The Reaction Mechanism of Claisen Rearrangement Obtained by Transition State Spectroscopy and Single Direct-Dynamics Trajectory

 and
and 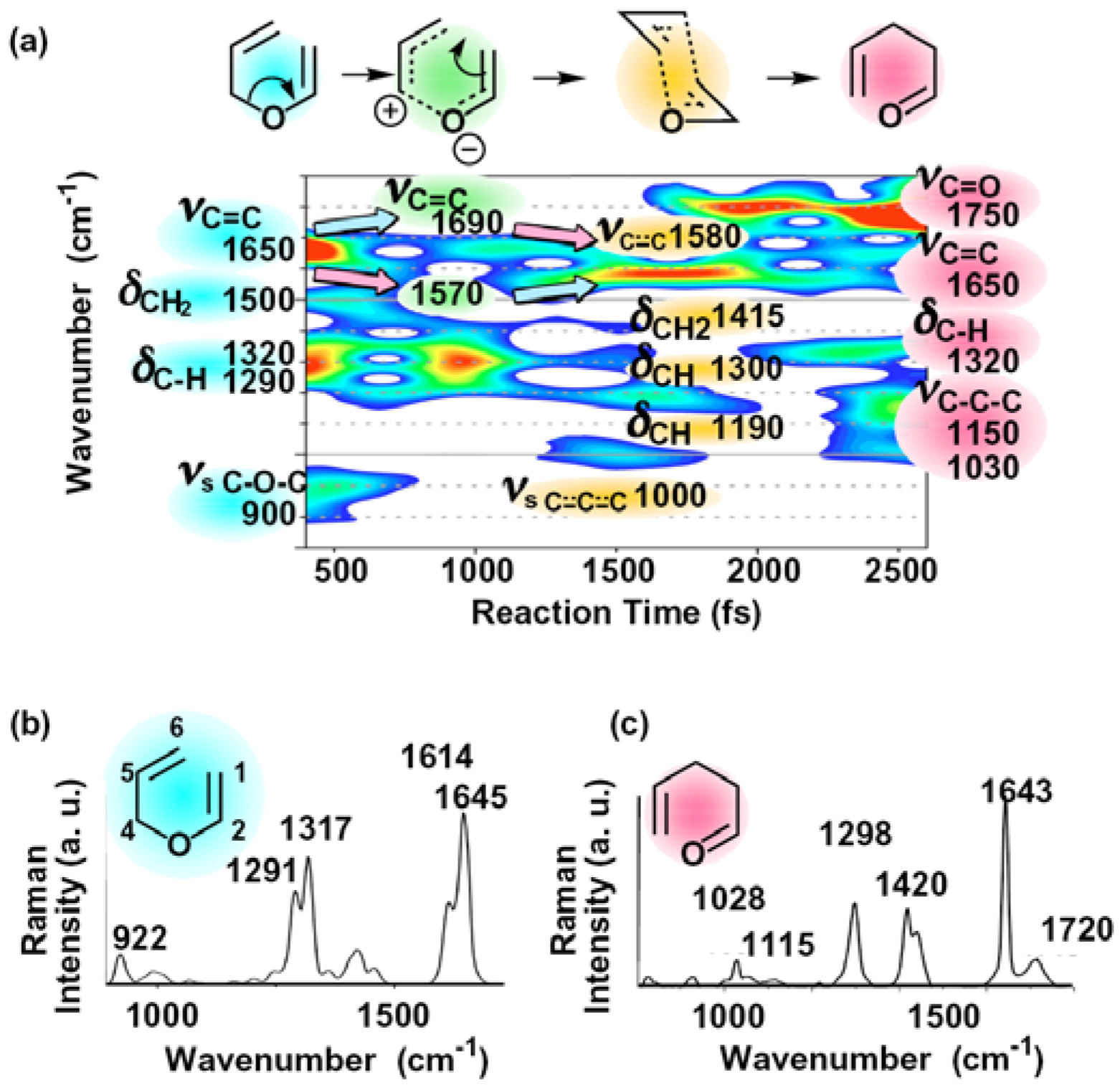{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussions
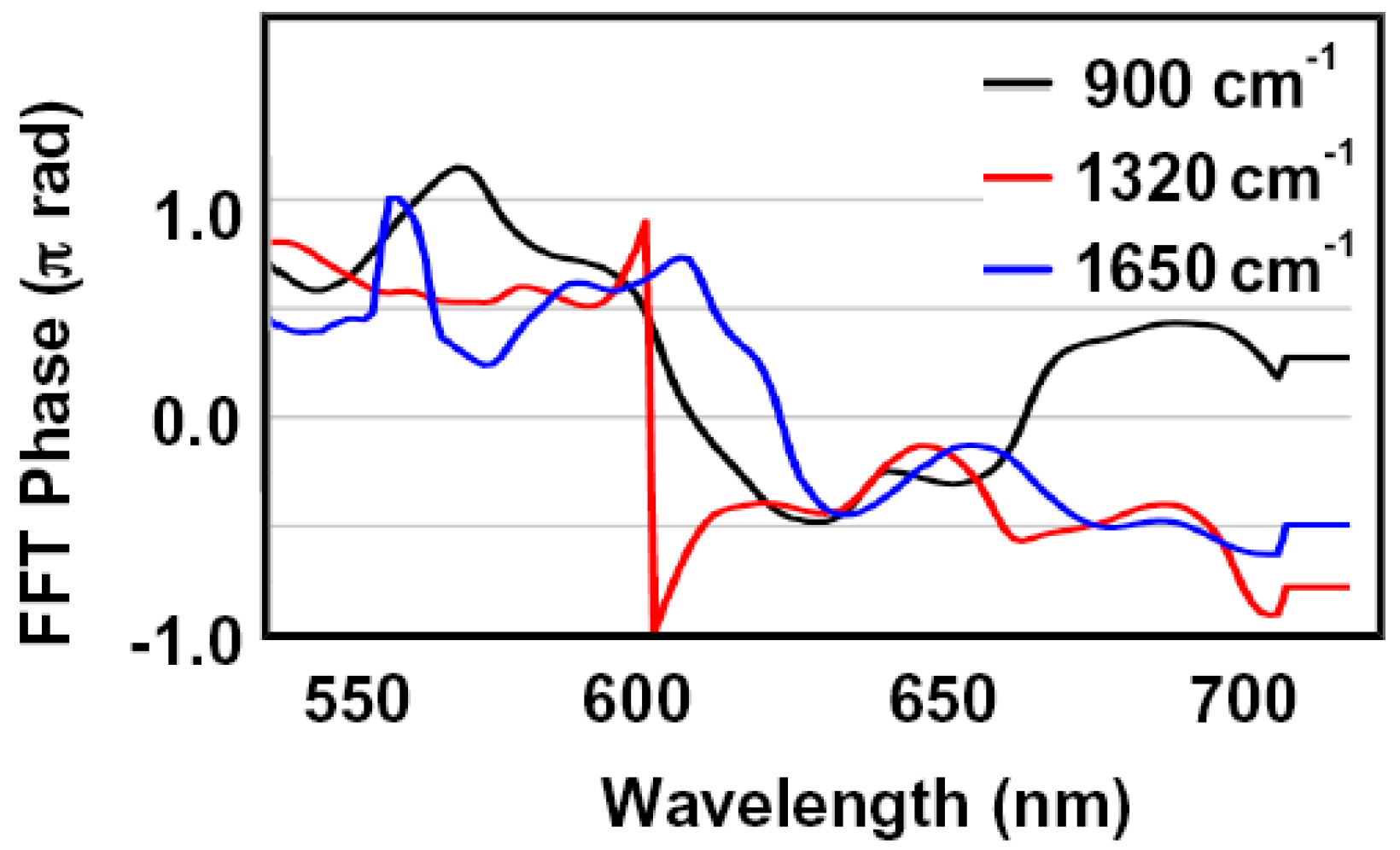
