Xanthene and Xanthone Derivatives as G-Quadruplex Stabilizing Ligands

Abstract
:1. Introduction


2. Results and Discussion
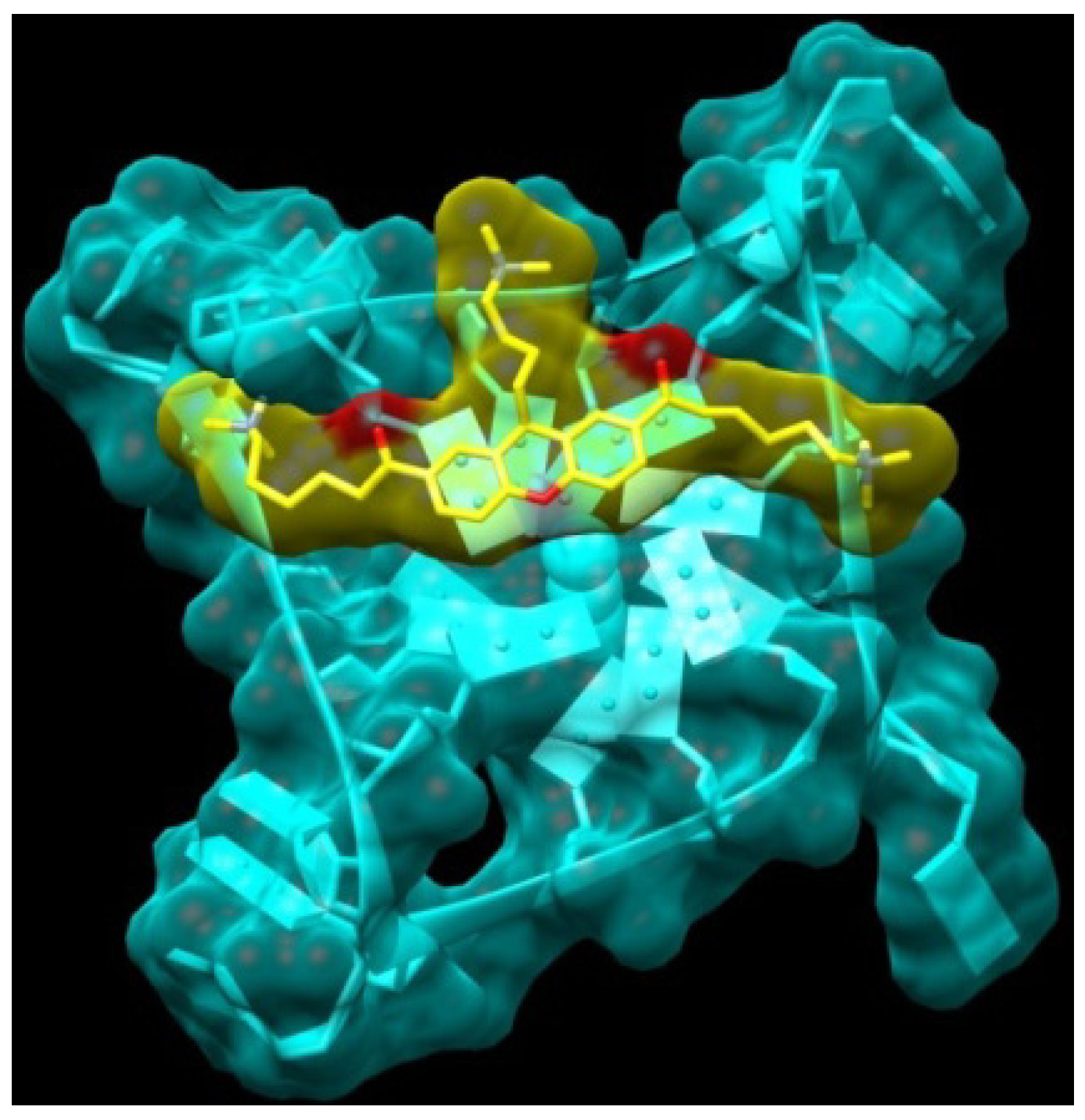
2.1. Design and Molecular Docking

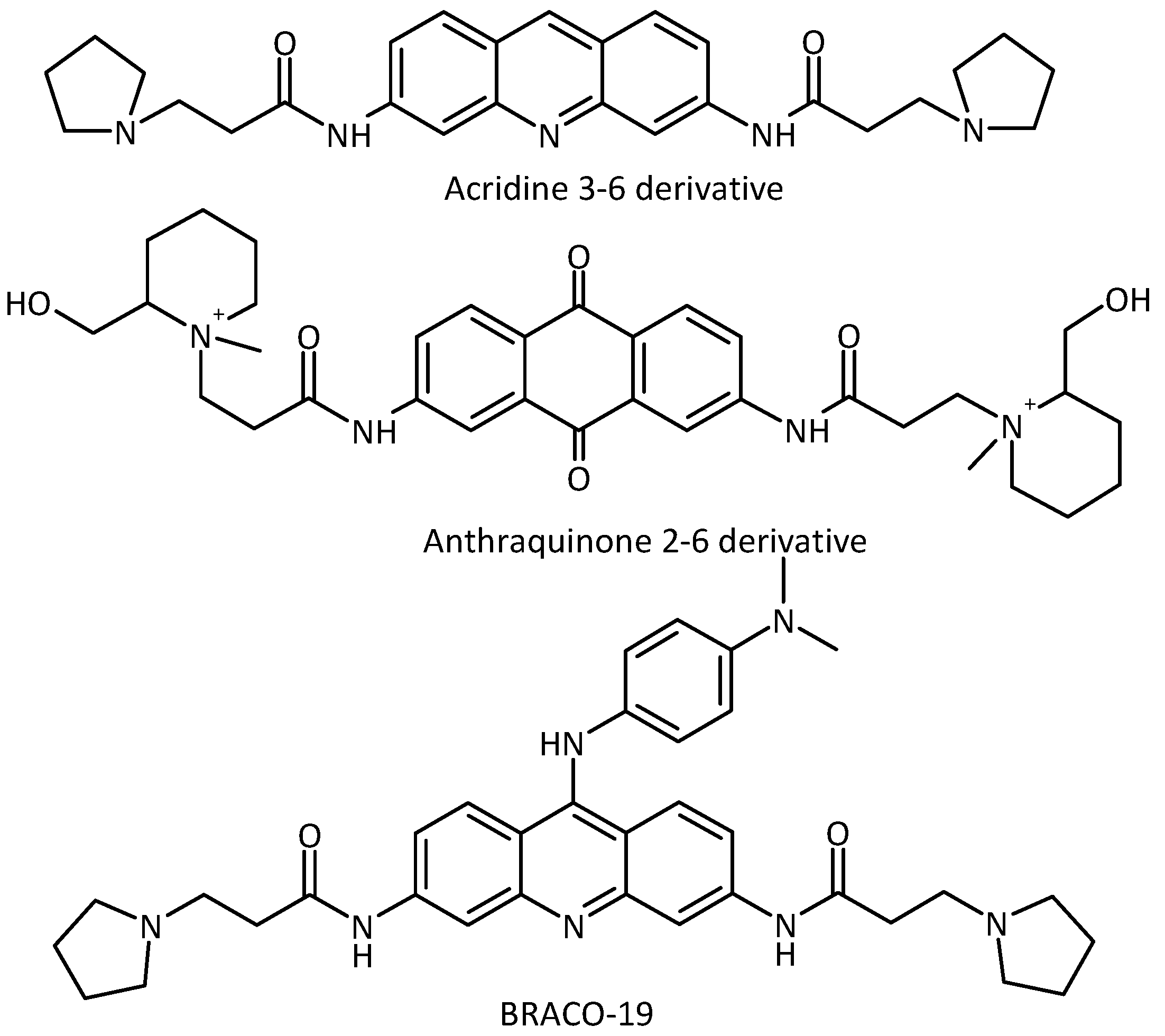{kind=link}
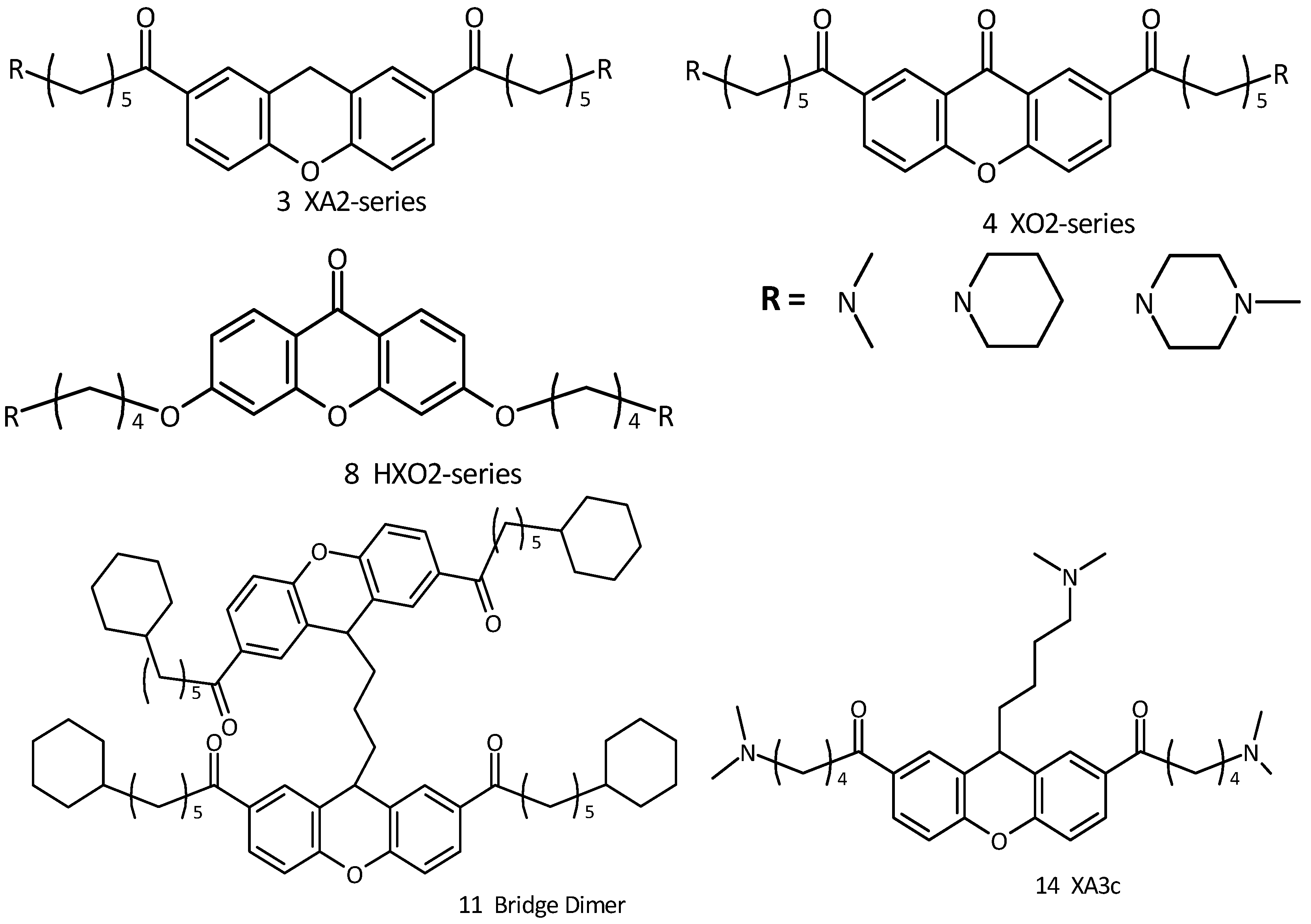{kind=link}
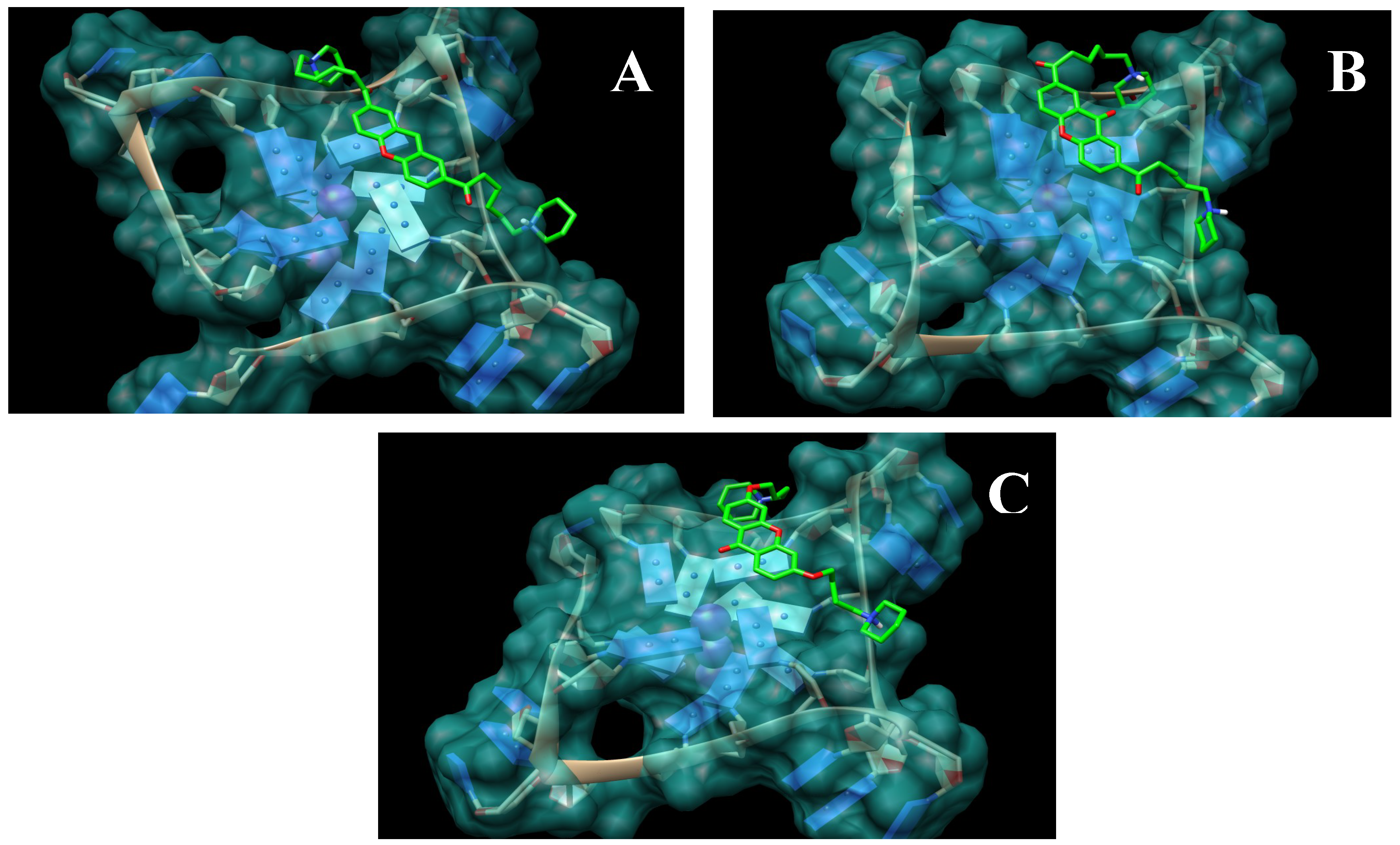{kind=link}
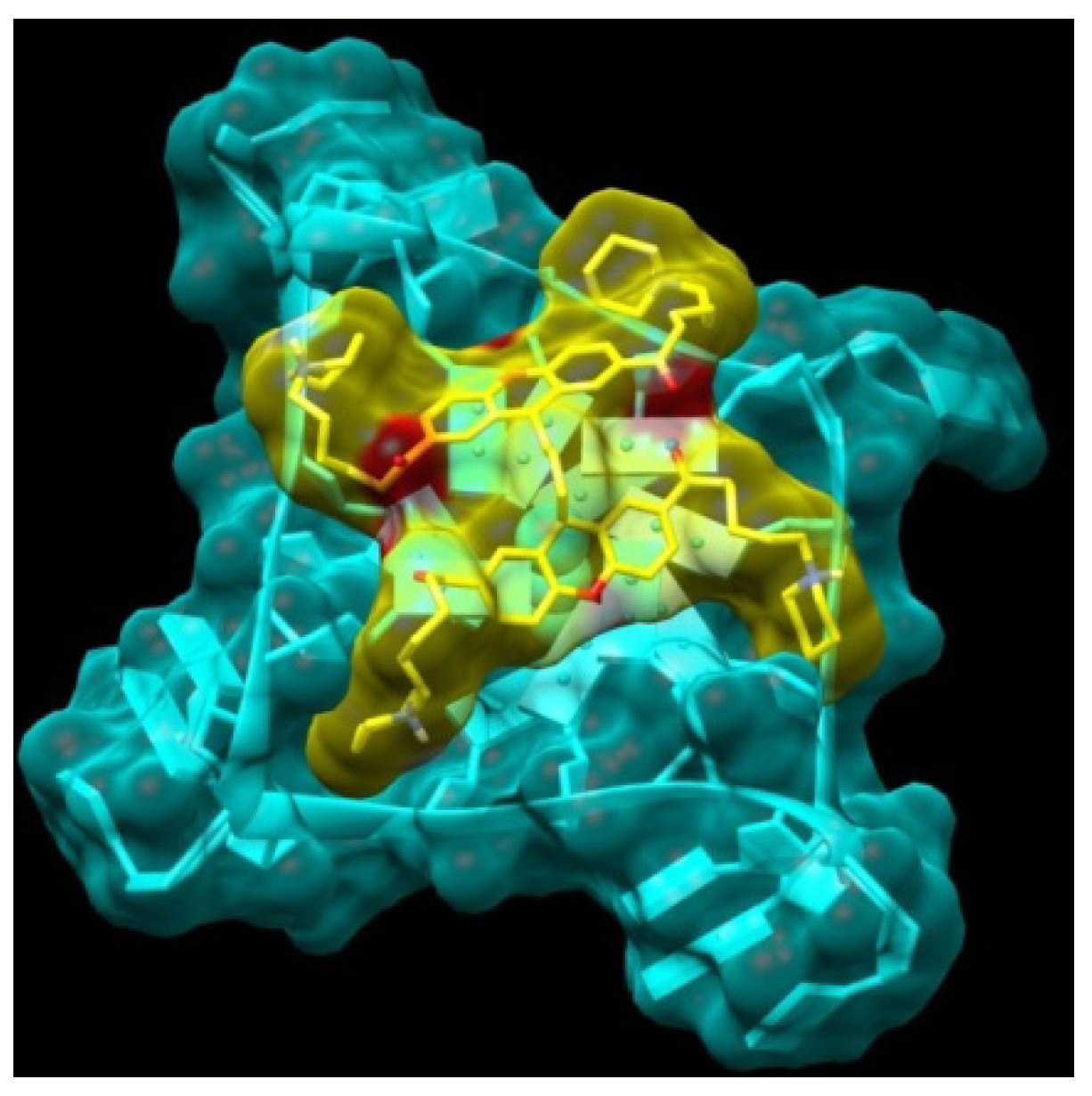{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Binding Energy |
|---|---|
| XA2dma 3a | −4.6 |
| XA2pip 3b | −4.4 |
| XA2mpz 3c | −4.5 |
| XO2dma 4a | −5.3 |
| XO2pip 4b | −5.0 |
| XO2mpz 4c | −5.4 |
| HXO2dma 8a | −5.8 |
| XO2pip 8b | −5.5 |
| XO2mpz 8c | −5.5 |
| Bridge Dimer 11 | −7.0 |
| XA3c 14 | −6.4 |
| Anthraquinone 2,6 derivative | −4.1 |
| Acridine 2-6 derivative | −4.6 |
| BRACO-19 | −7.0 |
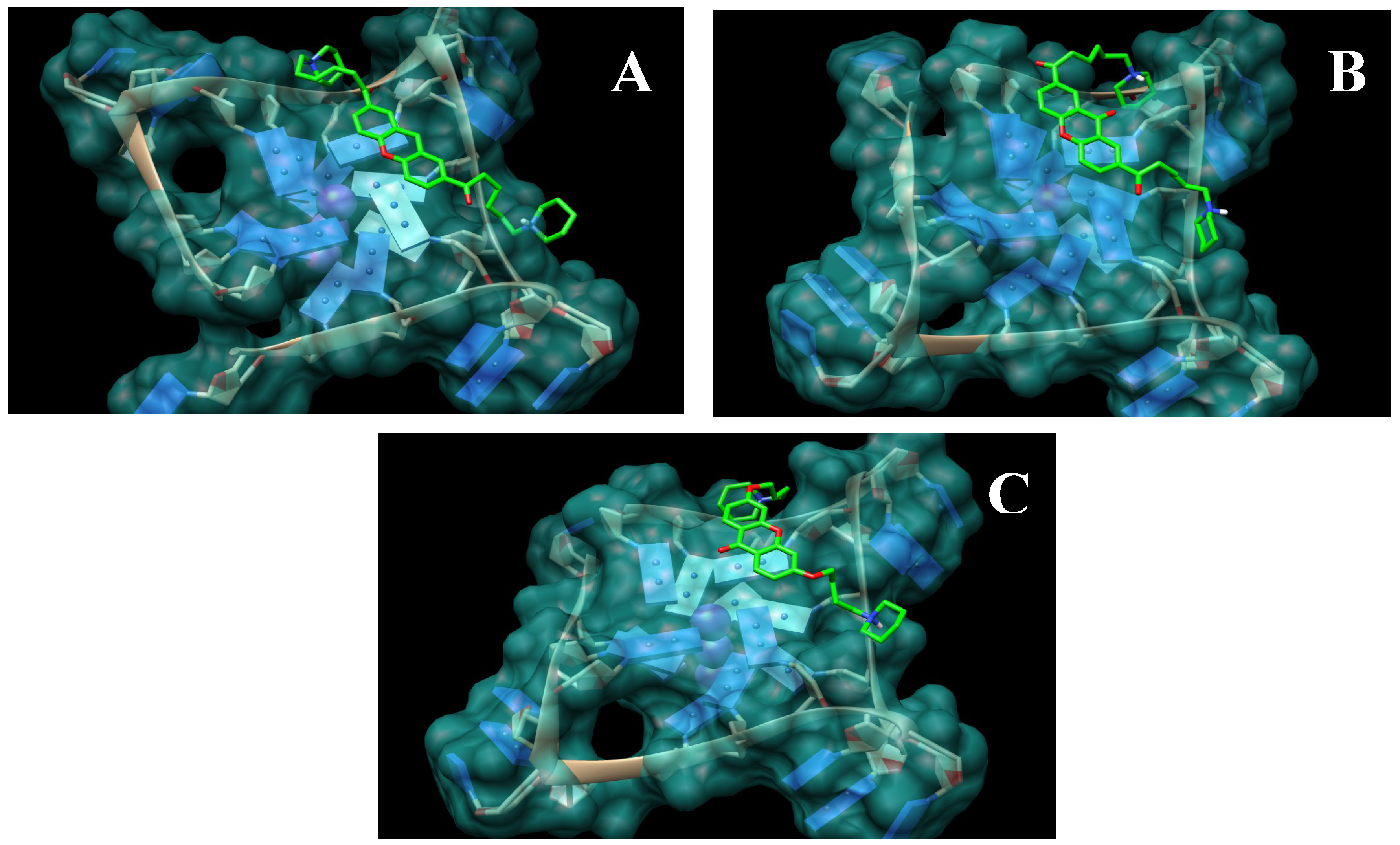


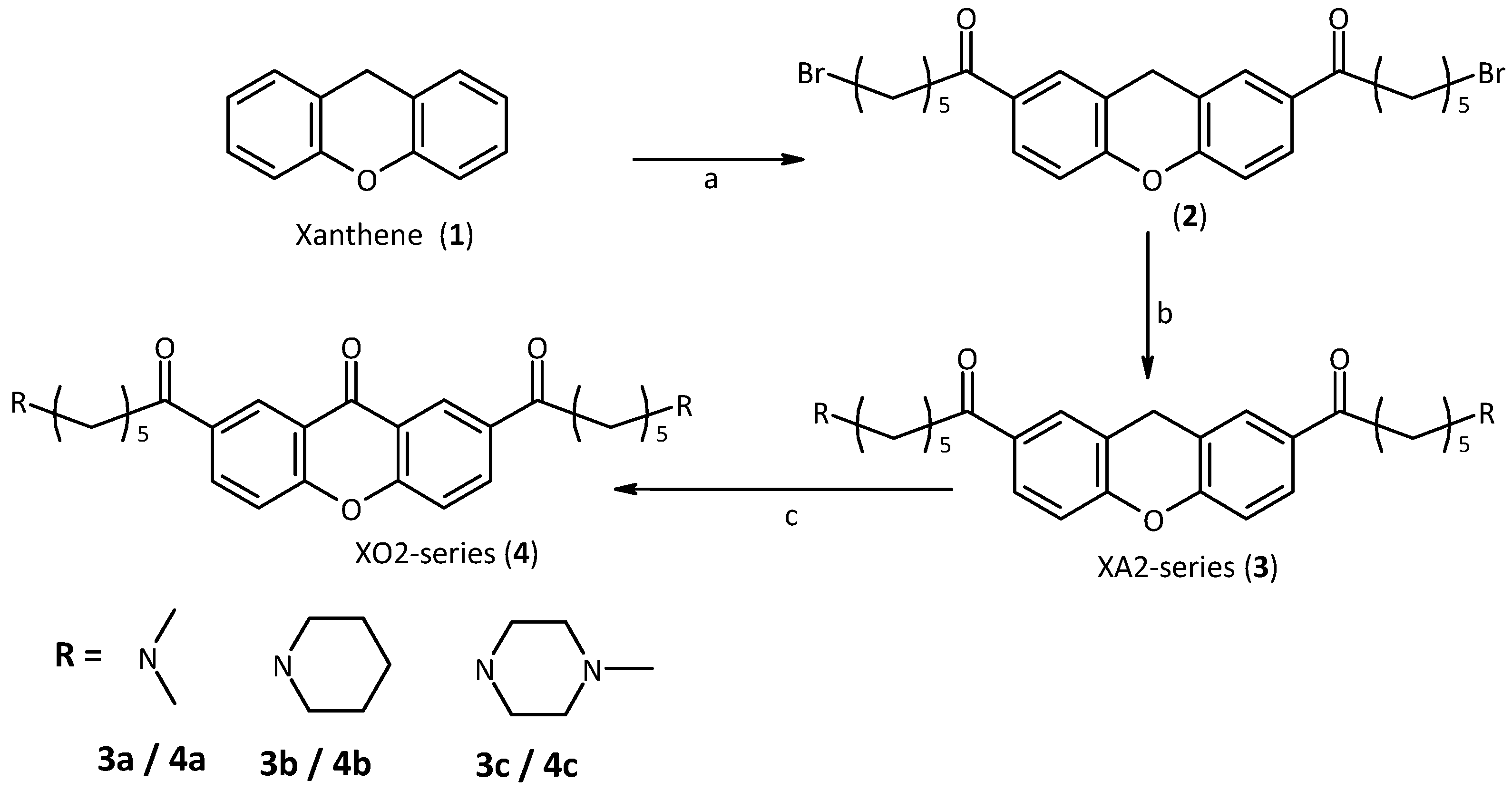
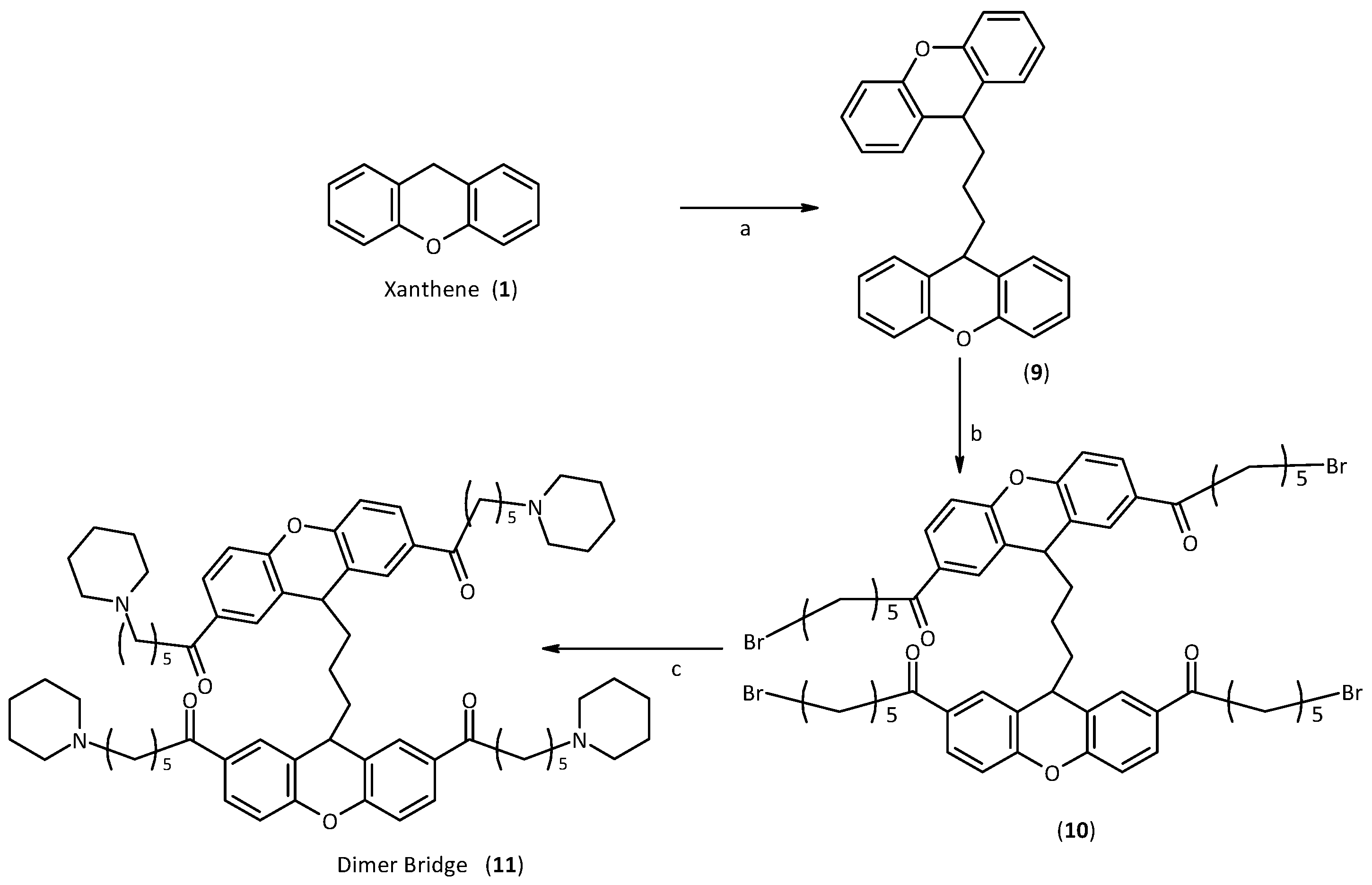
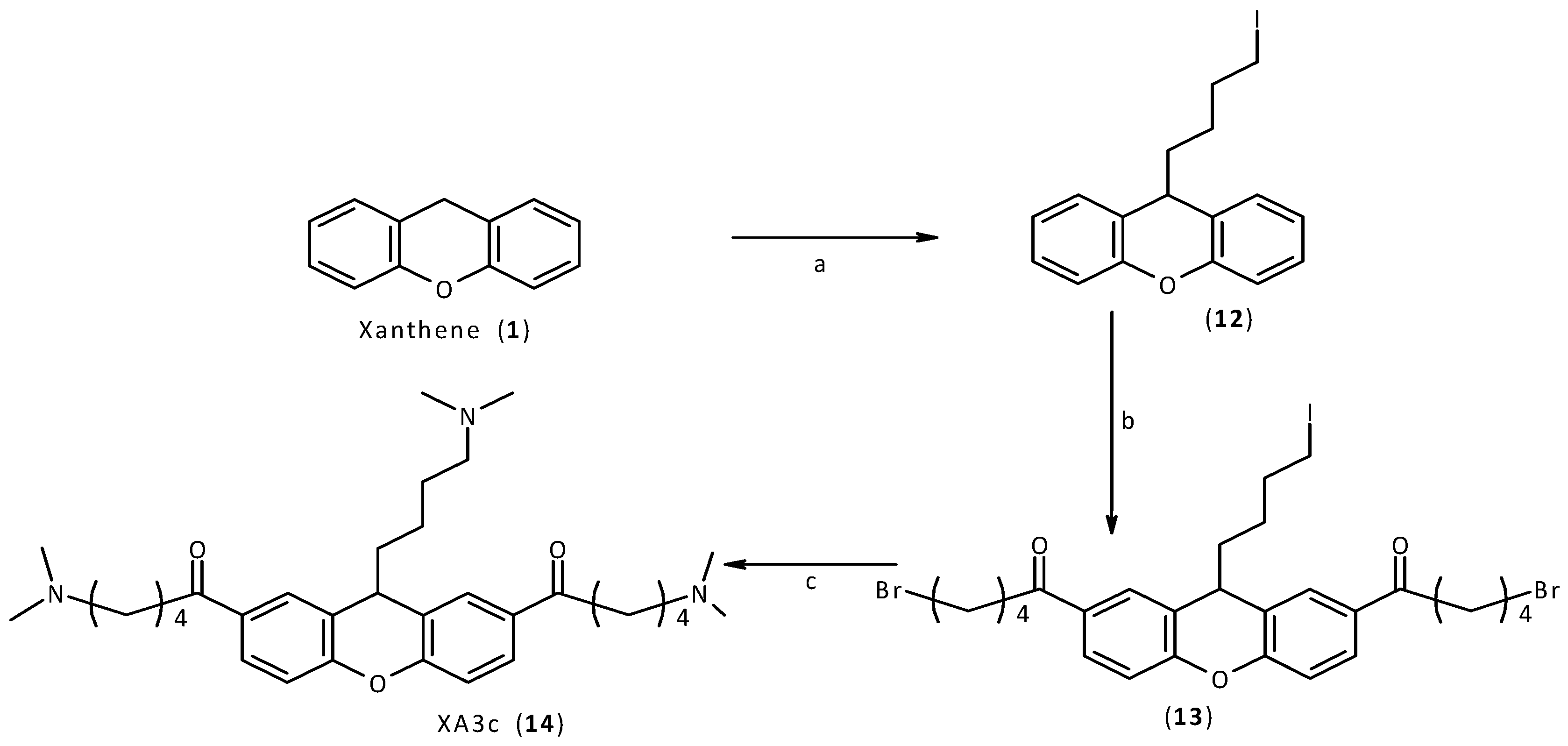
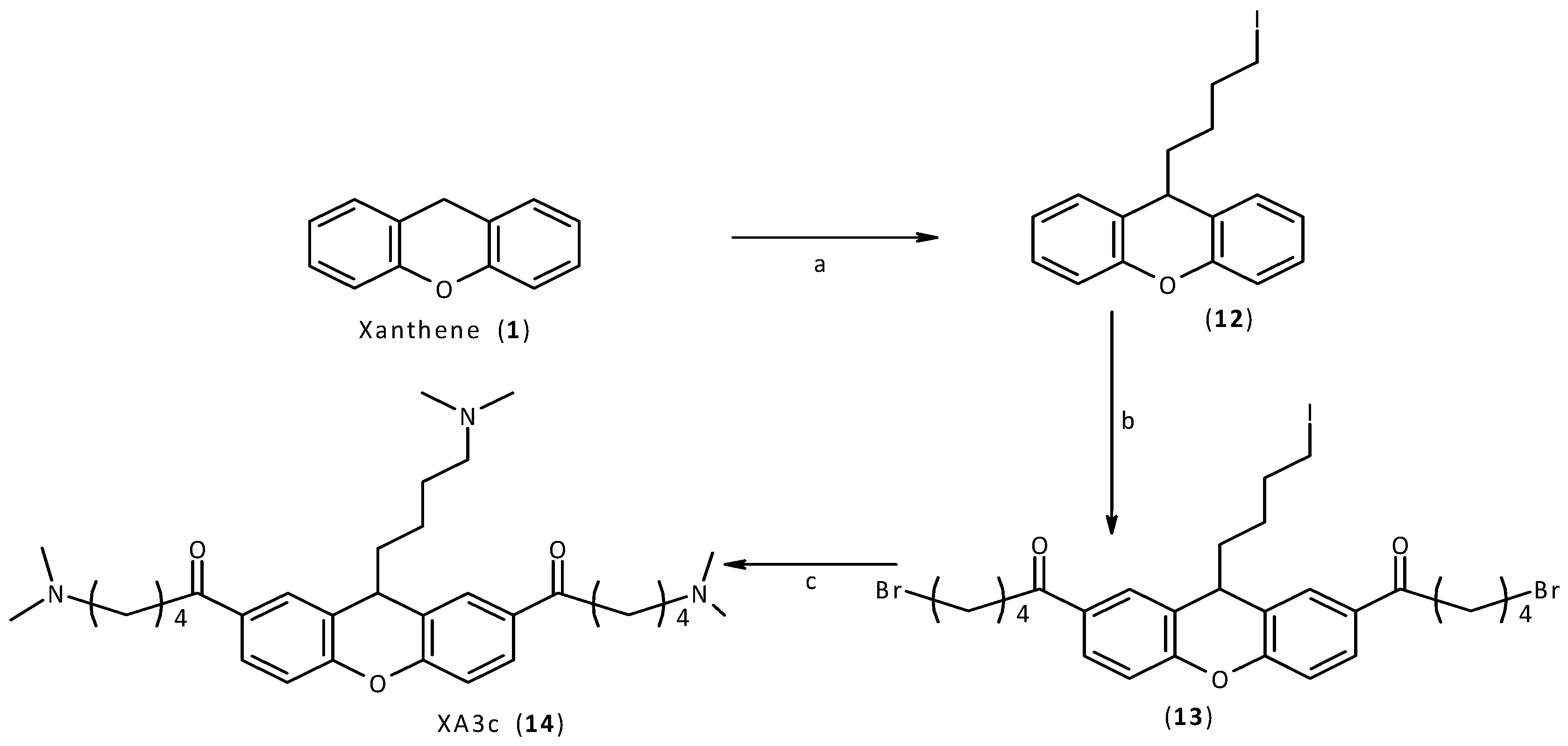
2.2. Synthesis



2.3. Studies of Ligands Interactions with G-Quadruplex and Duplex DNA by ESI-MS Experiments
| Compound | HTelo-21 | DK66 | |||||
|---|---|---|---|---|---|---|---|
| Log K1 | Log K2 | DNA bound 1:1 | DNA bound 1:2 | Log K1 | Log K2 | DNA bound 1:4 | |
| XA2dma (3a) | 4.6 ± 0.1 | 4.2 ± 0.1 | 23 ± 3 | 40 ± 4 | 3.4 ± 0.2 | 1.9 ± 0.2 | 9 ± 5 |
| XA2pip (3b) | 4.7 ± 0.1 | 4.6 ± 0.1 | 24 ± 3 | 41 ± 5 | 3.8 ± 0.2 | 2.1 ± 0.3 | 11 ± 5 |
| XA2mpz (3c) | 4.4 ± 0.1 | 3.9 ± 0.2 | 21 ± 4 | 39 ± 5 | 3.7 ± 0.1 | 2.0 ± 0.2 | 11 ± 4 |
| XO2dma (4a) | 4.0 ± 0.1 | 3.8 ± 0.1 | 17 ± 4 | 32 ± 4 | 3.3 ± 0.2 | 1.9 ± 0.3 | 9 ± 5 |
| XO2pip (4b) | 4.4 ± 0.1 | 4.2± 0.2 | 20 ± 3 | 39 ± 5 | 3.7 ± 0.1 | 2.5 ± 0.2 | 11 ± 6 |
| XO2mpz (4c) | 4.1 ± 0.1 | 3.7 ± 0.2 | 17 ± 4 | 33 ± 5 | 1.8 ± 0.2 | 1.1 ± 0.3 | 6 ± 5 |
| HXO2dma (8a) | 4.7 ± 0.1 | 3.6 ± 0.1 | 23 ± 5 | 35 ± 5 | 3.6 ± 0.1 | 3.4 ± 0.1 | 14 ± 5 |
| HXO2pip (8b) | 4.7 ± 0.1 | 4.6 ± 0.1 | 25 ± 3 | 41 ± 5 | 3.4 ± 0.1 | ‒ | 12 ± 5 |
| HXO2mpz (8c) | 4.3 ± 0.1 | 3.7 ± 0.2 | 21 ± 5 | 33 ± 4 | 3.5 ± 0.1 | 1.1 ± 0.1 | 13 ± 5 |
