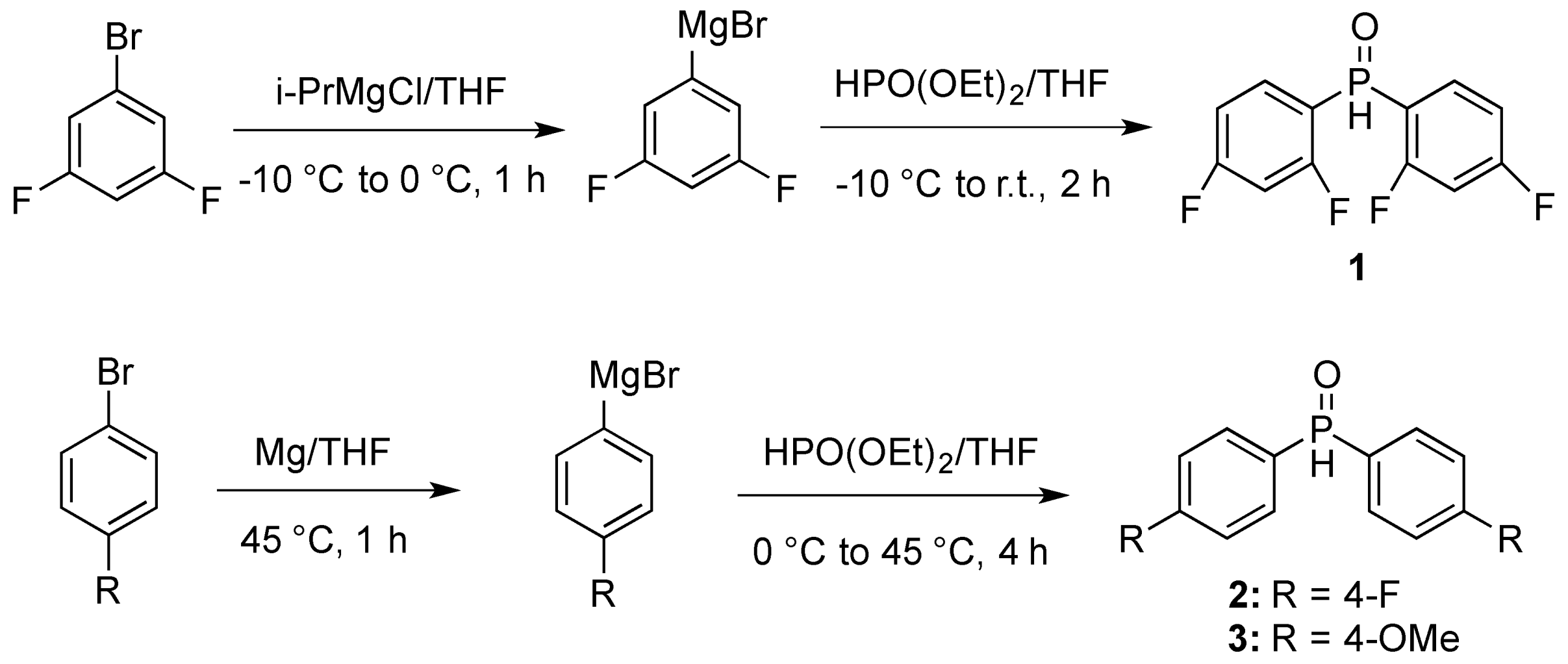
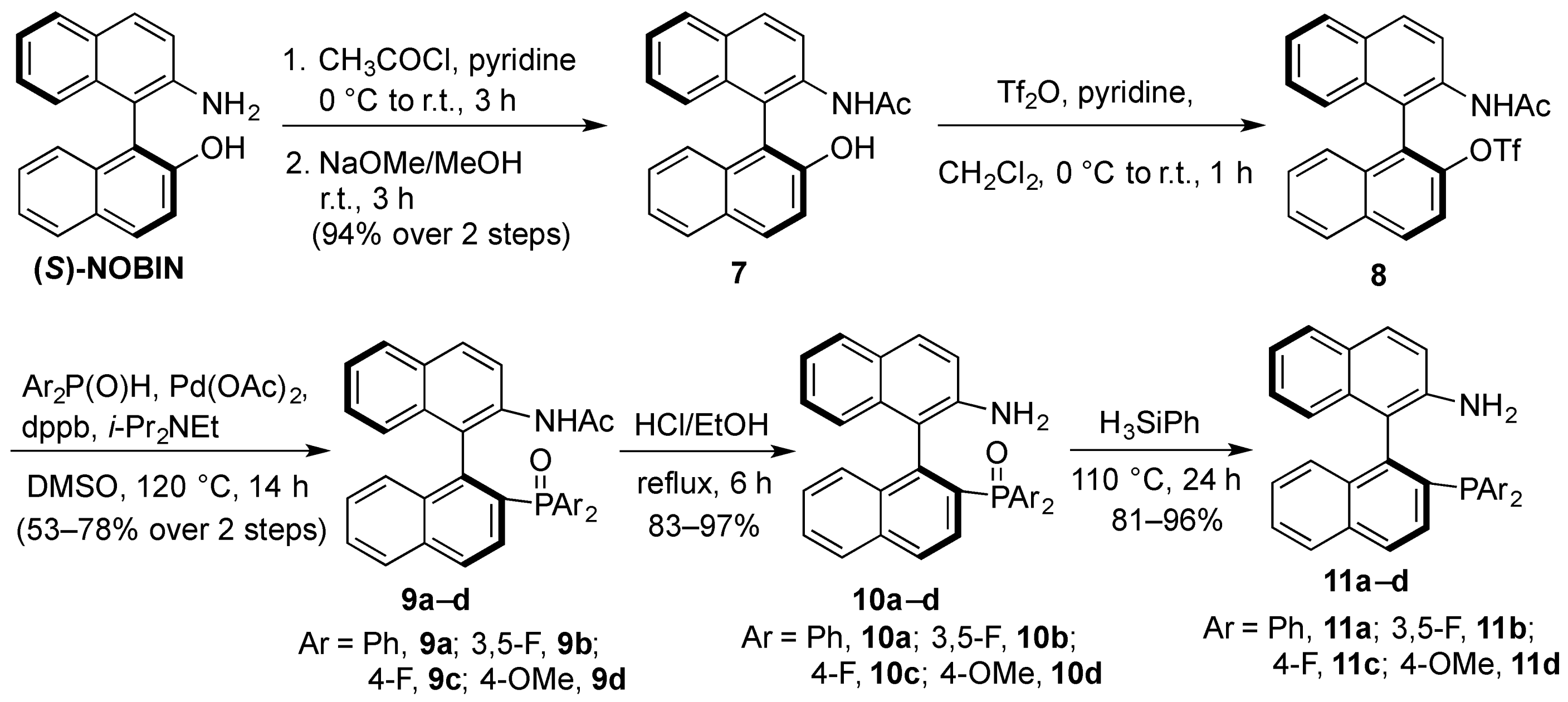
3.3. Preparations of Aminophosphine Oxides 10a–d and Aminophosphines 11a–d
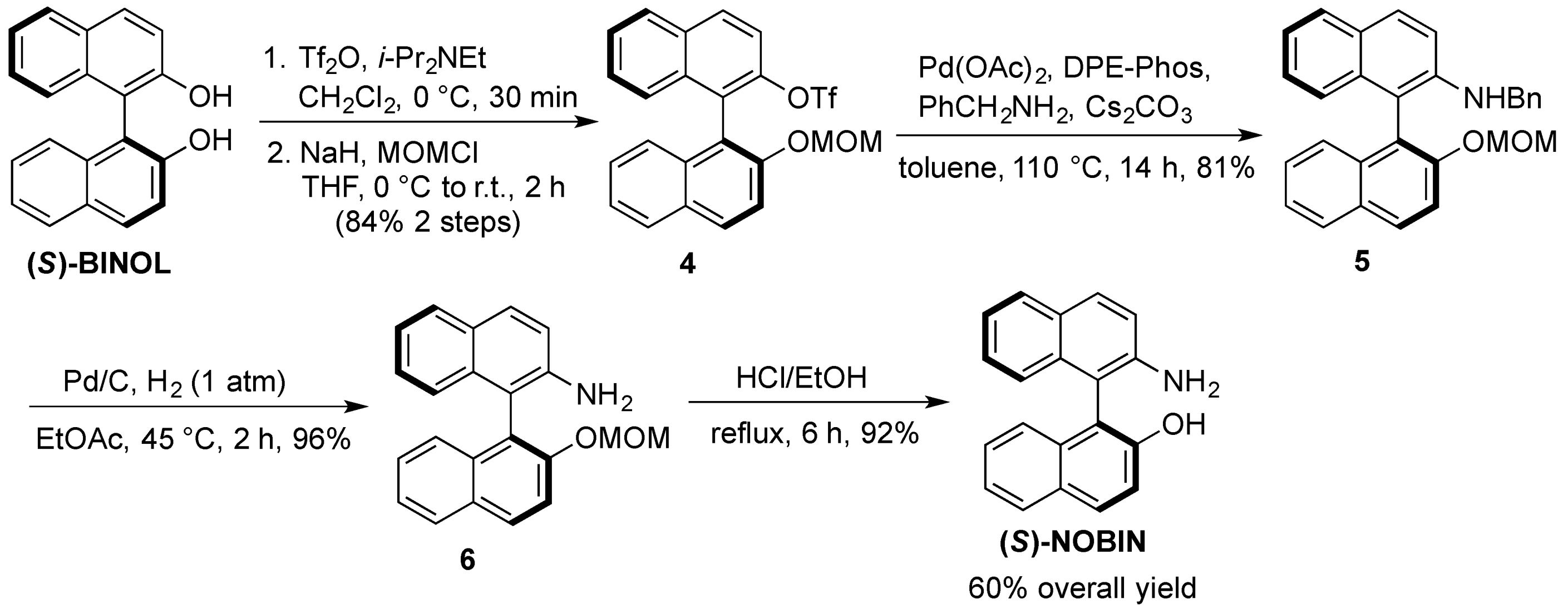
3.3.1. Multigram Synthesis of (S)-NOBIN from (S)-BINOL
(S)-2-Trifluoromethanesulfoxy-2′-methoxymethoxy-1,1′-binaphthyl (4). To a solution of (S)-BINOL(5.02 g, 17.5 mmol) and diisopropylethylamine (3.05 mL, 17.5 mmol) in dichloromethane (90 mL) was slowly added triflic anhydride (2.94 mL, 17.5 mmol) at 0 °C under nitrogen. After stirring at room temperature for 1 h, the reaction was quenched with saturated aqueous sodium bicarbonate and extracted twice with dichloromethane. The combined organic extracts were washed with brine, dried over anhydrous magnesium sulfate and evaporated under reduced pressure. The crude triflate was dissolved in tetrahydrofuran (80 mL) under nitrogen and cooled to 0 °C whereby sodium