2.1. Transition State Spectroscopy of the Claisen Rearrangement of Allyl Vinyl Ether


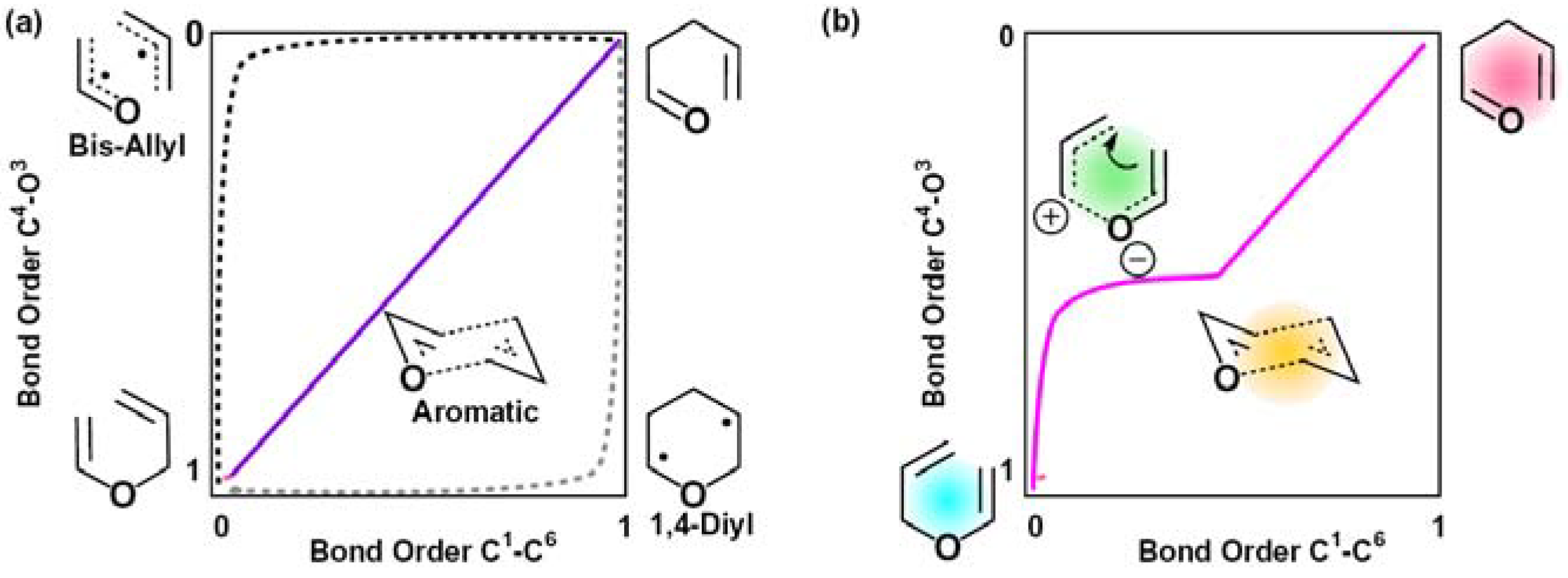
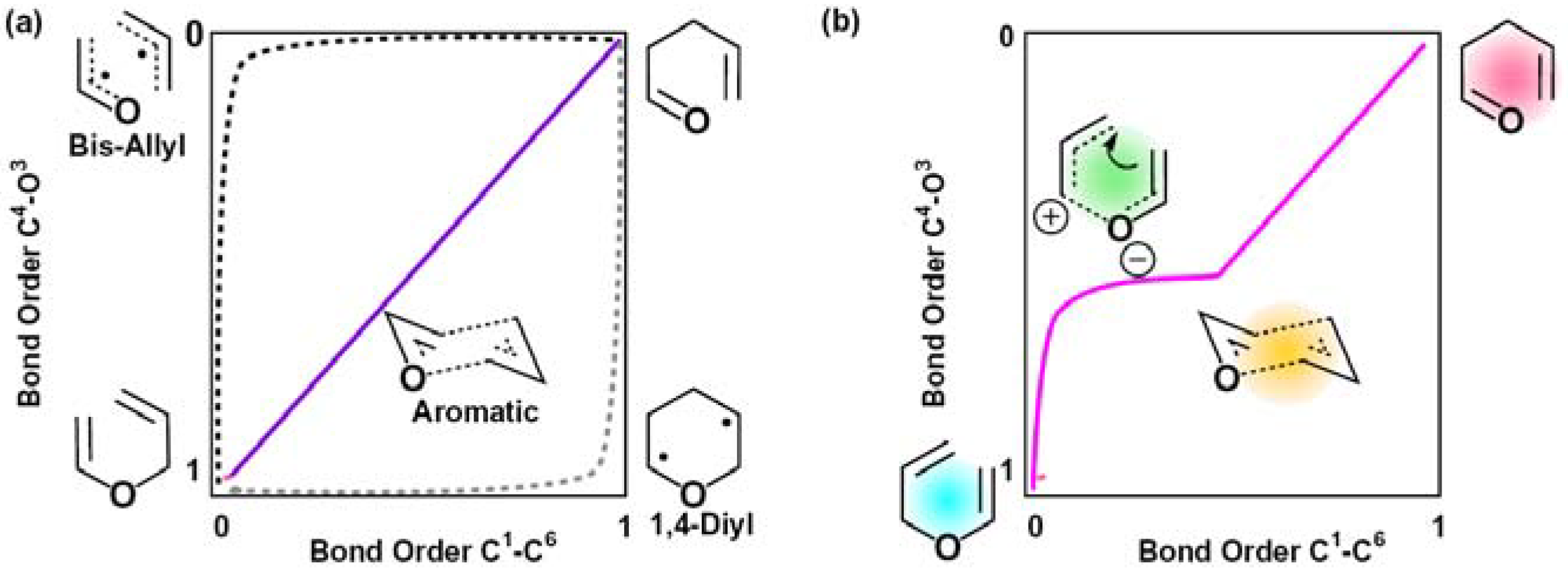
