The Passiflora tripartita (Banana Passion) Fruit: A Source of Bioactive Flavonoid C-Glycosides Isolated by HSCCC and Characterized by HPLC–DAD–ESI/MS/MS

 ,
,  ,
,
Abstract
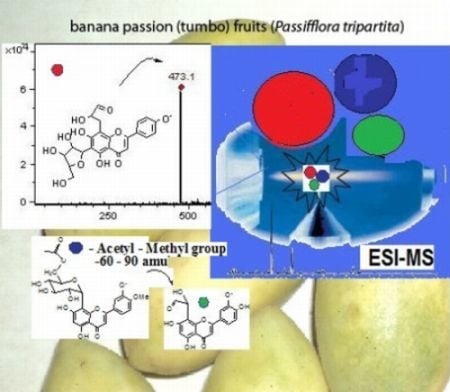
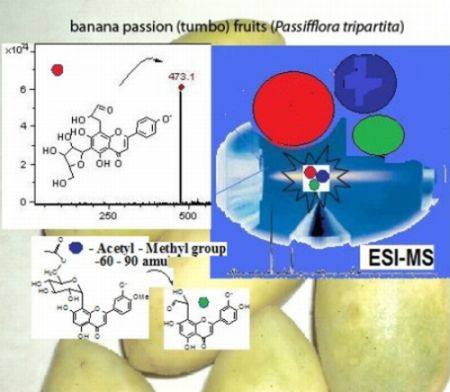
:
1. Introduction
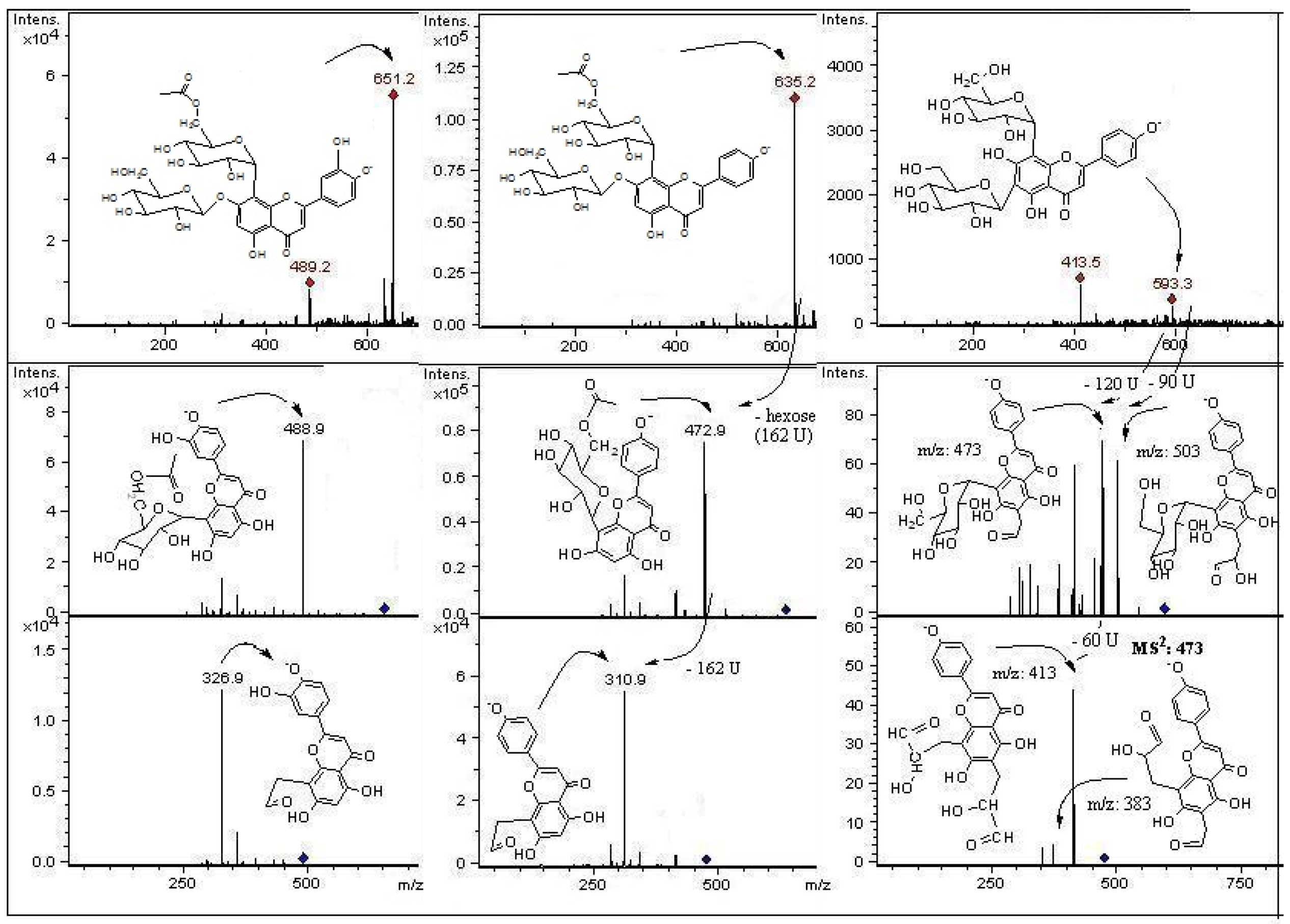
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DPPH a | FRAP b | TPC c | TFC d | Yield e | Fruit part |
|---|---|---|---|---|---|
| 10.41 ± 0.01 | 462.47 ± 0.49 | 56.03 ± 4.34 | 140.17 ± 4.23 | 5.24 | Peel |
| 12.89 ± 0.02 | 85.78 ± 0.12 | 22.57 ± 1.43 | 77.16 ± 8.4 | 4.07 | Pulp and juice |
| Compound | |||||
| 65.40 ± 0.24 | 393.97 ± 0.31 | - | - | 0.06 | 11 |
| 44.86 ± 0.13 | 438.23 ± 0.28 | - | - | 0.07 | 13 |
| 24.24 ± 0.11 | 543.72 ± 0.43 | - | - | 0.04 | 16 |
| 1.62 ± 0.01 | 637.84 ±0.65 | - | - | 0.13 | 19 |
| 3.69 ± 0.04 | 586.48 ± 0.18 | - | - | 0.14 | 31 |
| 1.16 ± 0.01 | 729.37 ± 0.48 | - | - | - | f Gallic acid |
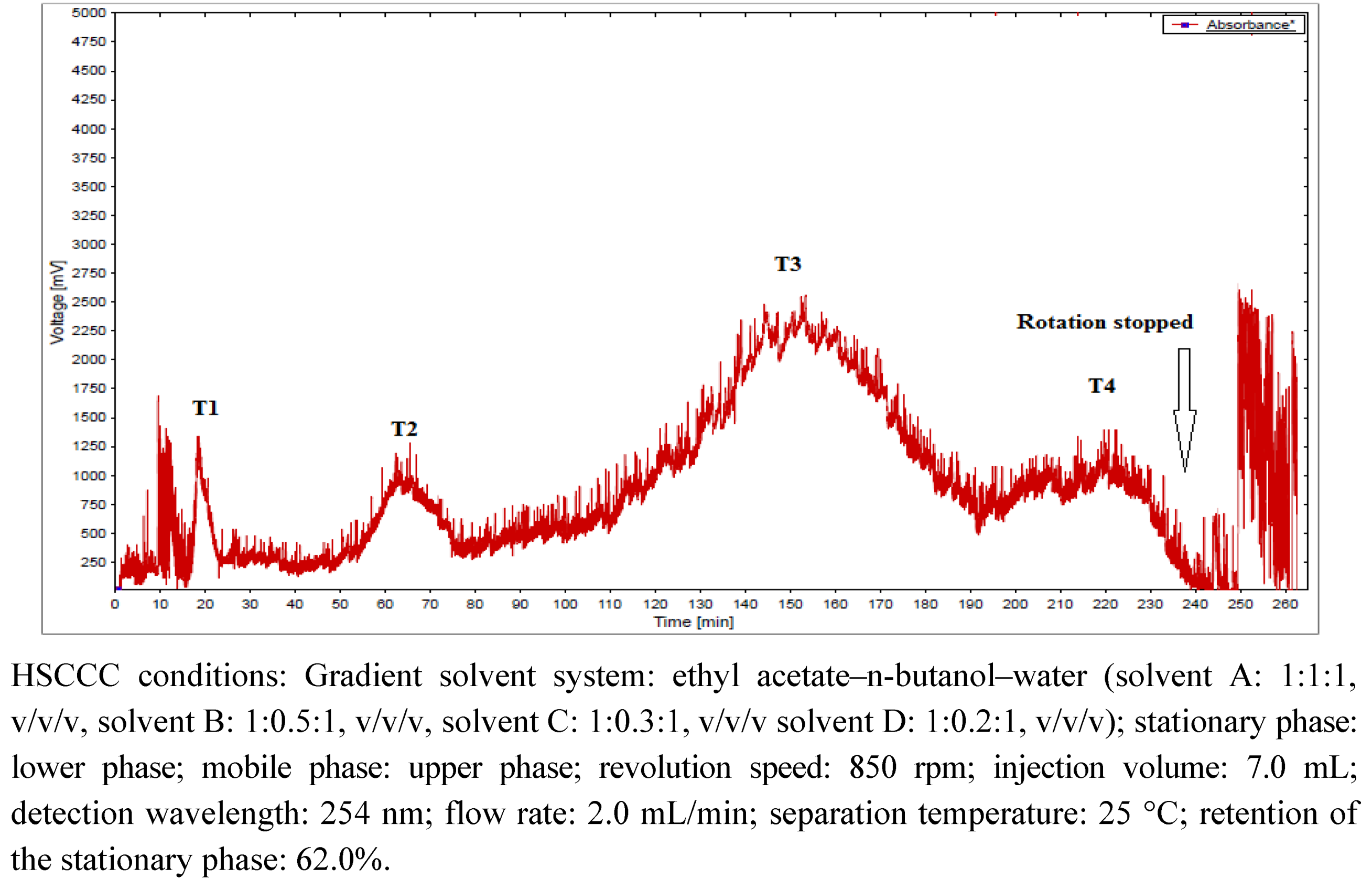
2.1. Isolation of Phenolic Compounds





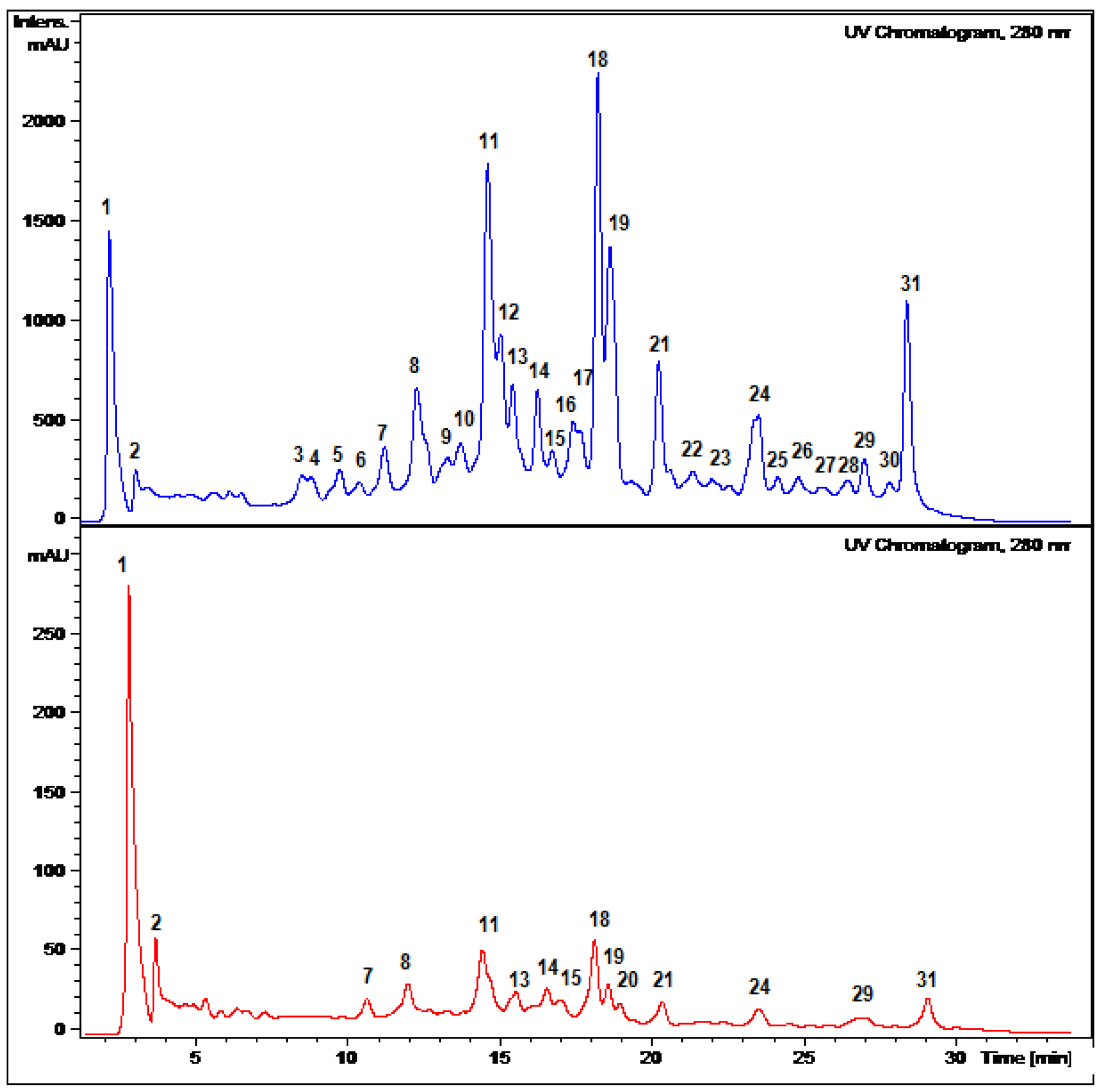
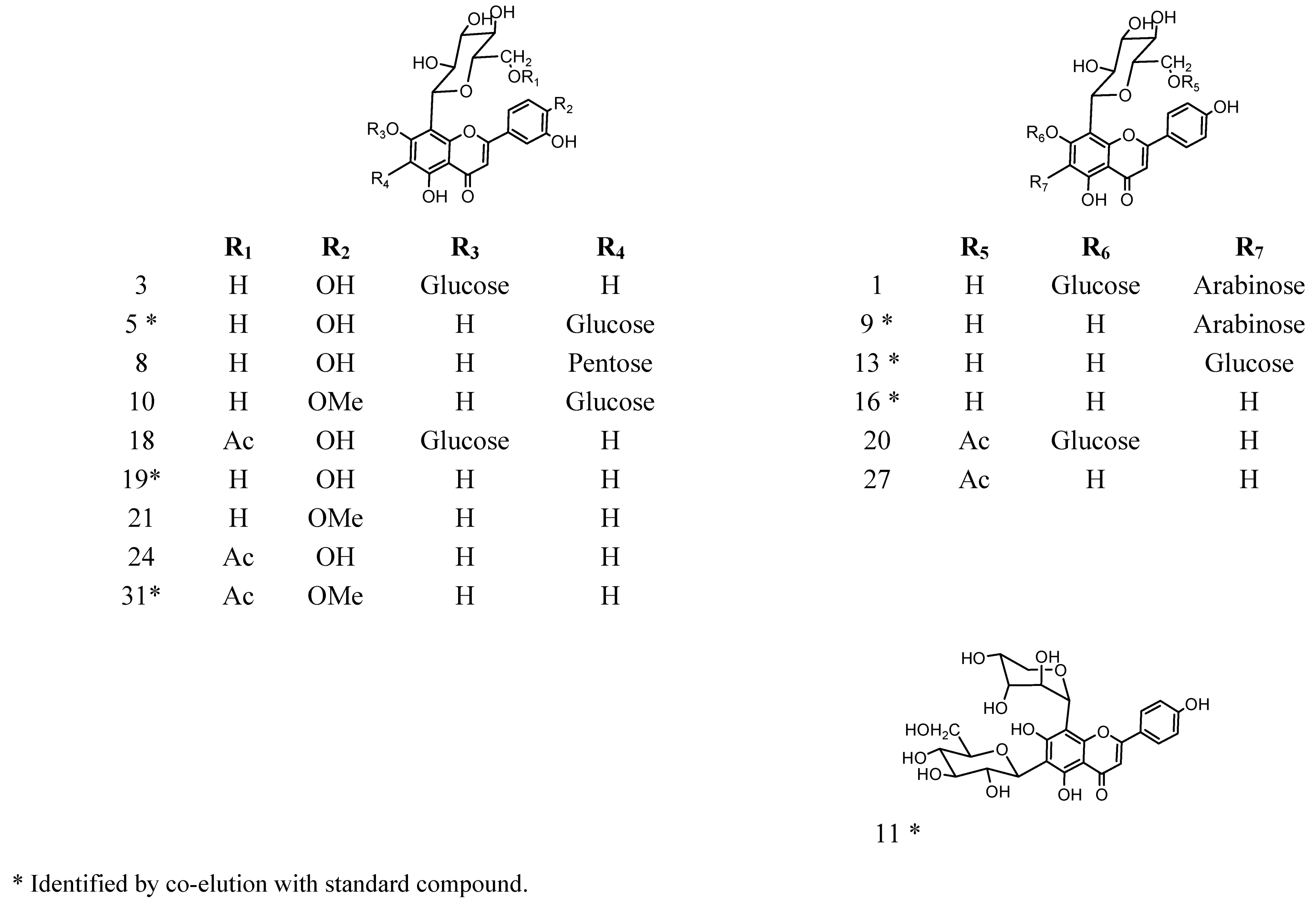
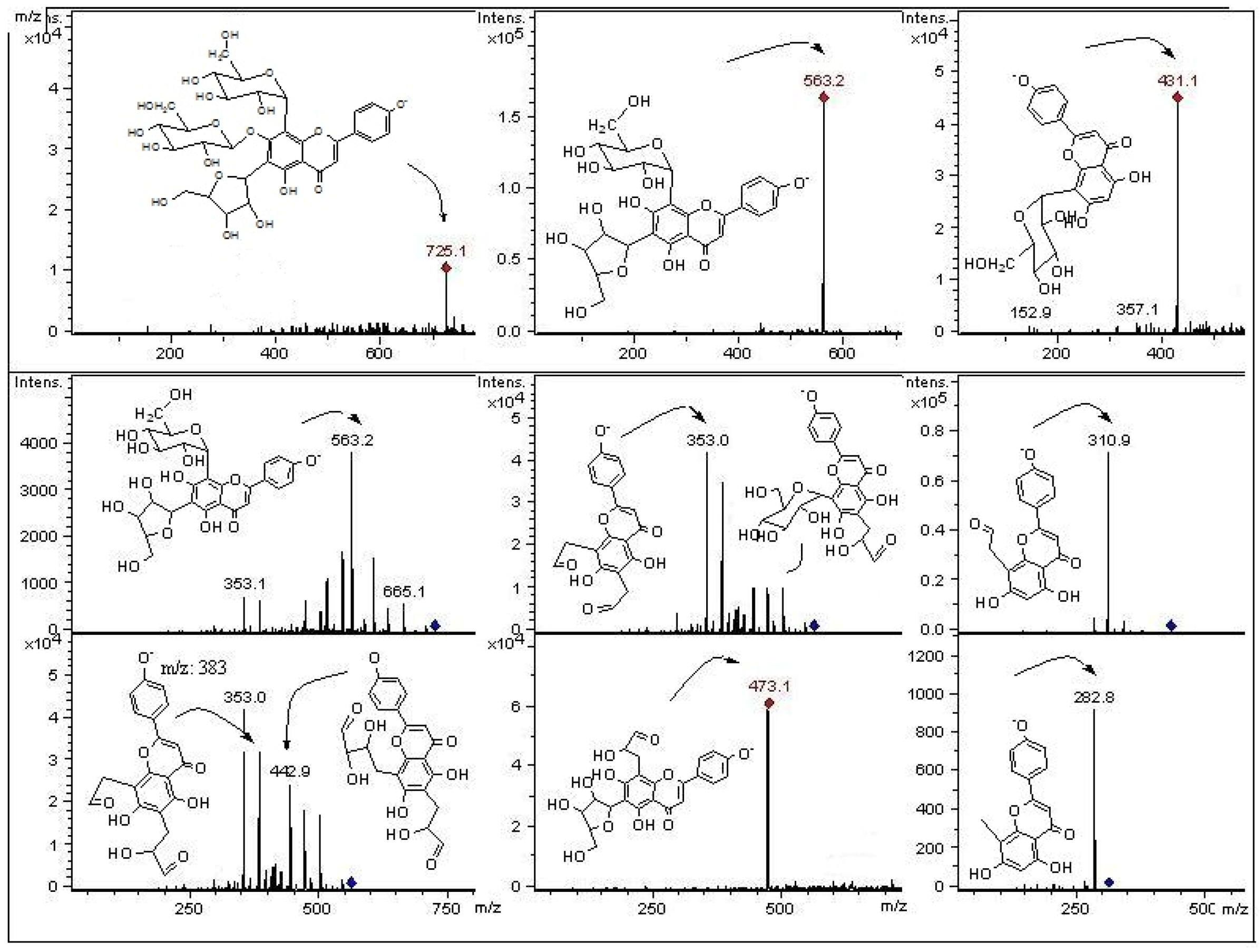
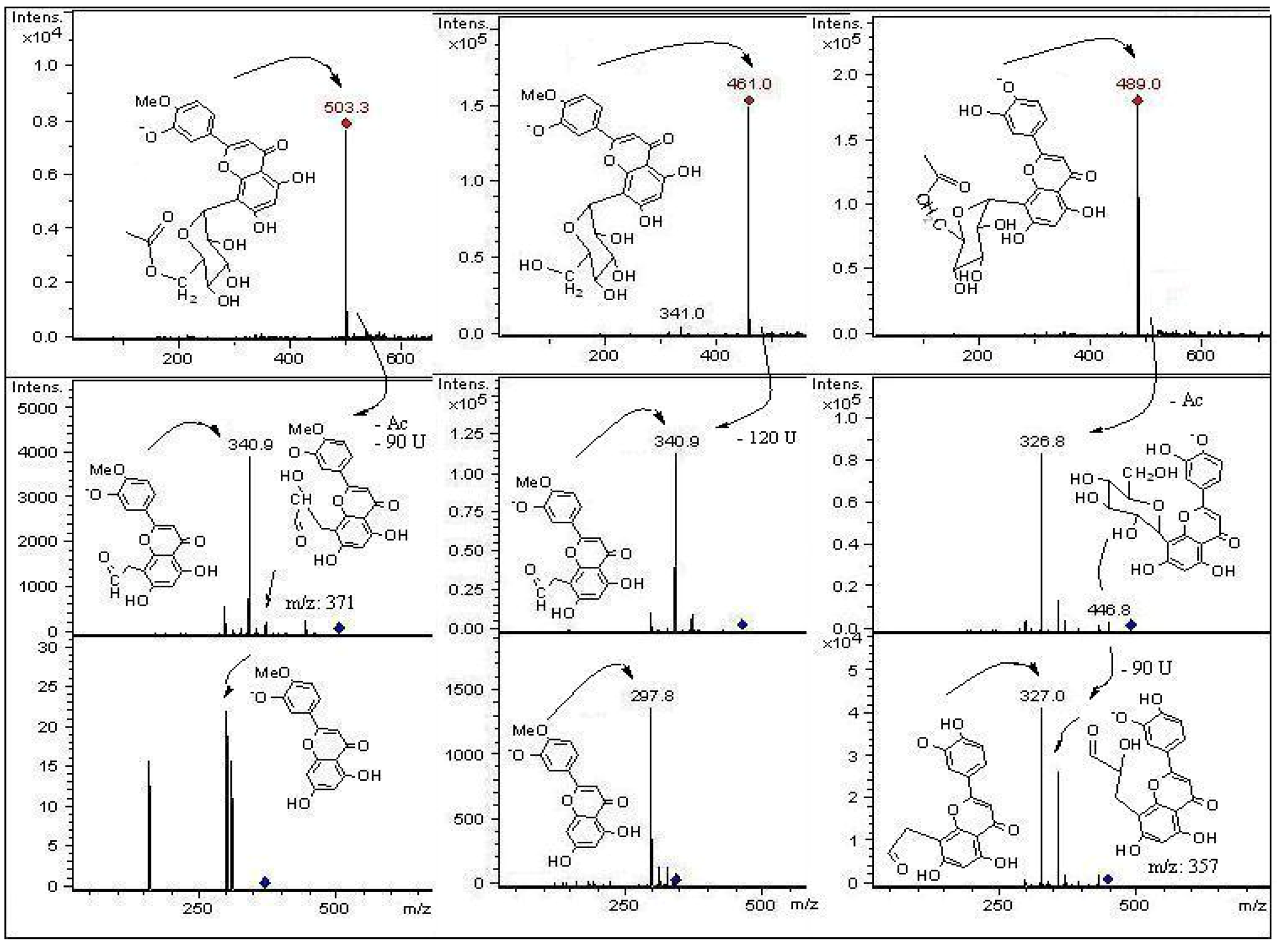
2.2. HPLC-DAD and ESI Identification of Phenolic Compounds in Tumbo Fruits