| Bridge Dimer (11) | 5.8 ± 0.1 | 5.6 ± 0.2 | 47 ± 2 | 57 ± 2 | 3.9 ± 0.1 | 3.3 ± 0.1 | 16 ± 2 |
| XA3c (14) | 5.2 ± 0.4 | 4.9 ± 0.1 | 41 ± 3 | 50 ± 2 | 3.6 ± 0.3 | 2.9 ± 0.2 | 13 ± 6 |
| Parameter | [CT] = 0 | [HTelo21]:[CT] 1:1 | [HTelo21]:[CT] 1:2 |
|---|---|---|---|
| Amount bound | 47.2 ± 2 | 41.2 ± 3 | 34.8 ± 3 |
| N | 1 | 87.3 | 73.7 |
| Compound | bcl2 | myc2345 | ||||||
|---|---|---|---|---|---|---|---|---|
| LogK1 | LogK2 | DNA bound 1:1 | DNA bound 1:2 | LogK1 | LogK2 | DNA bound 1:1 | DNA bound 1:2 | |
| XA2pip (3b) | 3.1 ± 0.1 | 3.0 ± 0.3 | 12 ± 3 | 26 ± 4 | 4.1 ± 0.1 | 4.1 ± 0.1 | 17 ± 4 | 33 ± 4 |
| XO2pip (4b) | 2.0 ± 0.3 | 1.6 ± 0.2 | 5 ± 2 | 15 ± 5 | 2.2 ± 0.1 | 2.0 ± 0.2 | 8 ± 2 | 17 ± 5 |
| XO2pip (8b) | 3.4 ± 0.1 | 2.9 ± 0.2 | 13 ± 4 | 28 ± 4 | 3.9 ± 0.1 | 3.7 ± 0.3 | 16 ± 4 | 30 ± 4 |
| Dimer (11) | 4.1 ± 0.3 | 3.2 ± 0.3 | 17 ± 2 | 32 ± 2 | 5.0 ± 0.2 | 5.0 ± 0.2 | 39 ± 2 | 48 ± 2 |
| XA3c (14) | 5.0 ± 0.1 | 4.8 ± 0.1 | 40 ± 3 | 49 ± 3 | 5.1 ± 0.1 | 4.7 ± 0.2 | 40 ± 3 | 49 ± 3 |
2.4. FRET Assays of Xanthene and Xanthone Derivatives on Quadruplex and Duplex DNA
| Compound | F21T | BCL2 | CKIT1 | T Loop |
|---|---|---|---|---|
| XA2pip (3b) | 1.3 | 0.7 | 1.0 | 0.4 |
| XO2pip (4b) | 2.4 | 0.8 | 1.9 | 1.3 |
| HXO2pip (8b) | 5.1 | 3.3 | 4.1 | 1.2 |
| XA3c (14) | 11.9 | 4.8 | 7.9 | 2.2 |
3. Experimental
3.1. General
3.2. Molecular Modelling
3.3. Synthesis
3.3.1. General Procedure for Nucleophilic Substitution
3.3.2. General Procedure for Xanthene Oxidation
3.4. Analysis of the DNA-Drug Interactions by ESI-MS
3.5. Analysis of the DNA-Drug Interactions by FRET
3.5.1. Sequence Preparation
- HTelo-21: 5’-FAM-GGGTTAGGGTTAGGGTTAGGG-TAMRA-3’
- c-kit-1: 5’-FAM-AGAGGGAGGGCGCTGGGAGGAGGGGCT-TAMRA-3’
- bcl2: 5’-FAM-5'-GGGCGCGGGAGGAAGGGGGCGGG-3'-TAMRA-3’
- T loop: 5’-FAM-TATAGCTATA-HEG-TATAGCTATA-TAMRA-3’
3.5.2. Sample Preparation and Measurement
