Biomedical Importance of Indoles



Abstract
:1. Introduction
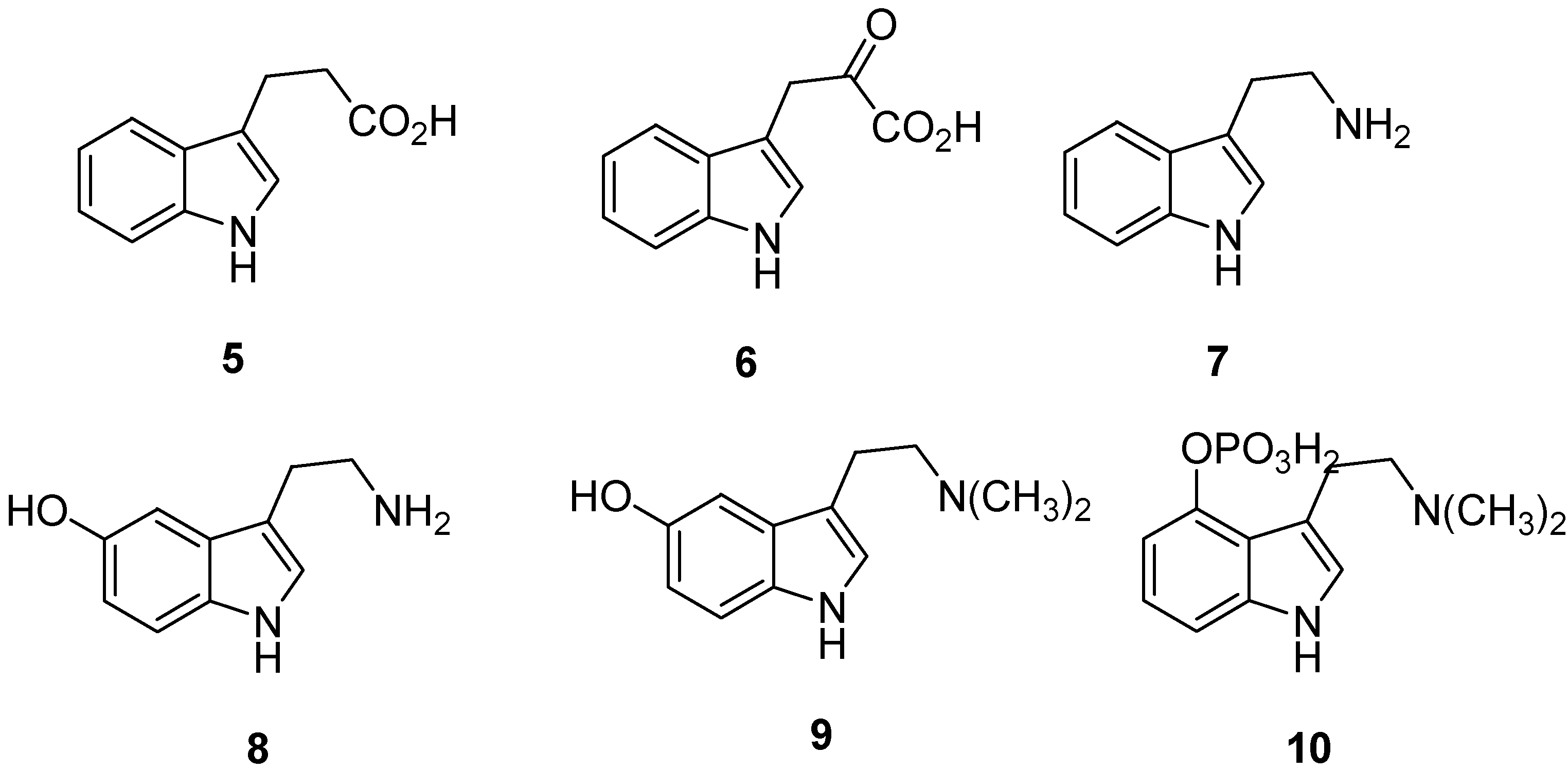
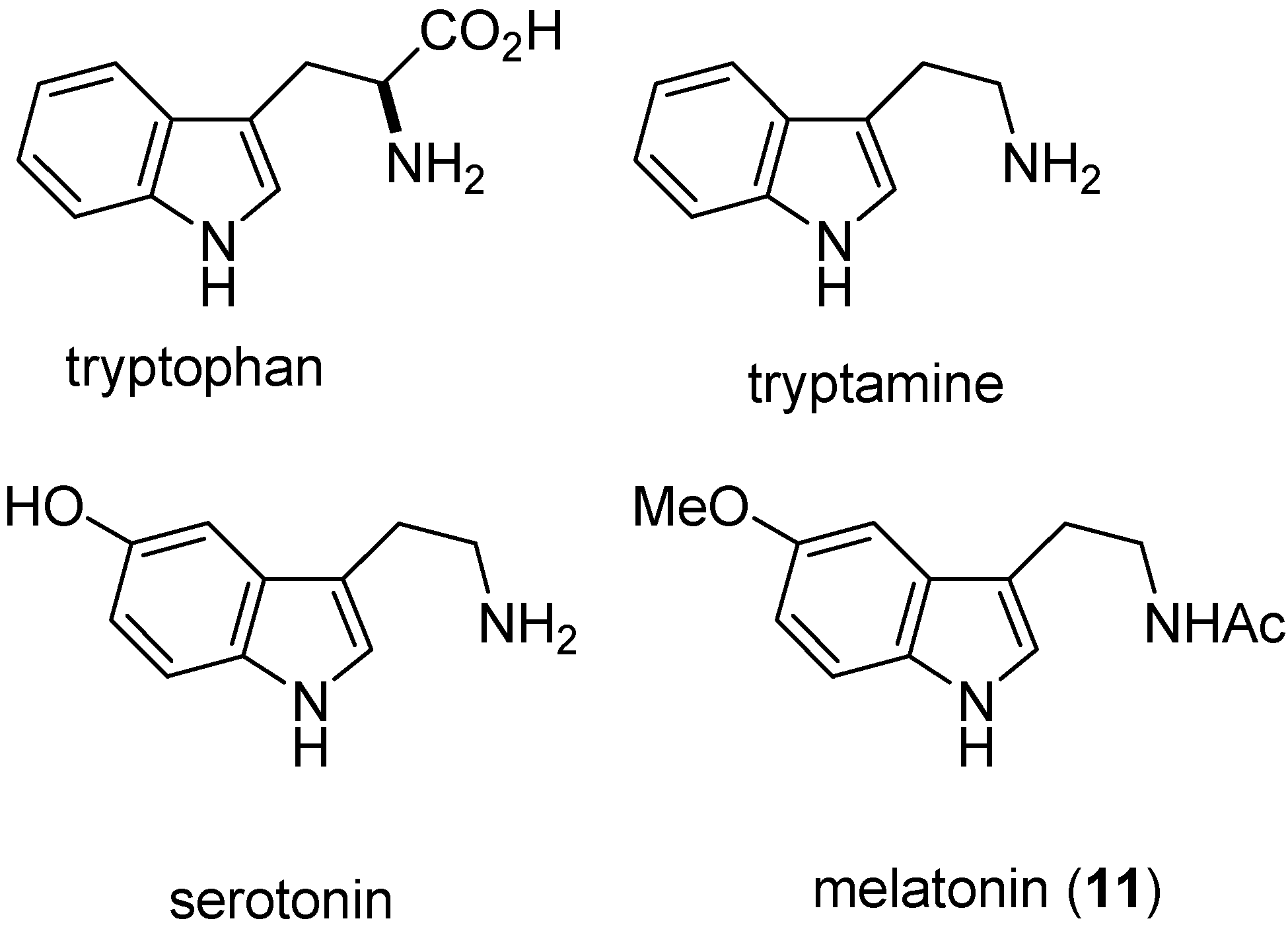
