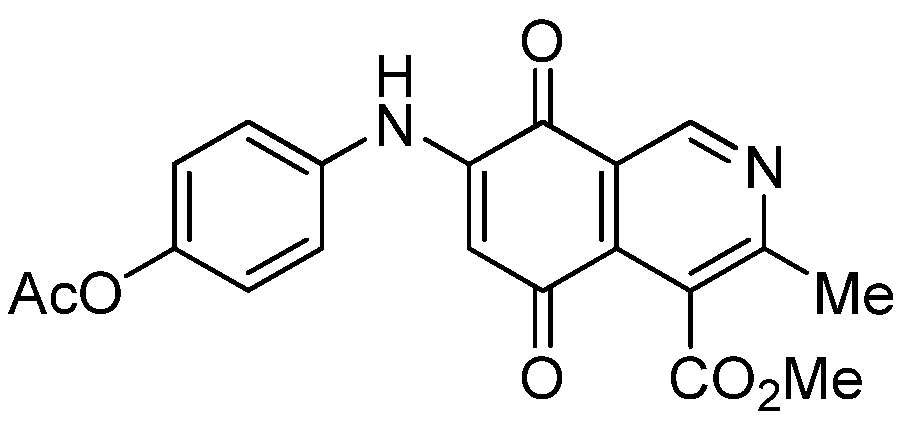 | 1.01 ± 0.04 | 0.98 ± 0.05 | 0.72 ± 0.19 | 521 |
| 7b |  | 0.29 ± 0.01 | 0.53 ± 0.04 | 1.34 ± 0.05 | 544 |
| 5a | 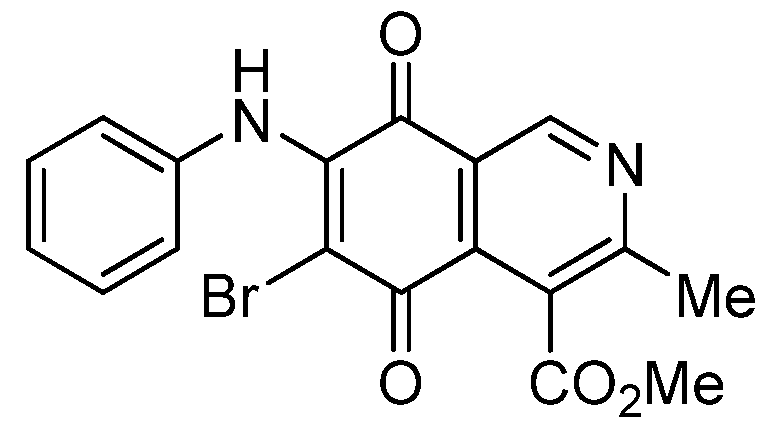 | 0.70 ± 0.04 | 0.31 ± 0.01 | 2.30 ± 0.10 | 373 |
| 5b | 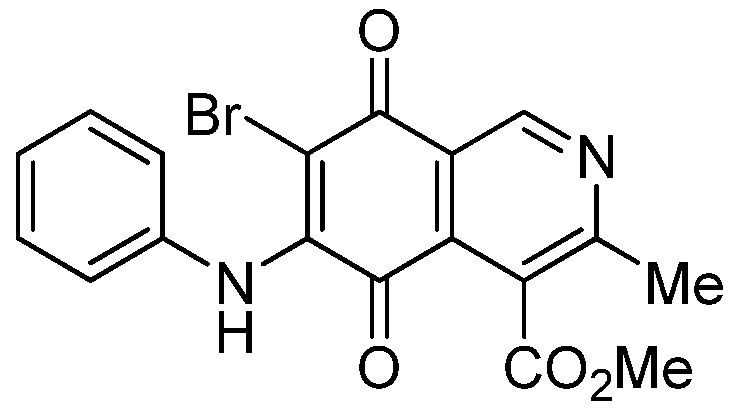 | 1.84 ± 0.14 | 4.72 ± 3.23 | 4.49 ± 0.31 | 518 |
| 6a |  | 1.11 ± 0.06 | 1.07 ± 0.04 | 9.72 ± 0.58 | 426 |
| 6b | 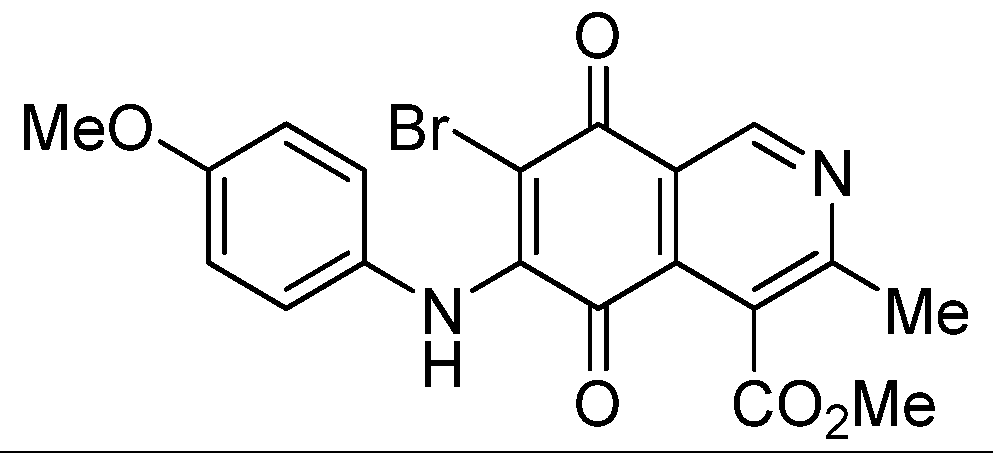 | 1.80 ± 0.09 | 1.79 ± 0.11 | 1.98 ± 0.13 | 373 |
| 8 | 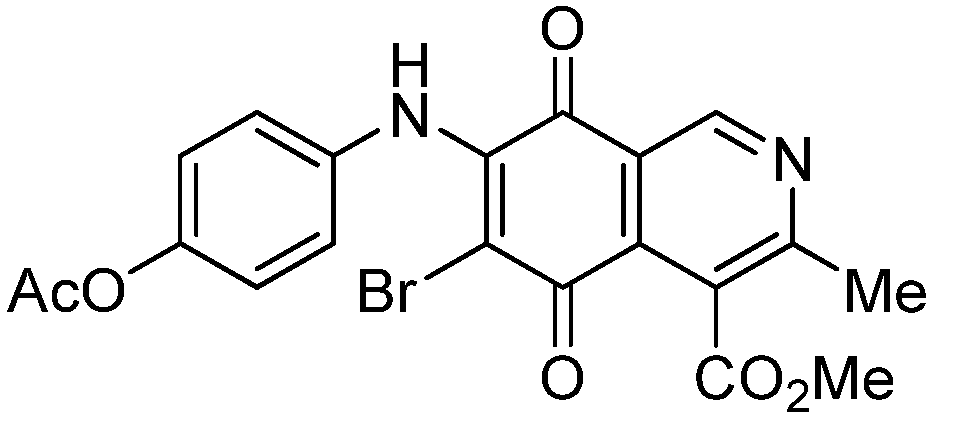 | 1.73 ± 0.12 | 3.91 ± 0.27 | 3.34 ± 0.23 | 386 |
| 9 |  | 1.10 ± 0.06 | 2.87 ± 0.14 | 2.35 ± 0.19 | 431 |
| - | etoposide | 0.58 ± 0.02 | 1.83 ± 0.09 | 3.49 ± 0.16 | - |
3. Experimental
3.1. General
3.2. Chemistry
3.3. General Procedure for the Synthesis of 6 and 7-Aminoisoquinolinequinone Derivatives
3.4. General Procedure for the Synthesis of 6- and 7-Bromo Derivatives
3.5. Antiproliferative Assay
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compounds are available from the authors.
References
- Rao, K.V.; Biemann, K.; Woodward, R.B. The structure of streptonigrin. J. Am. Chem. Soc. 1963, 85, 2532–2533. [Google Scholar] [CrossRef]
- Gould, S.J.; Weimeb, S.M. Streptonigrin. Fortschr. Chem. Org. Naturst. 1982, 41, 77–111. [Google Scholar]
- Balitz, D.M.; Bush, J.A.; Bradner, W.T.; Doyle, T.W.; O’Herron, F.A.; Nettleton, D.E. Isolation of Lavendamycin, a New Antibiotic from Streptomyces Lavendulae. J. Antibiot. (Tokyo) 1982, 35, 259–265. [Google Scholar]
- Doyle, T.W.; Balitz, D.M.; Grulich, R.E.; Nettleton, D.E.; Gould, S.J.; Tann, C.H.; Mews, A.E. Structure Determination of Lavendamycin—A New Antitumor Antibiotic from Streptomyces Lavendulae. Tetrahedron Lett. 1981, 22, 4595–4598. [Google Scholar] [CrossRef]
- Pettit, G.R.; Knight, J.C.; Collins, J.C.; Herald, D.L.; Pettit, R.K.; Boyd, M.R.; Young, V.G. Antineoplastic Agents 430. Isolation and Structure of Cribrostatins 3, 4, and 5 from the Republic of Maldives Cribrochalina Species. J. Nat. Prod. 2000, 63, 793–798. [Google Scholar]