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sun, D.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.M.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of human telomerase by a G-quadruplex interactive compound. J. Med. Chem. 1997, 40, 2113–2116. [Google Scholar] [CrossRef]
- Franceschin, M. G-quadruplex DNA structures and organic chemistry: More than one connection. Eur. J. Org. Chem. 2009, 2009, 2225–2238. [Google Scholar] [CrossRef]
- Biroccio, A.; Porru, M.; Rizzo, A.; Salvati, E.; D’Angelo, C.; Orlandi, A.; Passeri, D.; Franceschin, M.; Stevens, M.; Gilson, E.; et al. DNA damage persistence as determinant of tumor sensitivity to the combination of Topo I inhibitors and telomere-targeting agents. Clin. Cancer Res. 2011, 17, 2227–2236. [Google Scholar] [CrossRef]
- D’Ambrosio, D.; Reichenbach, P.; Micheli, E.; Alvino, A.; Franceschin, M.; Savino, M.; Lingner, J. Specific binding of telomeric G-quadruplexes by hydrosoluble perylene derivatives inhibits repeat addition processivity of human telomerase. Biochemie 2012, 94, 854–863. [Google Scholar] [CrossRef]
- Cummaro, A.; Fottichia, I.; Franceschin, M.; Giancola, C.; Petraccone, L. Binding properties of human telomeric quadruplex multimers: A new route for drug design. Biochemie 2011, 93, 1392–1400. [Google Scholar] [CrossRef]
- Micheli, E.; D’Ambrosio, D.; Franceschin, M.; Savino, M. Water soluble cationic perylene derivatives as possible telomerase inhibitors: The search for selective G-quadruplex targeting. Mini-Rev. Med. Chem. 2009, 9, 1622–1632. [Google Scholar] [CrossRef]
- Rossetti, L.; Franceschin, M.; Schirripa, S.; Bianco, A.; Ortaggi, G.; Savino, M. Selective interactions of perylene derivatives having different side chains with inter- and intramolecular G-quadruplex structures. A correlation with telomerase inhibition. Bioorg. Med. Chem. Lett. 2005, 15, 413–420. [Google Scholar] [CrossRef]
- Arola, A.; Ramon, V. Stabilisation of G-Quadruplex DNA by small molecules. Curr. Top. Med. Chem. 2008, 15, 1405–1415. [Google Scholar] [CrossRef]
- Perry, P.J.; Reszka, A.P.; Wood, A.A.; Read, M.A.; Gowan, S.M.; Dosanjh, H.S.; Trent, J.O.; Jenkins, T.C.; Kelland, L.R.; Neidle, S. Human telomerase inhibition by regioisomeric disubstituted amidoanthracene-9,10-diones. J. Med. Chem. 1998, 41, 4873–4884. [Google Scholar] [CrossRef]