2. Indole: Chemical and Biological Importance
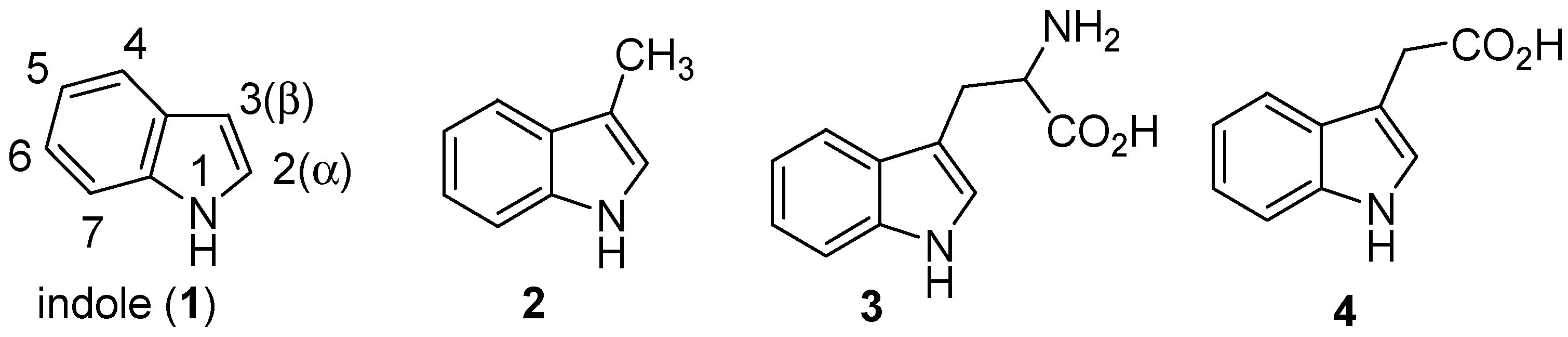
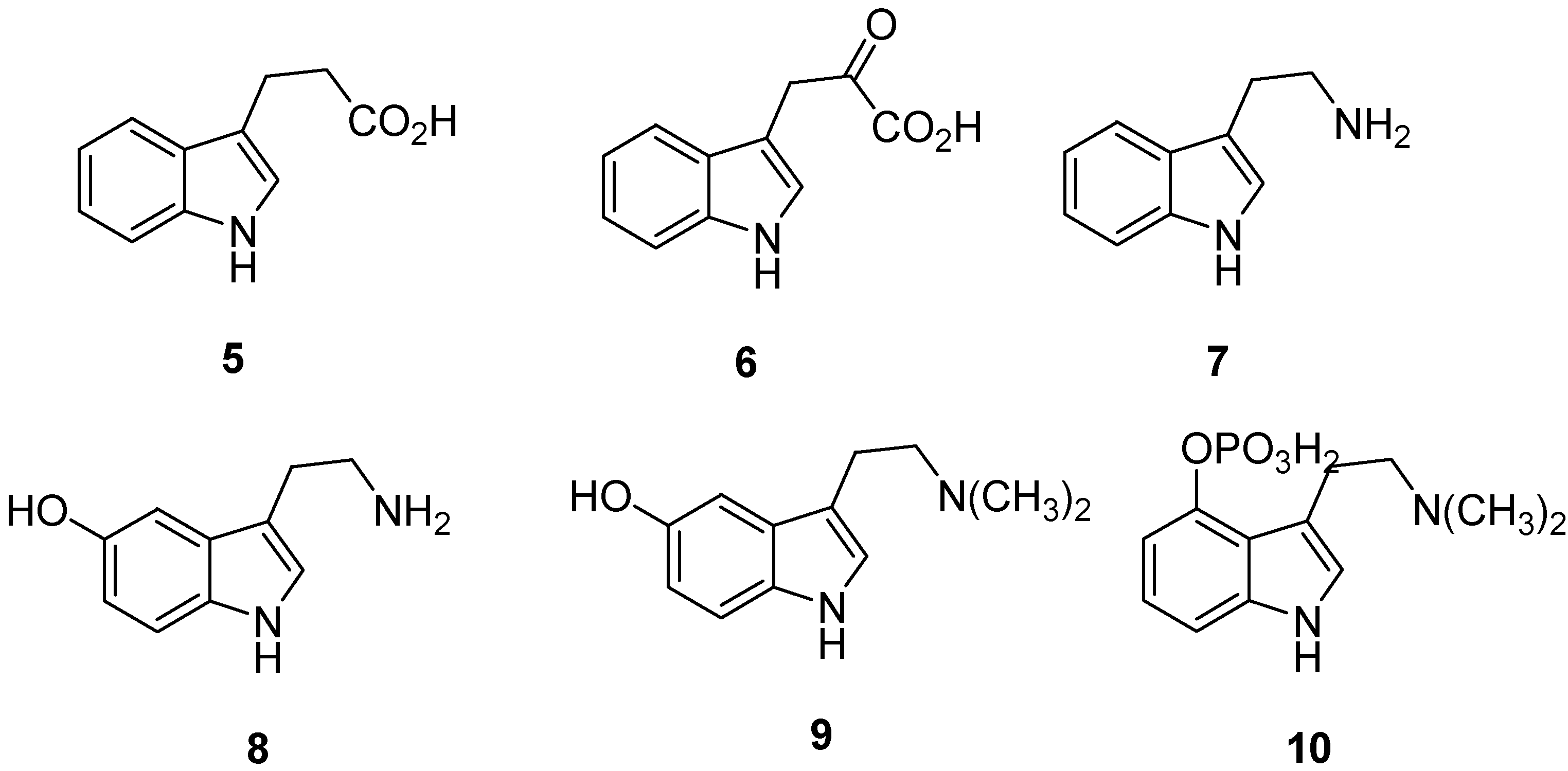