hydride (505 mg, 21 mmol) was added portionwise. After stirring for 15 min at 0 °C chloromethylmethyl ether (1.60 mL, 21 mmol) was added dropwise and the mixture was left to stir at room temperature for 2 h. The reaction was carefully quenched with water and extracted three times with ethyl acetate. The combined organic extracts were washed with brine, dried over anhydrous magnesium sulfate and evaporated under reduced pressure. The crude product was purified by flash chromatography on silica gel, hexane:dichloromethane (2:1) to yield 4 (6.84 g, 84%) as large white crystals. 1H-NMR (400 MHz, CDCl3) δ 3.23 (s, 3H), 5.04 (d, J = 7.0 Hz, 1H), 5.19 (d, J = 7.0 Hz, 1H), 7.05 (d, J = 8.5 Hz, 1H), 7.24 –7.29 (m, 1H), 7.33–7.40 (m, 3H), 7.52–7.57 (m, 1H), 7.58 (d, J = 9.0 Hz, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.99 (d, J = 8.0 Hz, 1H), 8.03 (d, J = 9.5 Hz, 1H), 8.05 (d, J = 9.5 Hz, 1H).
(S)-2-(N-Benzyl)amino-2′-methoxymethoxy-1,1′-binaphthyl (5). A suspension of 4 (6.5 g, 14.1 mmol), palladium acetate (316 mg, 1.41 mmol), DPE-Phos (1.52 g, 2.82 mmol), cesium carbonate (5.52 g, 16.9 mmol) and benzylamine (1.85 mL, 16.9 mmol) in toluene (5 mL) was heated under nitrogen at 110 °C. After 14 h the reaction was cooled, diluted with toluene (30 mL), filtered through Celite and evaporated under reduced pressure. The crude product was purified by flash chromatography on silica gel, hexane:ethyl acetate (9:1) to yield 5 (4.80 g, 81%) as a light yellow solid. 1H-NMR (400 MHz, CDCl3) δ 3.20 (s, 3H), 4.08 (t, J = 5.0 Hz, 1H), 4.42 (d, J = 5.0 Hz, 2H), 5.07 (dd, J = 21.5 Hz, 7.0 Hz, 2H), 6.97 (d, J = 8.0 Hz, 1H), 7.11–7.34 (m, 10H), 7.41 (t, J = 7.5 Hz, 1H), 7.63 (d, J = 9.0 Hz, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.81 (d, J = 9.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.99 (d, J = 9.0 Hz, 1H).