2.2.1. C-Glycosyl Flavones
2.2.2. C- and O-Glycosyl Flavones
| Peak | Rt(min) | λ max (nm) | [M−H]− | Fragment ions (m/z) | Compound identification | Fruit part |
|---|---|---|---|---|---|---|
| 1 | 2.1 | 269, 340 | 725 | 665, 563, 443, 383, 353 | (6-C-α-L-arabinopiranosyl)-7-O-glucosyl-8-C-β-D-glucopyranoside (7-O-glucosyl-isoschaftoside) | P, J |
| 2 | 2.8 | 288, 322 | 897 | 457, 451, 325, 305 | Feruloylated oligosaccharide | P, J |
| 3 | 9.4 | 270, 349 | 609 | 489, 369, 327 | Luteolin-(7-O-glucopyranosil)-8-C-glucopyranoside (Orientin-7-O-glucoside | P |
| 4 | 9.6 | 270, 349 | 645 | 447, 357, 327 | Luteolin-di-glycoside derivative | P |
| 5 | 9.9 | 269, 350 | 609 | 447, 357, 327 | Luteolin-6,8-di-C-β-D-glucopyranoside (Leucenin II) * | P |
| 6 | 10.2 | 269, 335 | 629 | 593 | Vicenin II derivative | P |
| 7 | 10.7 | - | 533 | 371 | 5′-Methoxy-demethylpiperitol-4-O-glucoside | P, J |
| 8 | 14.0 | 269, 349 | 579 | 489, 459, 399, 369 | Luteolin-(6-C-pentosyl)-8-C-β-D-glucopyranoside isomer | P, J |
| 9 | 14.1 | 269, 337 | 563 | 503, 473, 443, 383, 353 | (6-C-α-L-arabinopiranosyl)-8-C-β-D-glucopyranoside (Isoschaftoside) * | P |
| 10 | 14.3 | 271, 346 | 623 | 533, 503, 413, 383 | 4′-Methoxyluteolin -6,8-di-C-β-D-glucopyranoside(Leucenin II, 4′-methyl ether) | P |