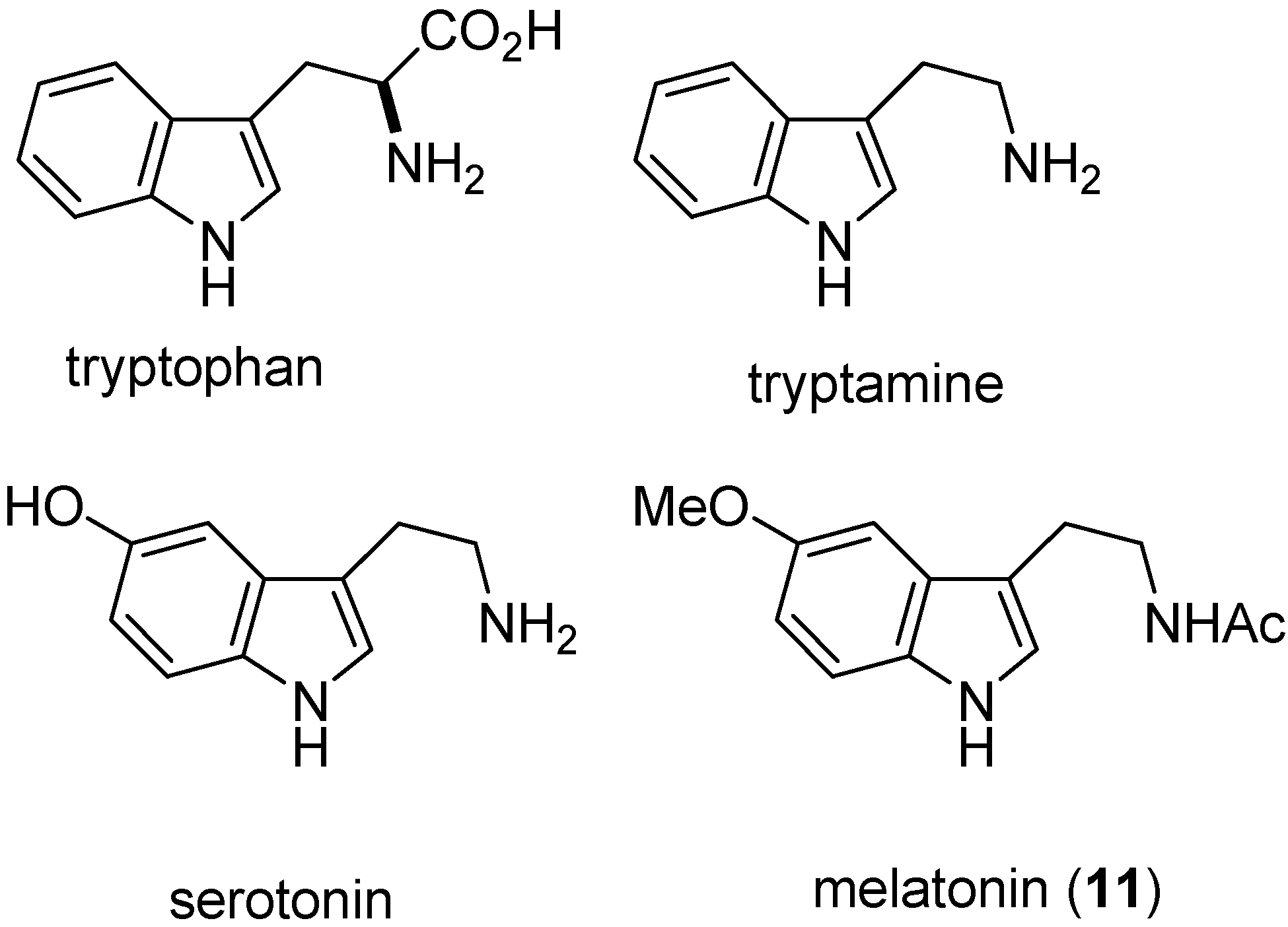
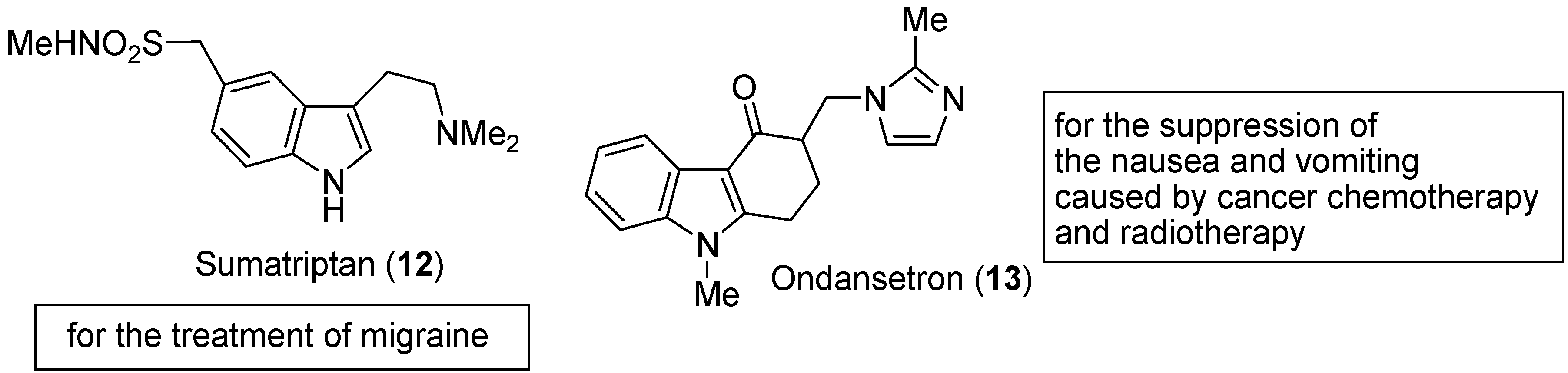