(S)-2-Amino-2′-methoxymethoxy-1,1′-binaphthyl (6). A suspension of 5 (4.51 g, 10.8 mmol) and palladium acetate (10% on carbon, 1.10 g, 1.08 mmol) in ethyl acetate (25 mL) was heated under hydrogen gas (1 atm) at 45 °C for 3 h. After filtration through Celite, the solvent was evaporated under reduced pressure to afford 6 (3.42 g, 96%) as a slightly yellow solid, which was used without further purification. 1H-NMR (400 MHz, CDCl3) δ 3.18 (s, 3H), 3.60 (bs, 2H), 5.05 (dd, J = 21.5 Hz, 7.0 Hz, 2H), 7.02 (d, J = 8.0 Hz, 1H), 7.10–7.31 (m, 5H), 7.39 (t, J = 7.5 Hz, 1H), 7.59 (d, J = 9.0 Hz, 1H), 7.75–7.84 (m, 2H), 7.90 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H).
(S)-NOBIN. To a solution of 6 (3.31 g, 10.1 mmol) in methanol (25 mL) and dichloromethane (25 mL) was added concentrated sulfuric acid (1.6 mL). After refluxing for 5 h, the reaction was cooled to room temperature, quenched with saturated aqueous sodium bicarbonate (pH 8–9) and extracted three times with dichloromethane. The combined organic extracts were washed with brine and dried over anhydrous magnesium sulfate and evaporated under reduced pressure to afford (S)-NOBIN (2.71 g, 94%) as a white solid which was used without further purification or recrystallized from benzene for spectroscopy. 1H-NMR (400 MHz, CDCl3) δ 3.70 (bs, 2H), 4.36 (bs, 1H), 7.06 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 9.0 Hz, 1H), 7.16–7.42 (m, 6H), 7.81 (d, J = 7.5 Hz, 1H), 7.84 (d, J = 9.0 Hz, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.93 (d, J = 9.0 Hz, 1H).
3.3.2. Synthesis of (S)-2-N-Acetyl-2′-hydroxy-1,1′-binaphthyl (7)
To a solution of (S)-NOBIN (4.94 g, 17.2 mmol) in pyridine (70 mL) was slowly added acetyl chloride (2.7 mL, 37.8 mmol) at 0 °C under nitrogen. After stirring at room temperature for 3 h, the reaction mixture was carefully poured onto ice cold water and extracted 3 times with dichloromethane. The extracts were washed twice with 5% aqueous hydrochloric acid, twice with water, twice with saturated aqueous sodium bicarbonate, brine, dried over anhydrous magnesium sulfate and evaporated under reduced pressure to afford the crude N,O-diacetate. To a solution of crude diacetate in methanol (450 mL) was added sodium methoxide (140 μL, 25% in methanol). After stirring at room temperature for 1 h, the solvent was evaporated under reduced pressure and the residue redissolved in dichloromethane. The organic layer was washed twice with water, dried over anhydrous magnesium sulfate and evaporated under reduced pressure to afford 7 (5.3 g, 94%) as an off-white solid that was used without further purification. 1H-NMR (400 MHz, CDCl3) δ 1.82 (s, 3H), 5.24 (bs, 1H), 6.91 (bs, 1H), 7.01 (d, J = 8.5 Hz, 1H), 7.14 (d, J = 8.5 Hz, 1H), 7.25–7.33 (m, 2H), 7.34–7.48 (m, 3H), 7.88–7.96 (m, 2H), 7.98 (d, J = 9.0 Hz, 1H), 8.04 (d, J = 9.0 Hz, 1H), 8.54 (d, J = 9.0 Hz, 1H); low resolution ESI [M+H+]: 328 (calcd. for (C22H18NO2)+, 328).
3.3.3. Synthesis of Aminophosphine Oxides 10a–d
To a solution of 7 (5.15 g, 15.7 mmol) and pyridine (3.82 mL, 47.1 mmol) in dichloromethane (80 mL) was slowly added triflic anhydride (2.91 mL, 17.3 mmol) at 0 °C under nitrogen. After stirring at room temperature for 3 h, the reaction was diluted with dichloromethane (80 mL). The organic layer was washed with 5% aqueous hydrochloric acid, saturated aqueous sodium bicarbonate, water, dried over anhydrous magnesium sulfate and evaporated under reduced pressure to afford the crude (S)-2-N-acetyl-2′-(trifluoromethanesulfonyloxy)1,1′-binaphthyl (8) which was used immediately for the phosphonylation reactions without further purification or characterization. To a suspension of the crude triflate (1.0 equiv.), phosphine oxide (2.1 equiv.), 1,4-bis(diphenylphosphino)butane (0.2 equiv.), palladium acetate (0.2 equiv.) in DMSO, 0.156 mmol/mL) was added diisopropylethylamine (3.7 equiv.). The reaction mixture was heated under nitrogen at 120 °C. After 12 h the reaction was cooled to room temperature, diluted with dichloromethane and the dark organic solution was washed twice with 5% aqueous hydrochloric acid, twice with water, saturated sodium bicarbonate, brine and dried (MgSO4). The filtrate was concentrated under reduced pressure and the residue was purified by flash chromatography on silica gel to afford the purified N-acetylphosphine oxide compounds 9a–d which were used as intermediates to the aminophosphine compounds 10a–d.