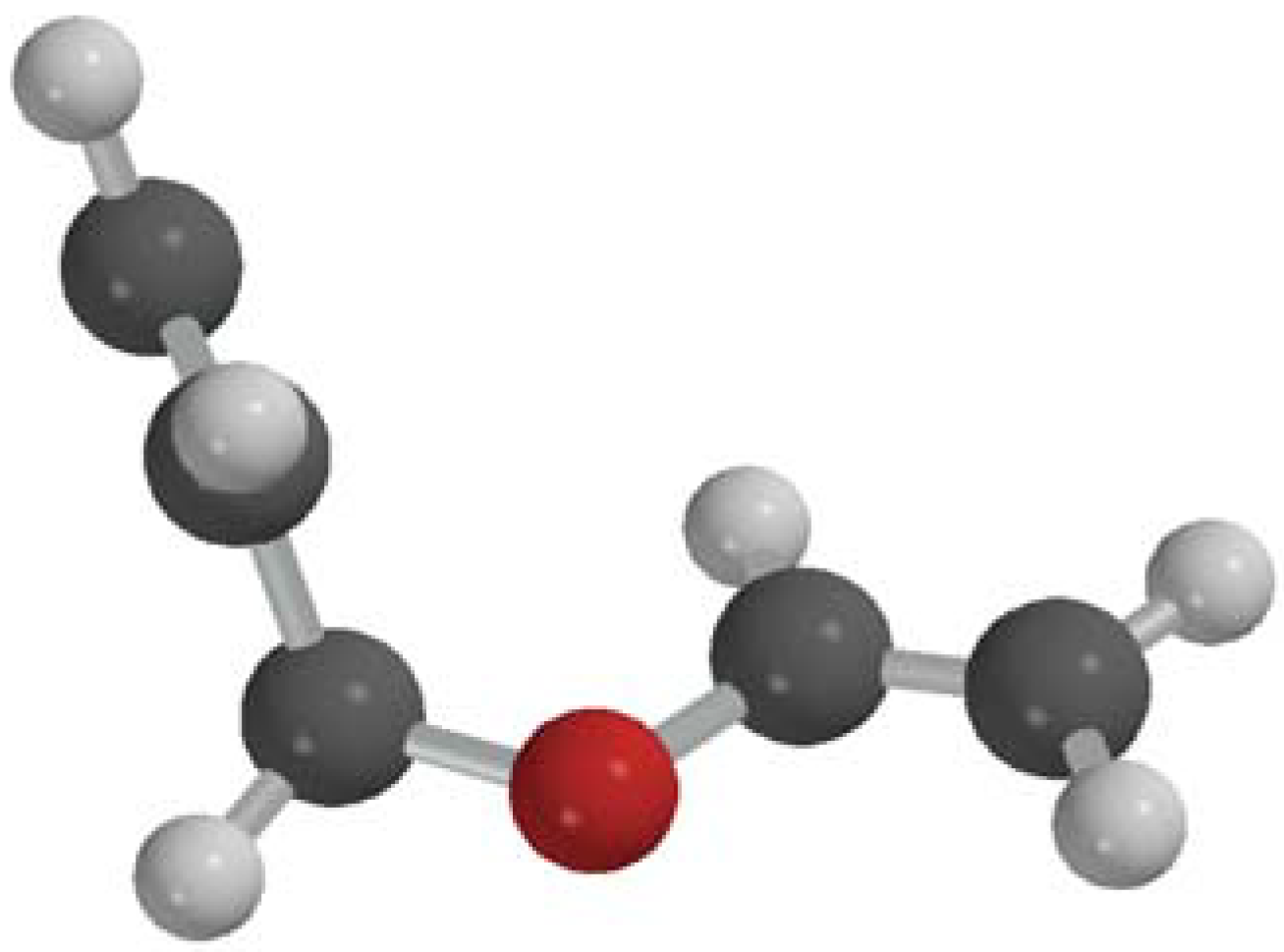
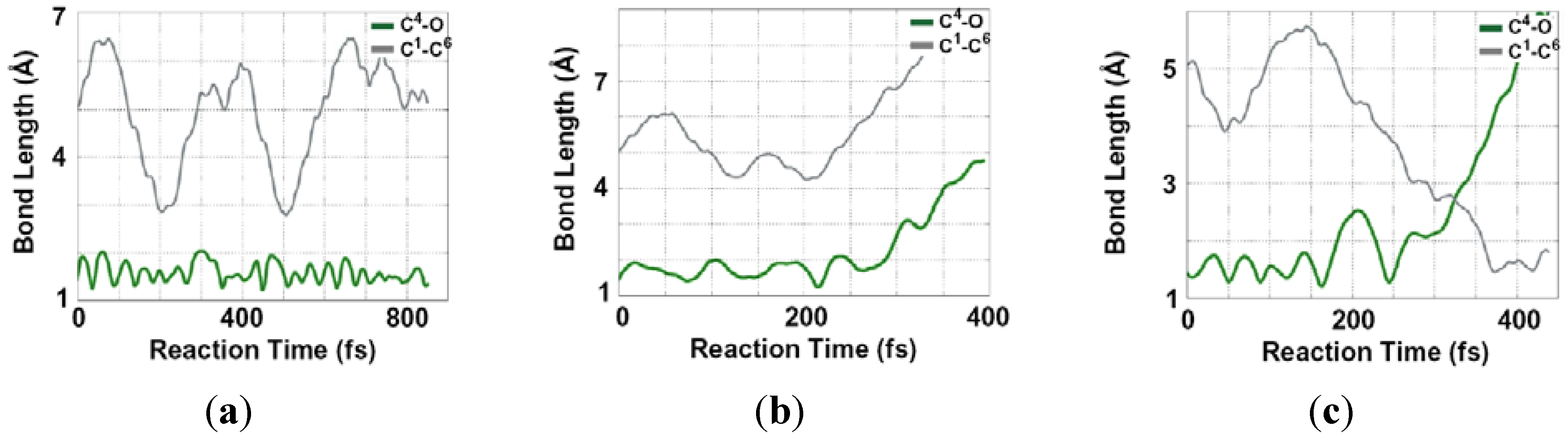
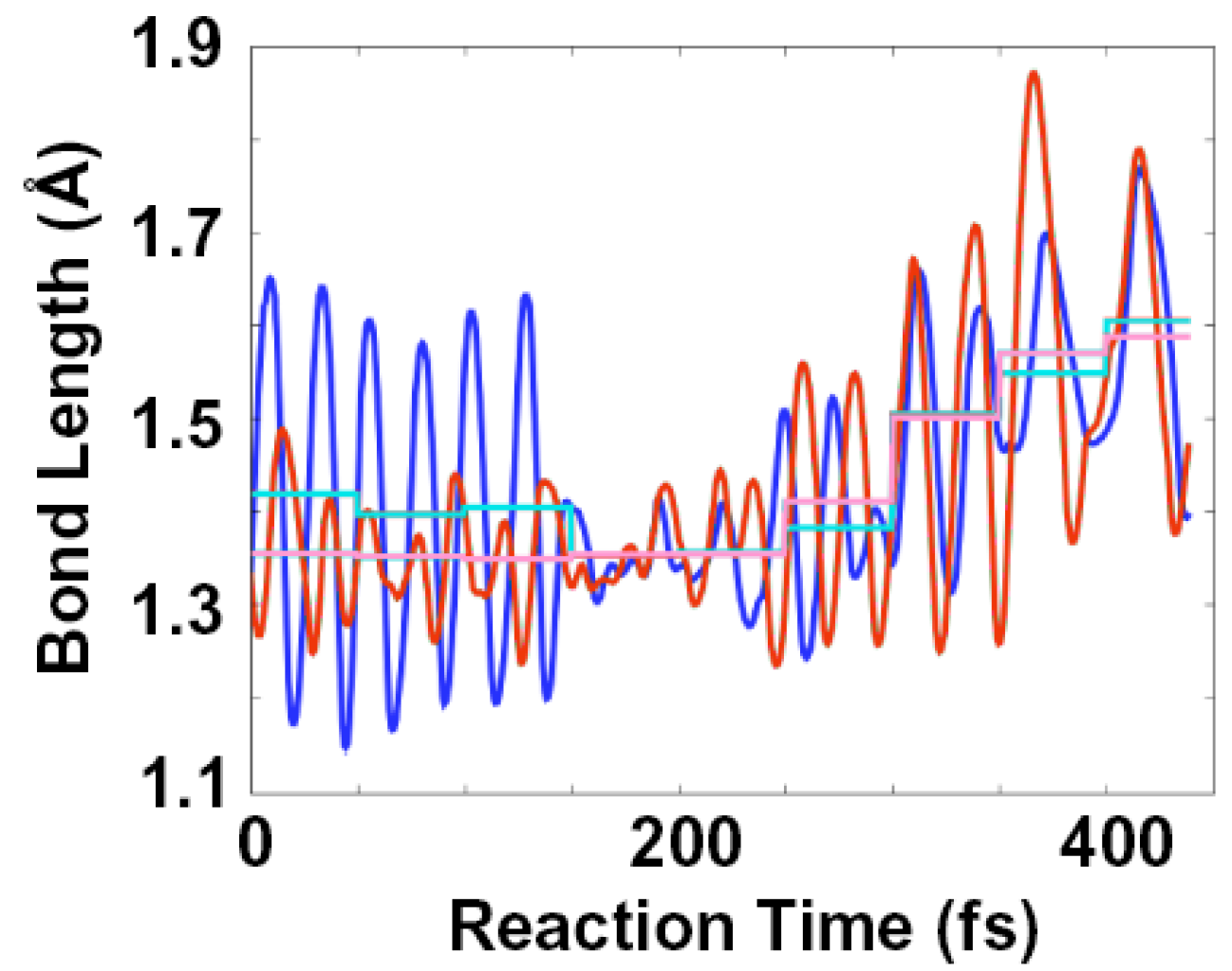
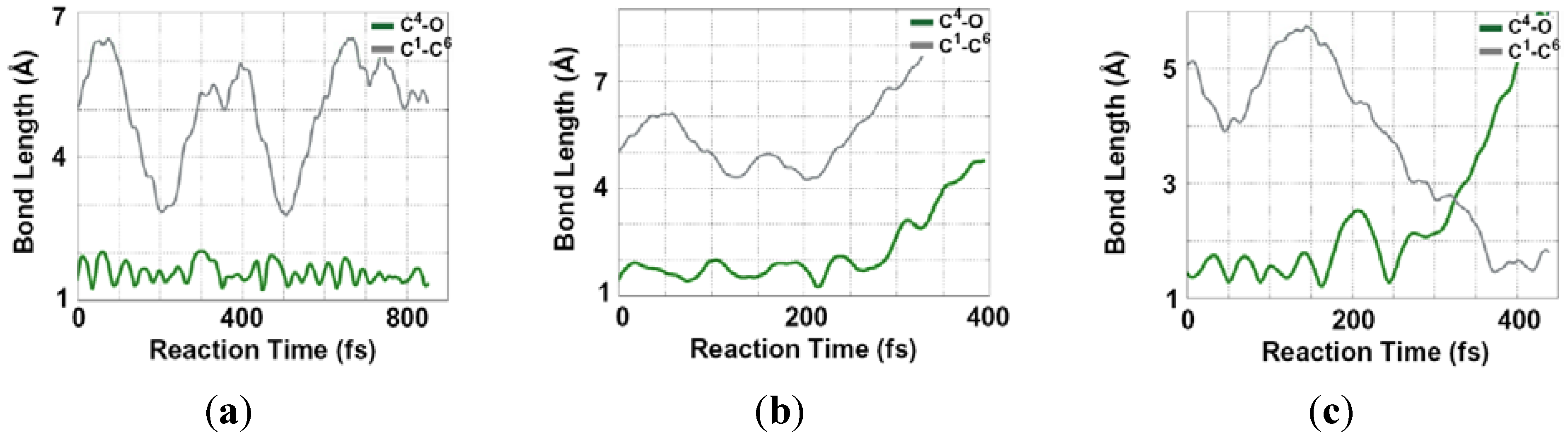
2.2. Single Direct-Dynamics Trajectory


3. Experimental
3.1. Visible 5-fs Laser System
3.2. “The Reaction in the Electronic Ground State”, Triggered by the Visible 5-fs Pulse
4. Conclusions
- Sample Availability: Samples of the compounds of allyl vibyl ether are available from the authors.