| 11 | 14.6 | 269, 337 | 563 | 473, 353 | Apigenin (6-C-β-D-glucopyranosyl) 8-C-α-L-arabinoside (Schaftoside) * | P, J |
| 12 | 14.7 | 270, 348 | 687 | 651,489, 327 | Luteolin-5-O-glucosyl-8-C-(6”acetyl)-β-D-glucopyranoside derivative | P |
| 13 | 15.6 | 269, 335 | 593 | 503, 473, 413, 383 | Apigenin-6,8-di-C-β-D-glucopyranoside (Vicenin II) * | P, J |
| 14 | 16.2 | 269, 335 | 593 | 503, 473, 413, 383 | Vicenin II isomer | P, J |
| 15 | 16.8 | 270, 340 | 523 | 361 | Unknown di- glucosyl flavonoid | P, J |
| 16 | 17.5 | 269, 338 | 431 | 357, 311, 283 | Apigenin-8-C-β-D-glucopyranoside (Vitexin) * | P |
| 17 | 18.0 | - | 611 | 593, 491, 429, 393, 369, 327 | Eriodictyol 6,8 di-C-glucoside | P |
| 18 | 18.3 | 270, 348 | 651 | 489, 327 | Luteolin-7-O -glucopyranosyl 8-C-(6”acetyl)-glucopyranoside | P, J |
| 19 | 18.6 | 270, 349 | 447 | 285 | Luteolin-8-C-β-D-glucopyranoside (Orientin) * | P,J |
| 20 | 17.0 | 269, 338 | 635 | 473, 311 | Apigenin-5-O-β-D-glucopyranosyl, 8-C-(6”acetyl)-β-D-glucopyrano- side | P, J |
| 21 | 20.2 | 270, 347 | 461 | 285, 216 | 4′-Methoxyluteolin-8-C-β-D-glucopyranoside | P, J |
| 22 | 20.4 | 257, 361 | 317 | 300, 179, 151 | Myricetin * | P |