- Milanowski, D.J.; Gustafson, K.R.; Kelley, J.A.; McMahon, J.B. Caulibugulones, A–F. Novel antiproliferative isoquinoline quinones and iminoquinones from the marine bryozoan caulibugula intermis. J. Nat. Prod. 2004, 67, 70–73. [Google Scholar] [CrossRef]
- Hawas, U.W.; Shaaban, M.; Shaaban, K.A.; Speitling, M.; Maier, A.; Kelter, G.; Fiebig, H.H.; Meiners, M.; Helmke, E.; Laatsch, H. Mansouramycins A–D, cytotoxic isoquinolinequinones from a marine streptomycete. J. Nat. Prod. 2009, 72, 2120–2124. [Google Scholar] [CrossRef]
- Boger, D.L.; Yasuda, M.; Mitscher, L.A.; Drake, S.D.; Kitos, P.A.; Thompson, S.C. Streptonigrin and lavendamycin partial structures. Probes for the minimum, potent pharmacophore of streptonigrin, lavendamycin, and synthetic quinoline-5,8-diones. J. Med. Chem. 1987, 30, 1918–1928. [Google Scholar]
- Fang, Y.; Linardic, C.M.; Richardson, D.A.; Cai, W.; Behforouz, M.; Abraham, R.T. Characterization of the cytotoxic activities of novel analogues of the antitumor agent, lavendamycin. Mol. Cancer Ther. 2003, 2, 517–526. [Google Scholar] [CrossRef]
- Hassani, M.; Cai, W.; Holley, D.C.; Lineswala, J.P.; Maharjan, B.R.; Ebrahimian, G.R.; Seradj, H.; Stocksdale, M.G.; Mohammadi, F.; Marvin, C.C.; et al. Novel Lavendamycin Analogues as Antitumor Agents: Synthesis, in Vitro Cytotoxicity, Structure-Metabolism, and Computational Molecular Modeling Studies with NAD(P)H: Quinone Oxidoreductase. J. Med. Chem. 2005, 48, 7733–7749. [Google Scholar]
- Verniest, G.; Wang, X.; de Kimpe, N.; Padwa, A. Heteroaryl Cross-Coupling as an Entry toward the Synthesis of Lavendamycin Analogues: A Model Study. J. Org. Chem. 2010, 75, 424–433. [Google Scholar] [CrossRef]
- Delgado, V.; Ibacache, A.; Theoduloz, C.; Valderrama, J.A. Synthesis and in vitro Cytotoxic Evaluation of Aminoquinones Structurally Related to Marine Isoquinolinequinones. Molecules 2012, 17, 7042–7056. [Google Scholar] [CrossRef]
- Boyd, V.A.; Sulikowski, G.A. Total Synthesis of the Angucycline AntibioticsUrdamycinone B and 104-2 via a Common Synthetic Intermediate. J. Am. Chem. Soc. 1995, 117, 8472–8473. [Google Scholar] [CrossRef]