References
- Norish, R.G.W.; Porter, G. Chemical reactions produced by very high light intensities. Nature 1949, 164, 658. [Google Scholar] [CrossRef]
- Mayman, T.H. Stimulated optical radiation in ruby. Nature 1960, 187, 493–494. [Google Scholar] [CrossRef]
- Hentschel, M.; Kienberger, R.; Spielmann, Ch.; Reider, G.A.; Milosevic, N.; Brabec, T.; Corkum, P.; Heinzmann, U.; Drescher, M.; Krausz, F. Attosecond metrology. Nature 2001, 414, 509–513. [Google Scholar] [CrossRef]
- Itatani, J.; Levesque, J.; Zeidler, D.; Niikura, H.; Pépin, H.; Kieffer, J.C.; Corkum, P.B.; Villeneuve, D.M. Tomographic imaging of molecular orbitals. Nature 2004, 432, 867–871. [Google Scholar] [CrossRef]
- Krausz, F.; Ivanov, M. Attosecond physics. Rev. Mod. Phys. 2009, 81, 163–234. [Google Scholar] [CrossRef]
- Zewail, A.H. Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond. J. Phys. Chem. A 2000, 104, 5660–5694. [Google Scholar] [CrossRef]
- Kobayashi, T.; Saito, T.; Ohtani, H. Real-time spectroscopy transition state in bacteriodhodopsin during retinal isomerization. Nature 2001, 414, 531–534. [Google Scholar] [CrossRef]
- Iwakura, I. The experimental visualization of molecular structural changes during both photochemical and thermal reactions by real-time vibrational spectroscopy. Phys. Chem. Chem. Phys. 2011, 13, 5546–5555. [Google Scholar] [CrossRef]
- Kobayashi, T.; Yabushita, A. Transition-state spectroscopy using ultrashort laser pulses. Chem. Rec. 2011, 11, 99–116. [Google Scholar] [CrossRef]
- Iwakura, I.; Yabushita, A.; Kobayashi, T. Direct observation of the molecular structural changes during the Claisen Rearrangement including the transition state. Chem. Lett. 2010, 39, 374–375. [Google Scholar] [CrossRef]
- Iwakura, I.; Yabushita, A.; Liu, J.; Okamura, K.; Kobayashi, T. Photo-impulsive reactions in the electronic ground state without electronic excitation: Non-photo, Non-thermal chemical reactions. Phys. Chem. Chem. Phys. 2012, 14, 9696–9701. [Google Scholar] [CrossRef]
- Vrakking, M.J.J.; Villeneuve, D.M.; Stolow, A. Observation of fractional revivals in molecular wavepackets. Phys. Rev. A 1996, 54, R37–R40. [Google Scholar]
- Claisen, L. Rearrangement of phenol allyl ethers into C-allylphenols. Chem. Ber. 1912, 45, 3157–3166. [Google Scholar] [CrossRef]
- Hurd, C.D.; Pollack, M.A. The rearrangement of vinyl allyl ethers. J. Am. Chem. Soc. 1938, 60, 1905–1911. [Google Scholar] [CrossRef]
- Schuler, F.W.; Murphy, G.W. The kinetics of the rearrangement of vinyl allyl ether. J. Am. Chem. Soc. 1950, 72, 3155–3159. [Google Scholar] [CrossRef]
- Castro, A.M.M. Claisen rearrangement over the past nine decades. Chem. Rev. 2004, 104, 2939–3002. [Google Scholar] [CrossRef]
- Gajewski, J.J.; Jurayj, J.; Kimbrough, D.R.; Gande, M.E.; Ganem, B.; Carpenter, B.K. The mechanism of rearrangement of chorismic acid and related compounds. J. Am. Chem. Soc. 1987, 109, 1170–1186. [Google Scholar]
- Vance, R.L.; Rondan, N.G.; Houk, K.N.; Jensen, F.; Borden, W.T.; Komornicki, A.; Wimmer, E. Transition structures for the Claisen rearrangement. J. Am. Chem. Soc. 1988, 110, 2314–2315. [Google Scholar]
- Wiest, O.; Black, K.A.; Houk., K.N. Density functional theory isotope effects and activation energies for the cope and Claisen rearrangements. J. Am. Chem. Soc. 1994, 116, 10336–10337. [Google Scholar] [CrossRef]
- Hu, H.; Kobrak, M.N.; Xu, C.; Hammes-Schiffer, S. Reaction path Hamiltonian analysis of dynamical solvent effects for a Claisen rearrangement and a Diels Alder reaction. J. Phys. Chem. A 2000, 104, 8058–8066. [Google Scholar] [CrossRef]
- Hill, J.G.; Karadakov, P.B.; Cooper, D.L. A spin-coupled study of the Claisen rearrangement of allyl vinyl ether. Theor. Chem. Acc. 2006, 115, 212–220. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Healy, E.F. Ground states of molecules. 68. MNDO study of the Claisen rearrangement. J. Am. Chem. Soc. 1984, 106, 7127–7131. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Jie, C. Mechanism of the Claisen rearrangement of allyl vinyl ethers. J. Am. Chem. Soc. 1989, 111, 511–519. [Google Scholar] [CrossRef]