- Perry, P.J.; Gowan, S.M.; Reszka, A.P.; Polucci, P.; Jenkins, T.C.; Kelland, L.R.; Neidle, S. 1,4- and 2,6-disubstituted amidoanthracene-9,10-dione derivatives as inhibitors of human telomerase. J. Med. Chem. 1998, 41, 3253–3260. [Google Scholar] [CrossRef]
- Harrison, R.J.; Gowan, S.M.; Kelland, L.R.; Neidle, S. Human telomerase inhibition by substituted acridine derivatives. Bioorg. Med. Chem. Lett. 1999, 9, 2463–2468. [Google Scholar] [CrossRef]
- Read, M.; Harrison, R.J.; Romagnoli, B.; Tanious, F.A.; Gowan, S.H.; Reszka, A.P.; Wilson, W.D.; Kelland, L.R.; Neidle, S. Structure-based design of selective and potent G-quadruplex-mediated telomerase inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 4844–4849. [Google Scholar] [CrossRef]
- Reed, J.; Gunaratnam, M.; Beltran, M.; Reszka, A.P.; Vilar, R.; Neidle, S. TRAP-LIG, a modified telomere repeat amplification protocol assay to quantitate telomerase inhibition by small molecules. Anal. Biochem. 2008, 390, 99–105. [Google Scholar]
- Gutierrez-Orozco, F.; Failla, M.L. Biological activities and bioavailability of mangosteen xanthones: A critical review of the current evidence. Nutrients 2013, 13, 3163–3183. [Google Scholar] [CrossRef]
- Al-Massarani, S.M.; El Gamal, A.A.; Al-Musayeib, N.M.; Mothana, R.A.; Basudan, O.A.; Al-Rehaily, A.J.; Farag, M.; Assaf, M.H.; El Tahir, K.H.; Maes, L. Phytochemical, antimicrobial and antiprotozoal evaluation of garcinia mangostana pericarp and α-mangostin, its major xanthone derivative. Molecules 2013, 18, 10599–10608. [Google Scholar] [CrossRef]
- Ginnari-Satriani, L.; Casagrande, V.; Bianco, A.; Ortaggi, G.; Franceschin, M. A hydrophilic three side-chained triazatruxene as a new strong and selective G-quadruplex ligand. Org. Biomol. Chem. 2009, 7, 2513–2516. [Google Scholar] [CrossRef]