| 23 | 21.3 | 257, 361 | 631 | 479, 317 | Myricetin-3-O- (6”-galloyl) glycoside | P |
| 24 | 24.5 | 270, 350 | 489 | 447, 327 | Luteolin-8-C-(6”acetyl)-β-D-glucopyranoside | P, J |
| 25 | 24.7 | 257, 361 | 691 | 631, 479, 335, 317, 273 | Myricetin-3-O-(6” galloyl) glycoside derivative | P |
| 26 | 25.1 | 257, 361 | 331 | 315, 300, 179, 151 | Myricetin 3′ methyl ether | P |
| 27 | 25.7 | 269, 338 | 473 | 413, 311 | Apigenin-8-C-(6”acetyl)-β-D-glucopyranoside | P |
| 28 | 27.6 | 270, 335 | 713 | 677, 533, 451, 337 | Unknown C-glycosyl derivative | P |
| 29 | 28.4 | 270, 335 | 723 | 677, 533, 451, 337 | Unknown C-glycosyl derivative | P, J |
| 30 | 28.7 | 272, 330 | 857 | 501, 337 | Unknown C-glycosyl derivative | P |
| 31 | 29.1 | 269, 346 | 503 | 371, 341, 299 | 4′- Methoxyluteolin-8-C-(6”acetyl)-β-D-glucopyranoside * | P, J |


2.2.3. Other Phenolic Compounds
2.2.4. Unidentified Compounds
2.3. Total Phenolic, Total Flavonoid Content and Antioxidant Power of Tumbo Fruits
3. Experimental
3.1. General
3.2. Plant Material
3.2.1. Extraction of Tumbo Fruits
3.2.2. Isolation and Characterization of Phenolics from the Peel Extract
3.3. Polyphenolic Content
3.4. Antioxidant Assessment
3.4.1. Bleaching of the 2,2-Diphenyl-1-picrylhydrazyl (DPPH) Radical Assay