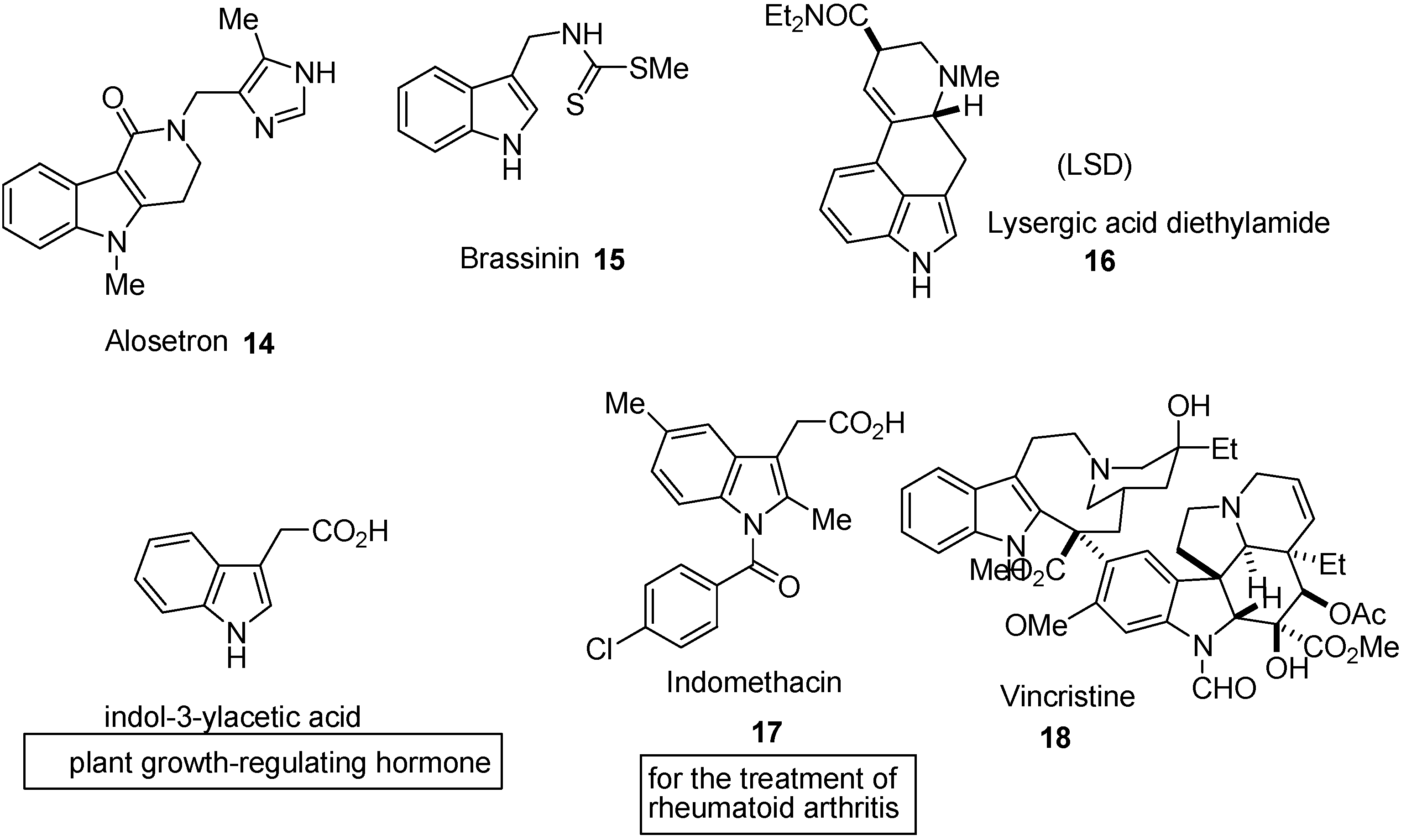
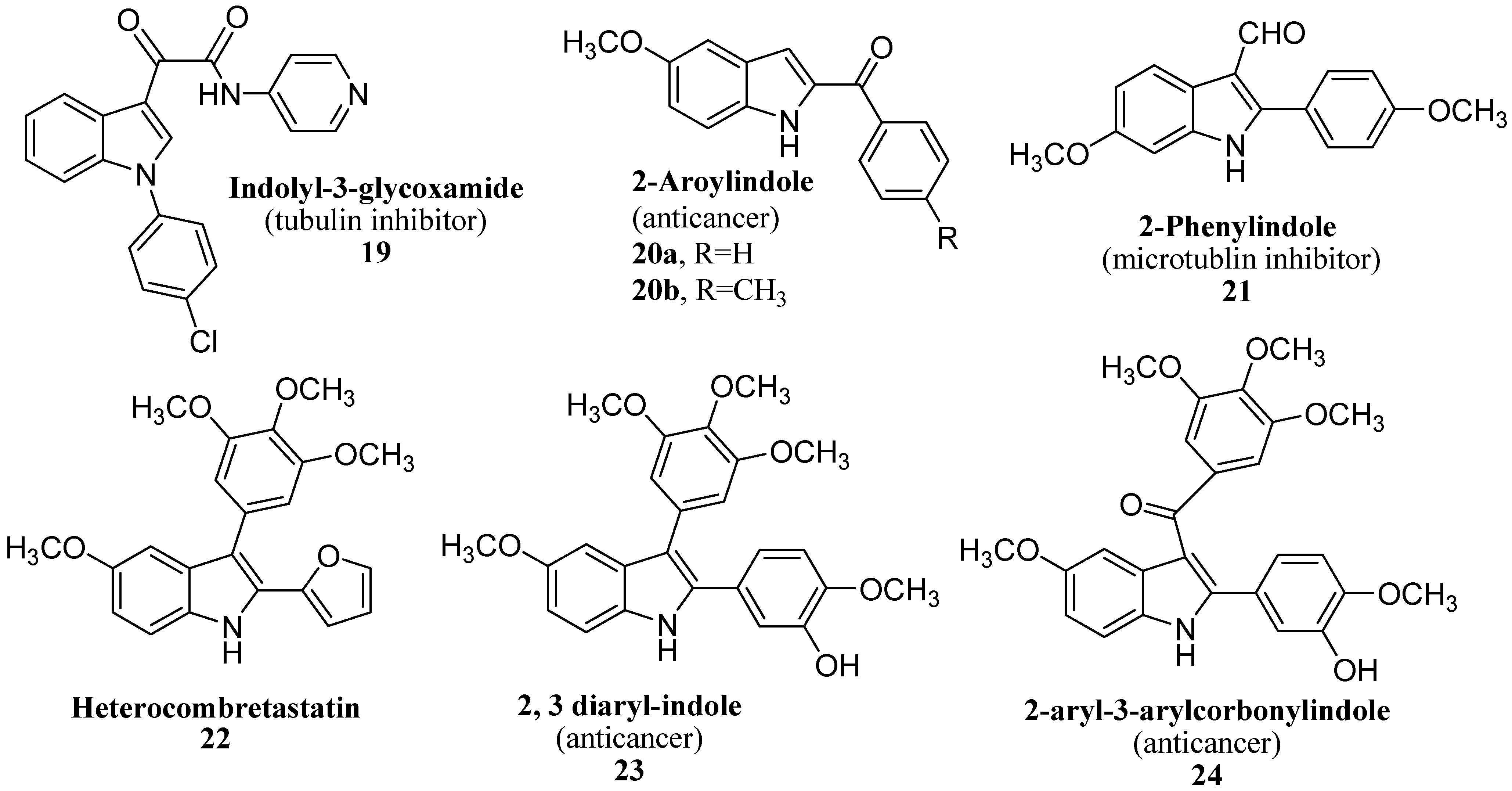