- Franceschin, M.; Rossetti, L.; D’Ambrosio, A.; Schirripa, S.; Bianco, A.; Ortaggi, G.; Savino, M.; Schultes, C.; Neidle, S. Natural and synthetic G-quadruplex interactive berberine derivatives. Bioorg. Med. Chem. Lett. 2006, 16, 1707–1711. [Google Scholar] [CrossRef]
- Franceschin, M.; Alvino, A.; Casagrande, V.; Mauriello, C.; Pascucci, E.; Savino, M.; Ortaggi, G.; Bianco, A. Specific interactions with intra- and intermolecular G-quadruplex DNA structures by hydrosoluble coronene derivatives: A new class of telomerase inhibitors. Bioorg. Med. Chem. 2007, 15, 1848–1858. [Google Scholar] [CrossRef]
- Jaumot, J.; Gargallo, R. Experimental Methods for studying the interactions between G-Quadruplex structures and ligands. Curr. Pharm. Des. 2012, 14, 1900–1916. [Google Scholar] [CrossRef]
- Murat, P.; Singh, Y.; Defrancq, E. Methods for investigating G-quadruplex DNA/ligand interactions. Chem. Soc. Rev. 2011, 40, 5293–5307. [Google Scholar] [CrossRef]
- Monchaud, D.; Teulade-Fichou, M.P. A hitchhiker’s guide to G-quadruplex ligands. Org. Biomol. Chem. 2008, 6, 627–636. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Cosconati, S.; Marinelli, L.; Trotta, R.; Virno, A.; Mayol, L.; Novellino, E.; Olson, A.J.; Randazzo, A. Tandem application of virtual screening and NMR experiments in the discovery of brand new DNA quadruplex groove binders. J. Am. Chem. Soc. 2009, 131, 16336–16337. [Google Scholar] [CrossRef]
- Lane, A.N.; Chaires, J.B.; Gray, R.D.; Trent, J.O. Stability and kinetics of G-quadruplex structures. Nucleic Acids. Res. 2008, 36, 5482–5515. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Lee, M.P.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef]
- Husby, J.; Todd, A.K.; Platts, J.A.; Neidle, S. Small-molecule G-quadruplex interactions: Systematic exploration of conformational space using multiple molecular dynamics. Biopolymers 2013, 99, 989–1005. [Google Scholar]
- Fedoroff, O.Y.; Salazar, M.; Han, H.; Chemeris, V.V.; Kerwin, S.M.; Hurley, L.H. NMR-Based model of a telomerase-inhibiting compound bound to G-quadruplex DNA. Biochemistry 1998, 37, 12367–12374. [Google Scholar] [CrossRef]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5414. [Google Scholar] [CrossRef]