(S)-2-N-Acetyl-2′-(diphenylphosphinoyl)-1,1′-binaphthyl (9a). The crude triflate (4.30 g, 9.36 mmol), diphenylphosphine oxide (3.98 g, 19.7 mmol), dppb (798 mg, 1.87 mmol), palladium acetate (419 mg, 1.87 mmol) were suspended in DMSO (60 mL). Diisopropylethylamine (6.03 mL, 34.6 mmol) was added and the reaction heated at 120 °C. After 16 h, the reaction was worked up according to the general procedure and the crude residue was purified by flash chromatography on silica gel toluene/ethyl acetate (2:1 to 1:1) to yield 9a (3.71 g, 78%) as an off white solid. 1H-NMR (400 MHz, CDCl3) δ 1.92 (s, 3H), 6.52 (d, J1 = 8.5 Hz, 1H), 6.60–6.67 (m, 2H), 6.77 (t, J = 7.5 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 7.11–7.25 (m, 5H), 7.41–7.75 (m, 8H), 7.86–8.00 (m, 4H); 31P-NMR (162 MHz, CDCl3) δ 28.9.
(S)-2-N-Acetyl-2′-(bis(3,5-difluorophenyl)phosphinoyl)-1,1′-binaphthyl (9b). The crude triflate (130 mg, 0.28 mmol), bis(3,5-difluorophenyl)phosphine oxide (115 mg, 0.42 mmol), 1,4-bis (diphenylphosphino)butane (36 mg, 0.08 mmol), palladium acetate (19 mg, 0.08 mmol) were suspended in DMSO (2.2 mL), to which was added diisopropylethylamine (1.17 mL, 6.72 mmol). The reaction mixture was heated under nitrogen at 120 °C. After 12 h the reaction was cooled to room temperature, diluted with dichloromethane and the dark organic solution was washed with saturated sodium bicarbonate, brine and dried (MgSO4). The filtrate was concentrated under reduced pressure and the residue was purified by flash chromatography on silica gel, ethyl acetate/hexanes (1:2 to 1:1) to afford 9b (86 mg, 53%) as a beige solid. 1H-NMR (400 MHz, CDCl3) δ 1.89 (3H, s), 6.20 (t, J = 9.0 Hz, 1H), 6.55 (d, J = 8.5 Hz, 1H), 6.51–6.67 (m, 2H), 7.04–7.35 (m, 5H), 7.43–7.86 (m, 5H), 7.79 (m, 2H), 7.95 (d, J = 8.5 Hz, 1H), 8.03 (dd, J = 8.5 Hz, 2.0 Hz, 1H), 9.16 (bs, 1H); 31P-NMR (162 MHz, CDCl3) δ 27.3.
(S)-2-N-Acetyl-2′-(bis(4-fluorophenyl)phosphinoyl)-1,1′-binaphthyl (9c). The crude triflate (327 mg, 0.71 mmol), bis(4-fluorophenyl)phosphine oxide (350 mg, 1.49 mmol), dppb (61 mg, 0.14 mmol), palladium acetate (31 mg, 0.14 mmol) were suspended in DMSO (4.6 mL). Diisopropylethylamine (458 μL, 2.63 mmol) was added and the reaction heated at 120 °C. After 16 h, the reaction was worked up according to the general procedure and the crude residue was purified by flash chromatography on neutral alumina eluting with methanol/dichloromethane/toluene (1:4:15) to yield 9c (283 mg, 73%) as a yellow solid. 1H-NMR (400 MHz, CDCl3) δ 1.91 (3H, s), 6.33 (t, J = 8.5 Hz, 2H), 6.51 (d, J = 8.5 Hz, 1H), 7.01 (t, J = 7.5 Hz, 1H), 7.08–7.28 (m, 6H), 7.44–7.77 (m, 6H), 7.89–7.99 (m, 4H), 9.49 (bs, 1H); 31P-NMR (162 MHz, CDCl3) δ 29.9.