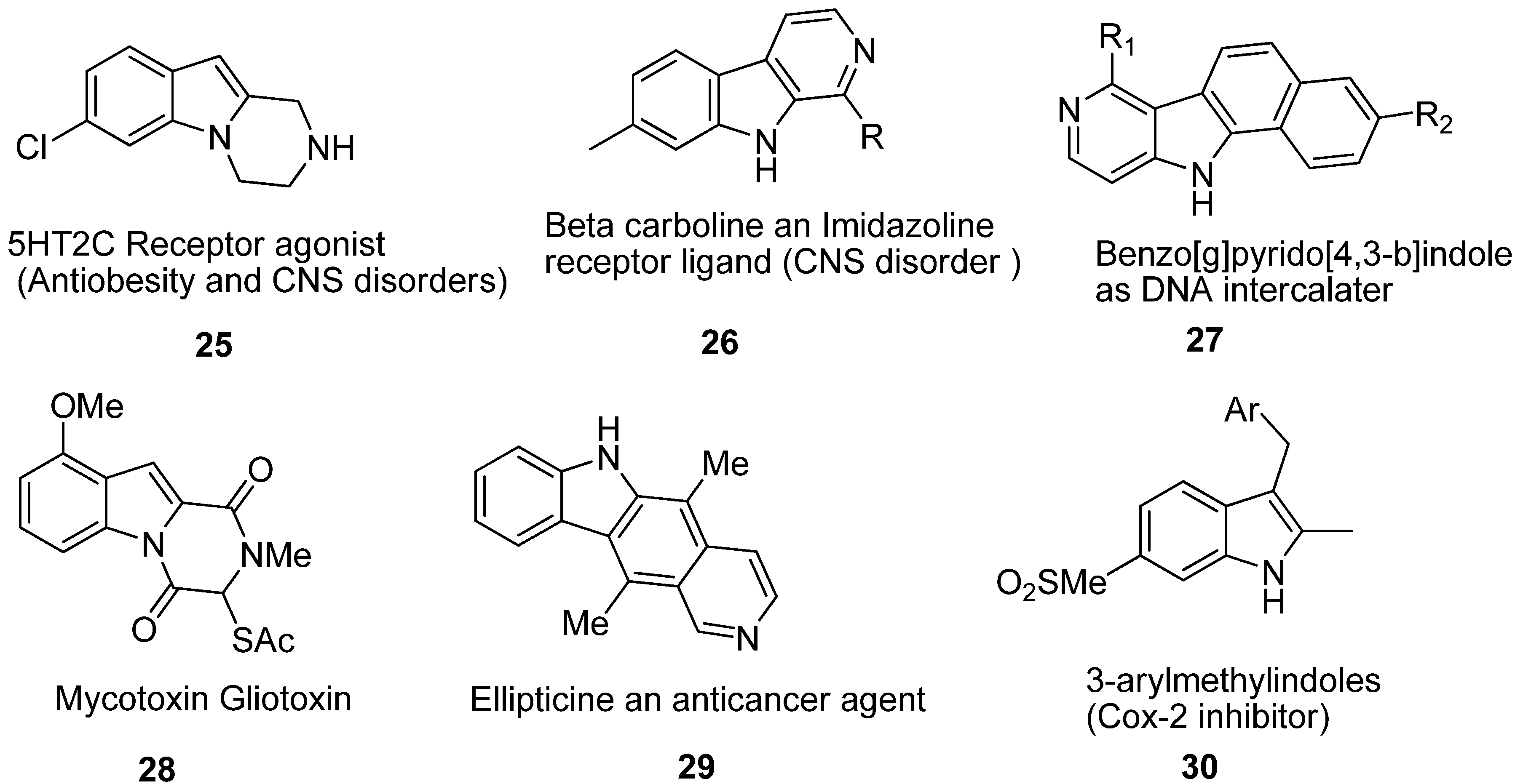