- Franceschin, M.; Lombardo, C.M.; Pascucci, E.; D’Ambrosio, D.; Micheli, E.; Bianco, A.; Ortaggi, G.; Savino, M. The number and distances of positive charges of polyamine side chains in a series of perylene diimides significantly influence their ability to induce Gquadruplex structures and inhibit human telomerase. Bioorg. Med. Chem. 2008, 16, 2292–2304. [Google Scholar] [CrossRef]
- Micheli, E.; Lombardo, C.M.; D’Ambrosio, D.; Franceschin, M.; Neidle, S.; Savino, M. Selective G-quadruplex ligands: The significant role of side chain charge density in a series of perylene derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 3903–3908. [Google Scholar] [CrossRef]
- Franceschin, M.; Alvino, A.; Ortaggi, G.; Bianco, A. New hydrosoluble perylene and coronene derivatives. Tetrahedron Lett. 2004, 45, 9015–9020. [Google Scholar] [CrossRef]
- Franceschin, M.; Pascucci, E.; Alvino, A.; D’Ambrosio, D.; Bianco, A.; Ortaggi, G.; Savino, M. New highly hydrosoluble and not self-aggregated perylene derivatives with three and four polar side chains as G-quadruplex telomere targeting agents and telomerase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 2515–2522. [Google Scholar] [CrossRef]
- Kenmoku, S.; Urano, Y.; Kanda, K.; Kojima, H.; Kikuchi, K.; Nagano, T. Rational design of novel photoinduced electron transfer type fluorescent probes for sodium cation. Tetrahedron 2004, 60, 11067–11074. [Google Scholar] [CrossRef]
- Gabelica, V.; de Pauw, E.; Rosu, F. Interaction between antitumor drugs and a double-stranded oligonucleotide studied by electrospray ionization mass spectrometry. J. Mass Spectrom. 1999, 34, 1328–1337. [Google Scholar] [CrossRef]
- Gabelica, V.; Rosu, F.; Houssier, C.; de Pauw, E. Gas phase thermal denaturation of an oligonucleotide duplex and its complexes with minor groove binders. Rapid Comm. Mass Spectrom. 2000, 14, 464–467. [Google Scholar] [CrossRef]