3.4.2. Ferric Reducing Antioxidant Power (FRAP) Assay
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
- Sample Availability: Samples of the isolated compounds and extracts are available from the authors.
References
- Jaroszewski, J.W.; Olafsdottir, E.S.; Wellendorph, P.; Christensen, J.; Franzyk, H.; Somanadhan, B.; Budnik, B.A.; Bolt Jørgensen, L.; Clausen, V. Cyanohydrin glycosides of Passiflora: Distribution pattern, A saturated cyclopentane derivative from P. guatemalensis, And formation of pseudocyanogenic α-hydroxyamides as isolation artefacts. Phytochemistry 2002, 59, 501–511. [Google Scholar]
- Vargas, A.J.; Geremias, D.S.; Provensi, G.; Fornari, P.E.; Reginatto, F.H.; Gosmann, G.; Schenkel, E.P.; Fröde, T.S. Passiflora alata and Passiflora edulis spray-dried aqueous extracts inhibit inflammation in mouse model of pleurisy. Fitoterapia 2007, 78, 112–119. [Google Scholar] [CrossRef]
- Deng, J.; Zhou, Y.; Bai, M.; Li, H.; Li, L. Anxiolytic and sedative activities of Passiflora edulis f. flavicarpa. J. Ethnopharmacol. 2010, 128, 148–153. [Google Scholar] [CrossRef]
- Li, H.; Zhou, P.; Yang, Q.; Shen, Y.; Deng, J.; Li, L.; Zhao, D. Comparative studies on anxiolytic activities and flavonoid compositions of Passiflora edulis ‘edulis’ and Passiflora edulis ‘flavicarpa’. J. Ethnopharmacol. 2011, 133, 1085–1090. [Google Scholar] [CrossRef]