(S)-2-N-Acetyl-2′-(bis(4-methoxyphenyl)phosphinoyl)-1,1′-binaphthyl (9d). The crude triflate (250 mg, 0.54 mmol), bis(4-methoxyphenyl)phosphine oxide (300 mg, 1.14 mmol), dppb (46 mg, 0.11 mmol), palladium acetate (24 mg, 0.11 mmol) were suspended in DMSO (3.5 mL). Diisopropylethylamine (351 μL, 2.01 mmol) was added and the reaction heated at 120 °C. After 16 h, the reaction was worked up according to the general procedure and the crude residue was purified by flash chromatography on silica gel toluene/ethyl acetate (1:2 to 1:99) to yield 9d (225 mg, 73%) as a yellow solid. 1H-NMR (400 MHz, CDCl3) δ 1.92 (s, 3H), 3.54 (s, 3H), 3.88 (s, 3H), 6.13 (dd, J = 8.5 Hz, 2.0 Hz, 2H), 6.50 (d, J = 8.5 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 7.00–7.14 (m, 4H), 7.21 (t, J = 7.50 Hz, 2H), 7.48–7.59 (m, 3H), 7.65–7.77 (m, 2H), 7.81–7.96 (m, 4H), 9.77 (bs, 1H); 31P-NMR (162 MHz, CDCl3) δ 31.3.
To a solution of the appropriate N-acetylphosphine oxide 9a–d in ethanol (0.1 mmol/mL) was added 5 M hydrochloric acid (0.025 mL of acid per mmol of phosphine oxide). After refluxing for 6 h, the reaction was cooled to room temperature, and quenched with saturated sodium bicarbonate to pH 9. The milky mixture was extracted three times with ether, and the combined organic layers were washed with brine and dried (MgSO4). The filtrate was evaporated under reduced pressure to afford the aminophosphine oxides 10a–d.
(S)-2-Amino-2′-(diphenylphosphinoyl)-1,1′-binaphthyl (10a). A solution of phosphine oxide (700 mg, 1.37 mmol) in ethanol (14 mL) was added 5M hydrochloric acid (3.5 mL) was heated to reflux. After 6 h, the reaction was worked up according to the general procedure to afford aminophosphine 10a (623 mg, 97%) as an off white solid. 1H-NMR (400 MHz, CDCl3) δ 3.88 (bs, 2H), 6.51 (d, J = 8.5 Hz, 1H), 6.76–6.82 (m, 2H), 6.87 (d, J = 8.8 Hz, 1H), 6.89–6.98 (m, 2H), 7.02 (t, J = 7.3 Hz, 1H), 7.20–7.30 (m, 4H), 7.35–7.50 (m, 5H), 7.55 (t, J = 7.3 Hz, 1H), 7.69–7.80 (m, 3H), 7.92 (d, J = 8.2 Hz, 1H), 7.96 (d, J = 8.8 Hz, 1H); 31P-NMR (162 MHz, CDCl3) δ 29.1.
(S)-2-Amino-2′-(bis(3,5-difluorophenyl)phosphinoyl)-1,1′-binaphthyl (10b). A solution of phosphine oxide (80 mg, 0.14 mmol) in ethanol (1.4 mL) was added 5M hydrochloric acid (350 μL) was heated to reflux. After 6 h, the reaction was worked up according to the general procedure to afford aminophosphine 10b (66 mg, 89%) as a dull yellow solid. [α]D20 = +131 (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 3.92 (bs, 2H), 6.37 (t, J = 8.7 Hz, 1H), 6.53 (d, J = 8.2 Hz, 1H), 6.73 (dd, J = 13.3, 5.3, 2H), 6.89–7.15 (m, 4H), 7.26–7.38 (m, 4H), 7.52 (d, J = 7.8 Hz, 1H), 7.48–7.76 (m, 3H), 7.98 (d, J = 8.3 Hz, 1H), 8.04 (dd, J = 8.8, 2.5 Hz, 1H); 13C-NMR (100 MHz, CDCl3) δ 106.7 (t, J = 25 Hz), 108.0 (t, J = 25 Hz), 113.4, 115.2, 118.9, 122.8, 124.5, 126.7, 127.5, 127.6, 128.1, 128.4, 128.6, 128.7, 129.2, 129.3, 129.5, 130.27, 131.1, 132.4, 133.5, 134.1, 135.1, 136.1, 137.3, 142.4, 145.0, 160.8, 161.8, 163.4, 164.4; 31P-NMR (162 MHz, CDCl3) δ 25.3 (p, J = 6.5 Hz). IR (cm−1) ν 1291, 3342; HRMS (ESI) [M+H+]: 542.1293, calcd for (C32H21NOPF4)+: 542.1297.