- Gajewski, J.J.; Conrad, N.D. Aliphatic Claisen rearrangement transition state structure from secondary .alpha.-deuterium isotope effects. J. Am. Chem. Soc. 1979, 101, 2747–2748. [Google Scholar] [CrossRef]
- Gajewski, J.J.; Gee, K.R.; Jurayj, J. Energetic and rate effects of the trifluoromethyl group at C-2 and C-4 on the aliphatic Claisen rearrangement. J. Org. Chem. 1990, 55, 1813–1822. [Google Scholar] [CrossRef]
- Kupczyk-Subotkowska, L.; Saunders, W.H., Jr.; Shine, H.J.; Subotkowski, W. Carbon kinetic isotope effects and transition structures in the rearrangements of allyl vinyl ethers. 2-(Trimethylsilyloxy)- and 2-(Methoxycarbonyl)-3-oxa-1,5-hexadiene. J. Am. Chem. Soc. 1994, 116, 7088–7093. [Google Scholar] [CrossRef]
- Davidson, M.M.; Hillier, I.H. Aqueous acceleration of the Claisen rearrangement of allyl vinyl ether: A hybrid, explicit solvent, and continuum model. J. Phys. Chem. 1995, 99, 6748–6751. [Google Scholar] [CrossRef]
- Gajewski, J.J. The Claisen rearrangement. Response to solvents and substituents: The case for both hydrophobic and hydrogen bond acceleration in water and for a variable transition state. Acc. Chem. Res. 1997, 30, 219–225. [Google Scholar] [CrossRef]
- Aviyente, V.; Yoo, H.Y.; Houk, K.N. Analysis of substituent effects on the Claisen rearrangement with Ab Initio and density functional theory. J. Org. Chem. 1997, 62, 6121–6128. [Google Scholar] [CrossRef]
- Yoo, H.Y.; Houk, K.N. Theory of substituent effects on pericyclic reaction rates: Alkoxy substituents in the Claisen rearrangement. J. Am. Chem. Soc. 1997, 119, 2877–2884. [Google Scholar] [CrossRef]
- Meyer, M.P.; DelMonte, A.J.; Singleton, D.A. Reinvestigation of the isotope effects for the Claisen and aromatic Claisen rearrangements: The nature of the Claisen transition states. J. Am. Chem. Soc. 1999, 121, 10865–10874. [Google Scholar] [CrossRef]
- Rehbein, J.; Hiersemann, M. Mechanistic Aspects of the Aliphatic Claisen Rearangement. In The Claisen Rearrangement, 1st; Hiersemann, M., Nubbemeyer, U., Eds.; Wiley-VCH: Winheim, Germany, 2007; pp. 525–557. [Google Scholar]
- Coates, R.M.; Rogers, B.D.; Hobbs, S.J.; Curran, D.P.; Peck, D.R. Synthesis and Claisen rearrangement of alkoxyallyl enolethers. Evidence for a dipolar transition state. J. Am. Chem. Soc. 1987, 109, 1160–1170. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Shirakawa, A.; Sakane, I.; Takasaka, M.; Kobayashi, T. Sub-5-fs visible pulse generation by pulse-front-matched noncollinear optical parametric amplifier. Appl. Phys. Lett. 1999, 74, 2268–2270. [Google Scholar] [CrossRef]
- Kobayashi, T.; Iwakura, I.; Yabushita, A. Excitonic and vibrational nonlinear processes in a polydiacetylene studied by a few-cycle pulse laser. New J. Phys. 2008, 10, 065016. [Google Scholar] [CrossRef]
- Du, J.; Teramoto, T.; Nakata, K.; Tokunaga, E.; Kobayashi, T. Real-time vibrational dynamics in chlorophyll a studied with a few-cycle pulse laser. Biophys. J. 2001, 101, 995–1003. [Google Scholar]
- Yagasaki, T.; Saito, S. Ultrafast intermolecular dynamics of liquid water: A theoretical study on two-dimensional infrared spectroscopy. J. Chem. Phys. 2008, 128, 154521. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iwakura, I.; Kaneko, Y.; Hayashi, S.; Yabushita, A.; Kobayashi, T. The Reaction Mechanism of Claisen Rearrangement Obtained by Transition State Spectroscopy and Single Direct-Dynamics Trajectory. Molecules 2013, 18, 1995-2004. https://doi.org/10.3390/molecules18021995
Iwakura I, Kaneko Y, Hayashi S, Yabushita A, Kobayashi T. The Reaction Mechanism of Claisen Rearrangement Obtained by Transition State Spectroscopy and Single Direct-Dynamics Trajectory. Molecules. 2013; 18(2):1995-2004. https://doi.org/10.3390/molecules18021995
Chicago/Turabian StyleIwakura, Izumi, Yu Kaneko, Shigehiko Hayashi, Atsushi Yabushita, and Takayoshi Kobayashi. 2013. "The Reaction Mechanism of Claisen Rearrangement Obtained by Transition State Spectroscopy and Single Direct-Dynamics Trajectory" Molecules 18, no. 2: 1995-2004. https://doi.org/10.3390/molecules18021995