- Bocco, A.; Cuvelier, M.E.; Richard, H.; Berset, C. Antioxidant activity and phenolic composition of Citrus peel and seed extracts. J. Agric. Food Chem. 1998, 46, 2123–2129. [Google Scholar] [CrossRef]
- Piccinelli, A.L.; Garcia Mesa, M.; Armenteros, D.M.; Alfonso, A.; Arevalo, A.C.; Campone, L.; Rastrelli, L. HPLC-PDA-MS and NMR characterization of C-glycosyl flavones in a hydroalcoholic extract of Citrus aurantifolia leaves with antiplatelet activity. J. Agric. Food Chem. 2008, 56, 1574–1581. [Google Scholar] [CrossRef]
- Prinza, S.; Ringla, A.; Huefnerb, A.; Pempa, E.; Kopp, B. 4′′′-Acetylvitexin-2′′-O-rhamnoside, isoorientin, orientin, and 8-methoxykaempferol-3-O-glucoside as markers for the differentiation of Crataegus monogyna and Crataegus pentagyna from Crataegus laevigata (Rosaceae). Chem. Biodivers. 2007, 4, 2920–2931. [Google Scholar] [CrossRef]
- Participants in the WHO Consultation on Medicinal Plants Commonly Used in NIS, Folium cum Flore Crataegi. In WHO Monographs on Medicinal Plants Commonly Used in the Newly Independent States; WHO Library Cataloguing-in-Publication Data: Paris, France, 2010; pp. 91–111.