(S)-2-Amino-2′-(bis(4-fluorophenyl)phosphinoyl)-1,1′-binaphthyl (10c). A solution of phosphine oxide (359 mg, 0.66 mmol) in ethanol (6.7 mL) was added hydrochloric acid (5 M, 1.7 mL) was heated to reflux. After 6 h, the reaction was worked up according to the general procedure to afford aminophosphine 10c (314 mg, 94%) as a light yellow solid. [α]D20 = +166 (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 4.01 (bs, 2H), 6.45–6.58 (m, 3H), 6.94 (d, J = 8.7 Hz, 1H), 6.98 (t, 7.6 Hz, 1H), 7.02–7.14 (m, 3H), 7.18–7.34 (m, 4H), 7.44–7.54 (m, 2H), 7.58 (t, J = 7.6 Hz, 1H), 7.66–7.77 (m, 3H), 7.95 (d, J = 8.2 Hz, 1H), 8.00 (dd, J = 8.7 Hz, 2.5 Hz, 1H); 13C-NMR (100 MHz, CDCl3) δ 114.9, 115.9, 119.2, 122.6, 124.8, 126.6, 126.8, 127.4, 128.0, 128.1, 128.2, 128.6, 128.9, 129.1, 129.7, 130.5, 130.7, 131.8, 132.9, 133.5, 134.3, 134.5, 135.9, 141.4, 144.0, 163.4, 166.1; 31P-NMR (162 MHz, CDCl3) δ 28.2; IR (cm−1) ν 1158, 3324; HRMS (ESI) [M+H+]: 506.1478, calcd for (C32H23NOPF2)+: 506.1485.
(S)-2-Amino-2′-(bis(4-methoxyphenyl)phosphinoyl)-1,1′-binaphthyl (10d). A solution of phosphine oxide (120 mg, 0.21 mmol) in ethanol (2.14 mL) was added hydrochloric acid (5 M, 536 μL) was heated to reflux. After 6 h, the reaction was worked up according to the general procedure to afford aminophosphine 10d (91 mg, 83%) as a light yellow solid. [α]D20 = +174 (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 3.65 (s, 3H), 3.84 (s, 3H), 3.94 (bs, 2H), 6.31 (dd, J = 8.7 Hz, 2.4 Hz, 2H), 6.52 (d, 8.7 Hz, 1H), 6.88 (dd, J = 8.7 Hz, 2.4 Hz, 2H), 6.91–7.00 (m, 2H), 7.06 (t, J = 7.5 Hz, 1H), 7.10–7.32 (m, 4H), 7.50 (t, J = 9.2 Hz, 2H), 7.55 (t, J = 7.5 Hz, 1H), 7.60-7.70 (m, 2H), 7.78 (dd, J = 11.2 Hz, 8.5 Hz, 1H), 7.93 (d, J = 8.5 Hz, 1H), 7.97 (dd, J = 8.5 Hz, 1.8 Hz, 1H); 13C-NMR (100 MHz, CDCl3) δ 113.3, 114.2, 119.43, 122.31, 122.7, 123.8, 124.3, 125.2, 126.3, 127.4, 127.8, 127.9, 128.3, 128.5, 128.7, 129.4, 129.6, 130.2, 132.0, 132.3, 132.4, 133.0, 133.5, 133.9, 134.0, 134.4, 135.7, 141.0, 143.8, 161.5, 162.3; 31P-NMR (162 MHz, CDCl3) δ 29.5; IR (cm−1) ν 1251, 3324; HRMS (ESI) [M+H+]: 530.1884, calcd for (C34H29NO3P)+: 530.1885.
3.3.4. Synthesis of aminophosphines 11a–d
The corresponding phosphine oxide 10a–d was dissolved in neat phenyl silane and heated at 120 °C for 24 h. The dark reaction was dried down under a gentle stream of N2 gas to remove most of the phenyl silane, and the crude residue was purified by flash chromatography on silica gel to afford the purified aminophosphine compounds 11a–d.
(S)-2-N-Amino-2′-(diphenylphosphino)-1,1′-binaphthyl (11a). The phosphine oxide 10a (450 mg, 0.96 mmol) was heated in neat phenyl silane (900 μL) at 120 °C for 24 h. The residue was purified by flash chromatography on silica gel, dichloromethane/hexanes (1:2 to 1:1) to afford 11a (418 mg, 96%) as a white solid. 1H-NMR (400 MHz, CDCl3) δ 3.29 (bs, 2H), 6.68 (d, J = 8.5 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 7.03 (d, J = 8.7 Hz, 1H), 7.05–7.21 (m, 7H), 7.24–7.36 (m, 6H), 7.45–7.53 (m, 2H), 7.74 (d, J = 8.0 Hz, 1H), 7.81 (d, J = 8.7 Hz, 1H), 7.90 (d, J = 8.5 Hz, 2H); 31P-NMR (162 MHz, CDCl3) δ −11.3.