- Kim, D.W.; Choi, H.Y.; Lee, K.-J.; Chi, D.Y. Facile Oxidation of fused 1,4-dimethoxybenzenes to 1,4-quinones using NBS: Fine-tuned Control over Bromination and Oxidation Reactions. Org. Lett. 2001, 3, 445–447. [Google Scholar] [CrossRef]
- Bukhtoyarova, D.; Ektova, L.V.; Alekseev, S.N.; Beregovaya, I.V. Synthesis and Spectral Properties of N-Aryl-5-hydroxy-1,4-naphthoquinone 4-Imines. Russ. J. Org. Chem. 2003, 39, 1309–1315. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Ibacache, J.A.; Arancibia, V.; Rodriguez, J.; Theoduloz, C. Studies on quinones. Part 45: Novel 7-aminoisoquinoline-5,8-quinone derivatives with antitumor properties on cancer cell lines. Bioorg. Med. Chem. 2009, 17, 2894–2901. [Google Scholar]
- Aguilar-Martinez, M.; Cuevas, G.; Jimenez-Estrada, M.; González, I.; Lotina-Hennsen, B.; Macias-Ruvalcaba, N. An Experimental and Theoretical Study of the Substituent Effectson the Redox Properties of2-[(R-phenyl)amine]-1,4-naphthalenediones in Acetonitrile. J. Org. Chem. 1999, 64, 3684–3694. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar]
- Van de Loosdrecht, A.A.; Beelen, R.H.; Ossenkoppele, G.J.; Broekhoven, M.G.; Langenhuijsen, M.M. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods 1994, 174, 311–320. [Google Scholar] [CrossRef]
- Scudiero, D.A.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Delgado, V.; Ibacache, A.; Arancibia, V.; Theoduloz, C.; Valderrama, J.A. Synthesis and in Vitro Antiproliferative Activity of New Phenylaminoisoquinolinequinones against Cancer Cell Lines. Molecules 2013, 18, 721-734. https://doi.org/10.3390/molecules18010721
Delgado V, Ibacache A, Arancibia V, Theoduloz C, Valderrama JA. Synthesis and in Vitro Antiproliferative Activity of New Phenylaminoisoquinolinequinones against Cancer Cell Lines. Molecules. 2013; 18(1):721-734. https://doi.org/10.3390/molecules18010721
Chicago/Turabian StyleDelgado, Virginia, Andrea Ibacache, Verónica Arancibia, Cristina Theoduloz, and Jaime A. Valderrama. 2013. "Synthesis and in Vitro Antiproliferative Activity of New Phenylaminoisoquinolinequinones against Cancer Cell Lines" Molecules 18, no. 1: 721-734. https://doi.org/10.3390/molecules18010721