- Figueirinha, A.; Paranhos, A.; Pérez-Alonso, J.J.; Santos-Buelga, C.; Batista, M.T. Cymbopogon citratus leaves: Characterisation of flavonoids by HPLC-PDA-ESI/MS/MS and an approach to their potential as a source of bioactive polyphenols. Food Chem. 2008, 718–728. [Google Scholar]
- Chassagne, D.; Crouzet, J.C.; Bayonove, C.L.; Baumes, R.L. Identification and quantification of Passion fruit cyanogenic glycosides. J. Agric. Food Chem. 1996, 44, 3817–3820. [Google Scholar] [CrossRef]
- Chassagne, D.; Crouzet, J.; Bayonove, C.L.; Baumes, R.L. Glycosidically bound eugenol and methyl salicylate in the fruit of edible Passiflora species. J. Agric. Food Chem. 1997, 45, 2685–2689. [Google Scholar] [CrossRef]
- Froehlich, O.; Duque, C.; Schreier, P. Volatile constituents of curuba (Passiflora mollissima) fruit. J. Agric. Food Chem. 1989, 37, 421–425. [Google Scholar] [CrossRef]
- Valente, A.; Albuquerque, T.G.; Sanches-Silva, A.; Costa, H.S. Ascorbic acid content in exotic fruits: A contribution to produce quality data for food composition databases. Food Res. Int. 2011, 44, 2237–2242. [Google Scholar] [CrossRef]
- Edwin, E.; Sheeja, E.; Dhanabal, S.P.; Suresh, B. Antihyperglycemic activity of Passiflora mollissima Bailey. Indian J. Pharm. Sci. 2007, 69, 570–571. [Google Scholar] [CrossRef]
- Kannan, S.; Parimala Devi, B.; Jayakar, B. In-vitro antibacterial activity of various extracts on the leaves of Passiflora mollissima. J. Chem. Pharm. Res. 2010, 2, 225–228. [Google Scholar]
- Muñoz Jáuregui, A.M.; Ramos-Escudero, D.F.; Alvarado-Ortiz Ureta, C.; Castañeda Castañeda, B. Evaluación de la capacidad antioxidante y contenido de compuestos fenólicos en recursos vegetales promisorios. Rev Soc. Quím. Perú. 2007, 73, 142–149. [Google Scholar]
- Simirgiotis, M.J.; Theoduloz, C.; Caligari, P.D. S.; Schmeda-Hirschmann, G. Comparison of phenolic composition and antioxidant properties of two native chilean and one domestic strawberry genotypes. Food Chem. 2009, 113, 377–385. [Google Scholar] [CrossRef]
- Simirgiotis, M.J.; Schmeda-Hirschmann, G. Direct identification of phenolic constituents in Boldo Folium (Peumus boldus Mol.) infusions by high-performance liquid chromatography with diode array detection and electrospray ionization tandem mass spectrometry. J. Chrom. A 2010, 1217, 443–449. [Google Scholar] [CrossRef]
- Ramos, F.A.R.; Castellanos, L.; Lopez, C.; Palacios, L.; Duque, C.; Pacheco, R.; Guzman, A. An Orientin derivative isolated from Passiflora tripartita var. mollissima. Lat. Am. J. Pharm. 2010, 29, 141–143. [Google Scholar]
- Nørbæk, R.; Brandt, K.; Kondo, T. Identification of flavone C-Glycosides Including a new flavonoid chromophore from Barley leaves (Hordeum vulgare L.) by improved NMR techniques. J. Agric. Food Chem. 2000, 48, 1703–1707. [Google Scholar] [CrossRef]
- Sato, S.; Koide, T. Synthesis of vicenin-1 and 3, 6,8- and 8,6-di-C-β-D-(glucopyranosyl-xylopyranosyl)- 40,5,7-trihydroxyflavones using two direct C-glycosylations of naringenin and phloroacetophenone with unprotected D-glucose and D-xylose in aqueous solution as the key reactions. Carbohydrate Res. 2010, 345, 1825–1830. [Google Scholar] [CrossRef]
- Choo, C.Y.; Sulong, N.Y.; Mana, F.; Wong, T.W. Vitexin and isovitexin from the Leaves of Ficus deltoidea with in vivo a-glucosidase inhibition. J. Ethnopharmacol. 2012, 142, 776–781. [Google Scholar] [CrossRef]
- Kumazawa, T.; Kimura, T.; Matsuba, S.; Sato, S.; Onodera, J.-I. Synthesis of 8-C-glucosylflavones. Carbohydrate Res. 2001, 334, 183–193. [Google Scholar] [CrossRef]
- Cuyckens, F.; Claeys, M. Optimization of a liquid chromatography method based on a simultaneous electrospray mass spectrometric and ultraviolet photodiode array detection for analysis of flavonoid glycosides. Rap. Comm. Mass Spec. 2002, 2341–2348. [Google Scholar] [CrossRef]
- Simirgiotis, M.J.; Silva, M.; Becerra, J.; Schmeda-Hirschmann, G. Direct characterisation of phenolic antioxidants in infusions from four Mapuche medicinal plants by liquid chromatography with diode array detection (HPLC-DAD) and electrospray ionisation tandem mass spectrometry (HPLC-ESI-MS). Food Chem. 2012, 131, 318–327. [Google Scholar] [CrossRef]
- Wang, X.; Liang, Y.; Zhu, L.; Xie, H.; Li, H.; He, J.; Pan, M.; Zhang, T.; Ito, Y. Preparative isolation and purification of flavone C-glycosides from the leaves of Ficus microcarpa L. f by medium-pressure liquid chromatography, high-speed countercurrent chromatography, and preparative liquid chromatography. J. Liq. Chrom. Rel. Tech. 2010, 33, 462–480. [Google Scholar] [CrossRef]