(S)-2-N-Amino-2′-(bis(3,5-difluorophenylphosphino)-1,1′-binaphthyl (11b). The phosphine oxide 10b (60 mg, 0.11 mmol) was heated in neat phenyl silane (150 μL) at 120 °C for 24 h. The residue was purified by flash chromatography on silica gel, dichloromethane/hexanes (1:2 to 1:1) to afford 11b (42 mg, 72%) as a white solid. [α]D20 = +27 (c 1.1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 3.51 (bs, 2H), 6.49–6.62 (m, 3H), 6.67 (t, J = 8.7 Hz, 1H), 6.73–6.84 (m, 3H), 6.98 (t, J = 7.5 Hz, 1H), 7.10 (d, J = 8.7 Hz, 1H), 7.16 (t, J = 7.5 Hz, 1H), 7.31–7.46 (m, 3H), 7.58 (t, J = 7.5 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 8.8 Hz, 1H), 7.92–8.04 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 104.8 (m), 116.3 (m), 118.3, 122.7, 124.3, 126.7, 126.8, 127.8, 128.2, 128.4, 128.7, 129.6, 130.1, 130.4, 133.2, 134.6, 134.8, 134.9, 140.1 (m), 142.3, 142.9, 143.3, 162.0 (m), 164.5 (m); 31P-NMR (162 MHz, CDCl3) δ −7.3; IR (cm−1) ν 3379; HRMS (ESI) [M+H+]: 526.1342, calcd for (C32H21NF4P)+: 526.1348.
(S)-2-N-Amino-2′-(bis(4-fluorophenylphosphino)-1,1′-binaphthyl (11c). The phosphine oxide 10c (115 mg, 0.23 mmol) was heated in neat phenyl silane (150 μL) at 120 °C for 24 h. The residue was purified by flash chromatography on silica gel, dichloromethane/hexanes (2:3 to 3:2) to afford 11b (90 mg, 81%) as a white solid. [α]D20 = +22 (c 0.95, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 3.25 (bs, 2H), 6.55 (d, J = 8.5 Hz, 1H), 6.78–6.89 (m, 2H), 6.93 (t, J = 7.5 Hz, 1H), 6.96–7.06 (m, 5H), 7.15 (t, J = 7.5 Hz, 1H), 7.20–7.37 (m, 4H), 7.41 (dd, J = 8.5, 2.8 Hz, 1H), 7.53 (t, J = 7.5 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.93 (d, J = 8.5 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 115.9 (m), 118.4, 122.6, 124.6, 125.6, 126.7, 127.5, 127.7, 128.3, 128.5, 129.1, 130.1, 132.6, 133.2, 133.7, 134.6, 135.9 (m), 137.3, 142.2, 162.3, 164.8; 31P-NMR (162 MHz, CDCl3) δ −13.2; IR (cm−1) ν 3379; HRMS (ESI) [M+H+]: 490.1528, calcd for (C32H23NF2P)+: 490.1536.
(S)-2-N-Amino-2′-(bis(4-methoxyphenylphosphino)-1,1′-binaphthyl (11d). The phosphine oxide 10d (70 mg, 0.13 mmol) was heated in neat phenyl silane (100 μL) at 120 °C for 24 h. The residue was purified by flash chromatography on silica gel, 85% dichloromethane/hexane to afford 11d (54 mg, 81%) as a white solid. [α]D20 = +31 (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 3.22 (bs, 2H), 3.76 (s, 3H), 3.82 (s, 3H), 6.63 (d, J = 8.5 Hz, 1H), 6.70 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.5 Hz, 2H), 6.91–7.03 (m, 3H), 7.06 (d, J = 8.5 Hz, 1H), 7.14 (t, J = 7.5 Hz, 1H), 7.18–7.35 (m, 4H), 7.43–7.55 (m, 2H), 7.75 (d, J = 8.5 Hz, 1H), 7.81 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.5 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 55.6, 55.7, 114.4, 118.6, 122.5, 124.9, 126.5, 126.6, 127.3, 127.4, 128.2, 128.3, 128.5, 128.7, 129.1, 129.2, 129.9, 130.4, 133.3, 134.4, 134.6, 135.6, 138.5, 141.0, 141.4, 142.4, 160.3, 160.4; 31P-NMR (162 MHz, CDCl3) δ −14.2; IR (cm−1) ν 3374; HRMS (ESI) [M+H+]: 512.1774, calcd for (C34H27NO2P)+: 512.1779.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