- Rosu, F.; Gabelica, V.; Houssier, C.; Colson, P.; de Pauw, E. Triplex and quadruplex DNA structures studied by electrospray mass spectrometry. Rapid Comm. Mass Spectrom. 2002, 16, 1729–1736. [Google Scholar] [CrossRef]
- Rosu, F.; de Pauw, E.; Gabelica, V. Electrospray mass spectrometry to study drug-nucleic acids interactions. Biochemie 2008, 90, 1074–1087. [Google Scholar] [CrossRef]
- Baker, E.S.; Bernstein, S.L.; Gabelica, V.; de Pauw, E.; Bowers, M.T. G-quadruplexes in telomeric repeats are conserved in a solvent-free environment. Int. J. Mass. Spectrom. 2006, 253, 225–237. [Google Scholar] [CrossRef]
- Casagrande, V.; Alvino, A.; Bianco, A.; Ortaggi, G.; Franceschin, M. Study of binding affinity and selectivity of perylene and coronene derivatives towards duplex and quadruplex DNA by ESI-MS. J. Mass Spectrom. 2009, 4, 530–540. [Google Scholar]
- Altieri, A.; Franceschin, M.; Nocioni, D.; Alvino, A.; Casagrande, V.; Scarpati, M.L.; Bianco, A. Total synthesis of taspine and a symmetrical analogue: Study of binding to G-quadruplex DNA by ESI-MS. Eur. J. Org. Chem. 2013, 2013, 191–196. [Google Scholar] [CrossRef]
- Casagrande, V.; Salvati, E.; Alvino, A.; Bianco, A.; Ciammaichella, A.; D’Angelo, C.; Ginnari-Satriani, L.; Serrilli, A.M.; Iachettini, S.; Leonetti, C. N-cyclic bay-substituted perylene Gquadruplex ligands have selective antiproliferative effects on cancer cells and induce telomere damage. J. Med. Chem. 2011, 54, 1140–1156. [Google Scholar] [CrossRef]
- Franceschin, M.; Rizzo, A.; Casagrande, V.; Salvati, E.; Alvino, A.; Altieri, A.; Ciammaichella, A.; Iachettini, S.; Leonetti, C.; Ortaggi, G. Aromatic core extension in the series of N-cyclic bay-substituted perylene G-quadruplex ligands: Increased telomere damage, antitumor activity, and strong selectivity for neoplastic over healthy cells. Chem. Med. Chem. 2012, 7, 2144–2154. [Google Scholar]
- Guyen, B.; Schultes, C.M.; Hazel, P.; Mann, J.; Neidle, S. Synthesis and evaluation of analogues of 10H-indolo[3,2-b]quinoline as G-quadruplex stabilising ligands and potential inhibitors of the enzyme telomerase. Org. Biomol. Chem. 2004, 2, 981–988. [Google Scholar]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graphics Mod. 1999, 17, 57–61. [Google Scholar]