- Djoukeng, J.D.; Arbona, V.; Argamasilla, R.; Gomez-Cadenas, A. Flavonoid profiling in leaves of Citrus genotypes under different environmental situations. J. Agric. Food Chem. 2008, 56, 11087–11097. [Google Scholar] [CrossRef]
- Simirgiotis, M.J.; Schmeda-Hirschmann, G. Determination of phenolic composition and antioxidant activity in fruits, rhizomes and leaves of the white strawberry (Fragaria chiloensis spp. chiloensis form chiloensis) using HPLC-DAD-ESI-MS and free radical quenching techniques. J. Food Comp. Anal. 2010, 23, 545–553. [Google Scholar] [CrossRef]
- Kite, G.C.; Stoneham, C.A.; Veitch, N.C. Flavonol tetraglycosides and other constituents from leaves of Styphnolobium japonicum (Leguminosae) and related taxa. Phytochemistry 2007, 68, 1407–1416. [Google Scholar] [CrossRef]
- Simirgiotis, M.J.; Caligari, P.D. S.; Schmeda-Hirschmann, G. Identification of phenolic compounds from the fruits of the mountain papaya Vasconcellea pubescens A. DC. grown in Chile by liquid chromatography-UV detection-mass spectrometry. Food Chem. 2009, 115, 775–784. [Google Scholar] [CrossRef]
- Romani, A.; Campo, M.; Pinelli, P. HPLC/DAD/ESI-MS analyses and anti-radical activity of hydrolyzable tannins from different vegetal species. Food Chem. 2012, 130, 214–221. [Google Scholar] [CrossRef]
- Levigne, S.; Ralet, V.M.-C.J.; Quemener, B.C.; Pollet, B.N. L.; Lapierre, C.; Thibault, J.F.J. Isolation from sugar beet cell walls of arabinan oligosaccharides esterified by two ferulic acid monomers. Plant Phys. 2004, 134, 1173–1180. [Google Scholar] [CrossRef]
- Ye, M.; Yan, Y.; Guo, D.-A. Characterization of phenolic compounds in the Chinese herbal drug Tu-Si-Zi by liquid chromatography coupled to electrospray ionization mass spectrometry. Rap. Comm. Mass Spec. 2005, 19, 1469–1484. [Google Scholar]
- Ding, S.; Dudley, E.; Plummer, S.; Tang, J.; Newton, R.P.; Brenton, A.G. Fingerprint profile of Ginkgo biloba nutritional supplements by LC/ESI-MS/MS. Phytochemistry 2008, 69, 1555–1564. [Google Scholar]
- Gardana, C.; Nalin, F.; Simonetti, P. Evaluation of flavonoids and furanocoumarins from Citrus bergamia (Bergamot) Juice and Identification of new compounds. Molecules 2008, 13, 2220–2228. [Google Scholar] [CrossRef]
- Contreras-Calderón, J.; Calderón-Jaimes, L.; Guerra-Hernández, E.; García-Villanova, B. Antioxidant capacity, phenolic content and vitamin C in pulp, Peel and seed from 24 exotic fruits from Colombia. Food Res. Int. 2011, 44, 2047–2053. [Google Scholar] [CrossRef]
- Leong, A.C.-N.; Kinjo, Y.; Tako, M.; Iwasaki, H.; Oku, H.; Tamaki, H. Flavonoid glycosides in the shoot system of Okinawa Taumu (Colocasia esculenta S.). Food Chem. 2010, 119, 630–635. [Google Scholar] [CrossRef]
- Yildirim, A.; Mavi, A.; Kara, A. Determination of antioxidant and antimicrobial activities of Rumex crispus L. extracts. J. Agric. Food Chem. 2001, 49, 4083–4089. [Google Scholar] [CrossRef]
- Simirgiotis, M.J.; Adachi, S.; To, S.; Yang, H.; Reynertson, K.A.; Basile, M.J.; Gil, R.R.; Weinstein, I.B.; Kennelly, E.J. Cytotoxic chalcones and antioxidants from the fruits of Syzygium samarangense (Wax Jambu). Food Chem. 2008, 107, 813–819. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “Antioxidant Power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Simirgiotis, M.J.; Schmeda-Hirschmann, G.; Bórquez, J.; Kennelly, E.J. The Passiflora tripartita (Banana Passion) Fruit: A Source of Bioactive Flavonoid C-Glycosides Isolated by HSCCC and Characterized by HPLC–DAD–ESI/MS/MS. Molecules 2013, 18, 1672-1692. https://doi.org/10.3390/molecules18021672
Simirgiotis MJ, Schmeda-Hirschmann G, Bórquez J, Kennelly EJ. The Passiflora tripartita (Banana Passion) Fruit: A Source of Bioactive Flavonoid C-Glycosides Isolated by HSCCC and Characterized by HPLC–DAD–ESI/MS/MS. Molecules. 2013; 18(2):1672-1692. https://doi.org/10.3390/molecules18021672
Chicago/Turabian StyleSimirgiotis, Mario J., Guillermo Schmeda-Hirschmann, Jorge Bórquez, and Edward J. Kennelly. 2013. "The Passiflora tripartita (Banana Passion) Fruit: A Source of Bioactive Flavonoid C-Glycosides Isolated by HSCCC and Characterized by HPLC–DAD–ESI/MS/MS" Molecules 18, no. 2: 1672-1692. https://doi.org/10.3390/molecules18021672