- Franceschin, M.; Borbone, N.; Oliviero, G.; Casagrande, V.; Scuotto, M.; Coppola, T.; Borioni, S.; Mayol, L.; Ortaggi, G.; Bianco, A.; et al. Synthesis of a dibromoperylene phosphoramidite building block and its incorporation at the 5' end of a G-quadruplex forming oligonucleotide: Spectroscopic properties and structuralstudies of the resulting dibromoperylene conjugate. Bioconjug Chem. 2011, 22, 1309–1319. [Google Scholar] [CrossRef]
- Di Fabio, G.; D’Onofrio, J.; Chiapparelli, M.; Hoorelbeke, B.; Montesarchio, D.; Balzarini, J.; de Napoli, L. Discovery of novel anti-HIV active G-quadruplex-forming oligonucleotides. Chem. Commun. 2011, 47, 2363–2365. [Google Scholar] [CrossRef] [Green Version]
- D’Onofrio, J.; Petraccone, L.; Erra, E.; Martino, L.; di Fabio, G.; de Napoli, L.; Giancola, C.; Montesarchio, D. 5'-Modified G-quadruplex forming oligonucleotides endowed with anti-HIV activity: Synthesis and biophysical properties. Bioconjug Chem. 2007, 18, 1194–1204. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all the compounds described here are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Altieri, A.; Alvino, A.; Ohnmacht, S.; Ortaggi, G.; Neidle, S.; Nocioni, D.; Franceschin, M.; Bianco, A. Xanthene and Xanthone Derivatives as G-Quadruplex Stabilizing Ligands. Molecules 2013, 18, 13446-13470. https://doi.org/10.3390/molecules181113446
Altieri A, Alvino A, Ohnmacht S, Ortaggi G, Neidle S, Nocioni D, Franceschin M, Bianco A. Xanthene and Xanthone Derivatives as G-Quadruplex Stabilizing Ligands. Molecules. 2013; 18(11):13446-13470. https://doi.org/10.3390/molecules181113446
Chicago/Turabian StyleAltieri, Alessandro, Antonello Alvino, Stephan Ohnmacht, Giancarlo Ortaggi, Stephen Neidle, Daniele Nocioni, Marco Franceschin, and Armandodoriano Bianco. 2013. "Xanthene and Xanthone Derivatives as G-Quadruplex Stabilizing Ligands" Molecules 18, no. 11: 13446-13470. https://doi.org/10.3390/molecules181113446