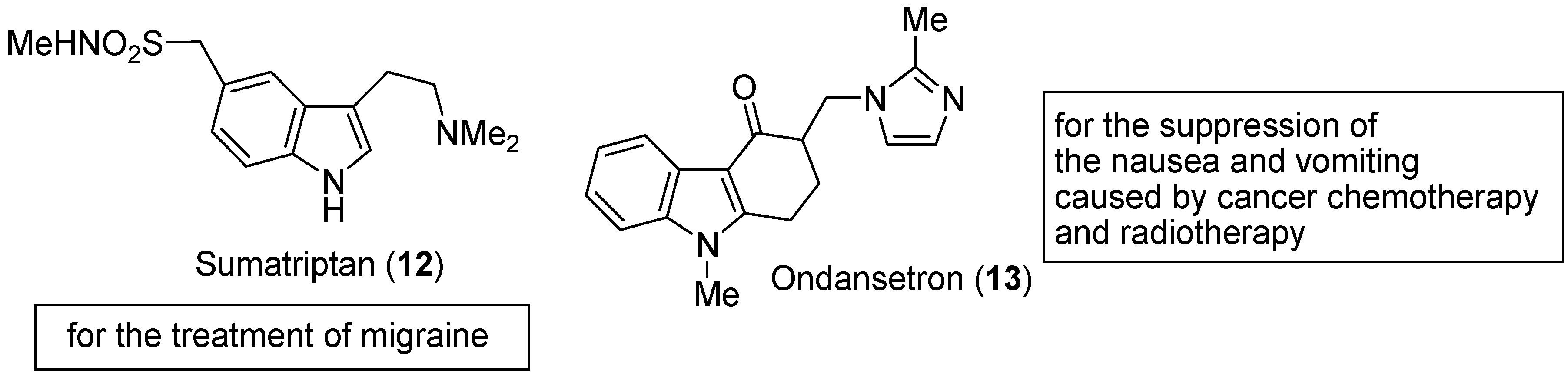
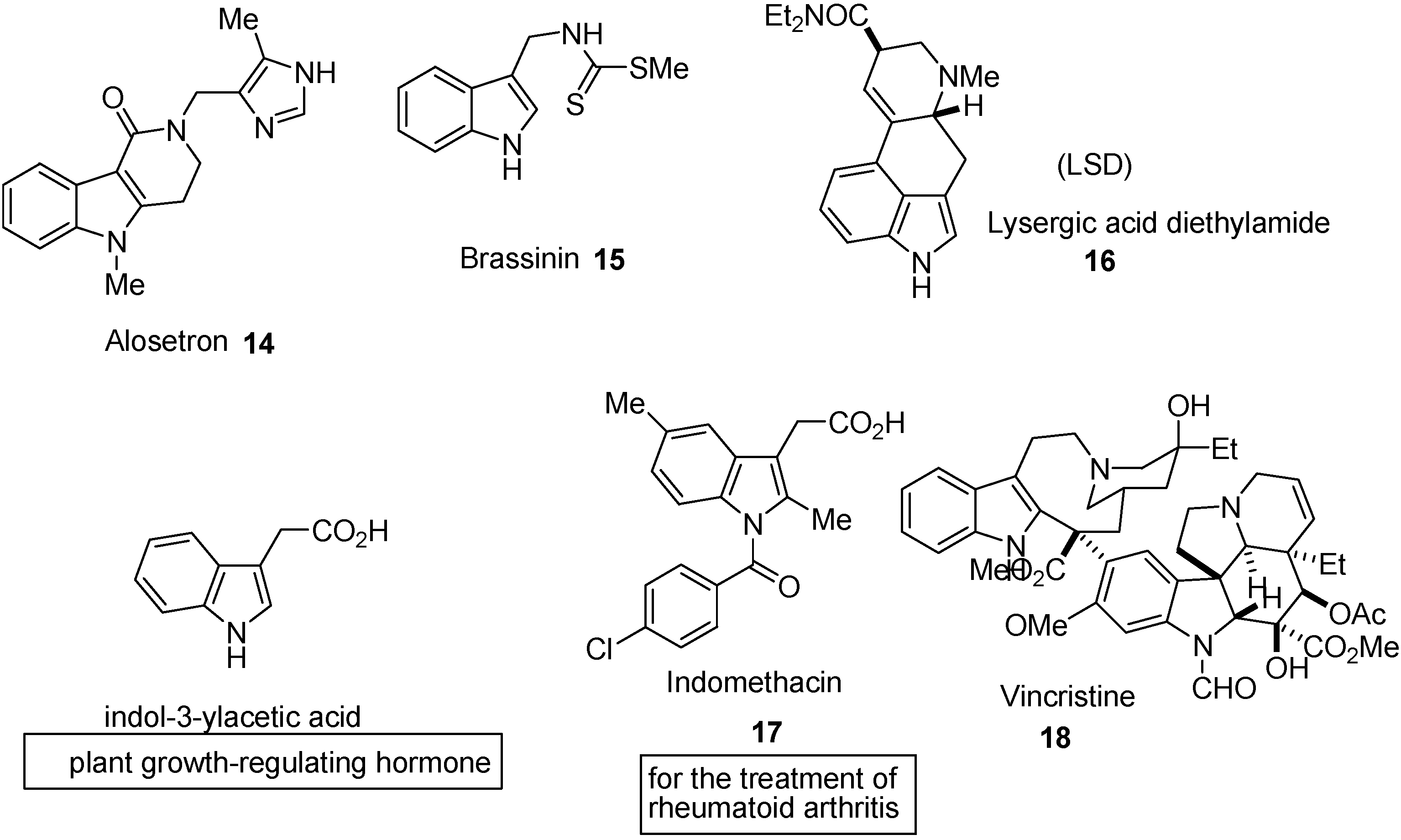
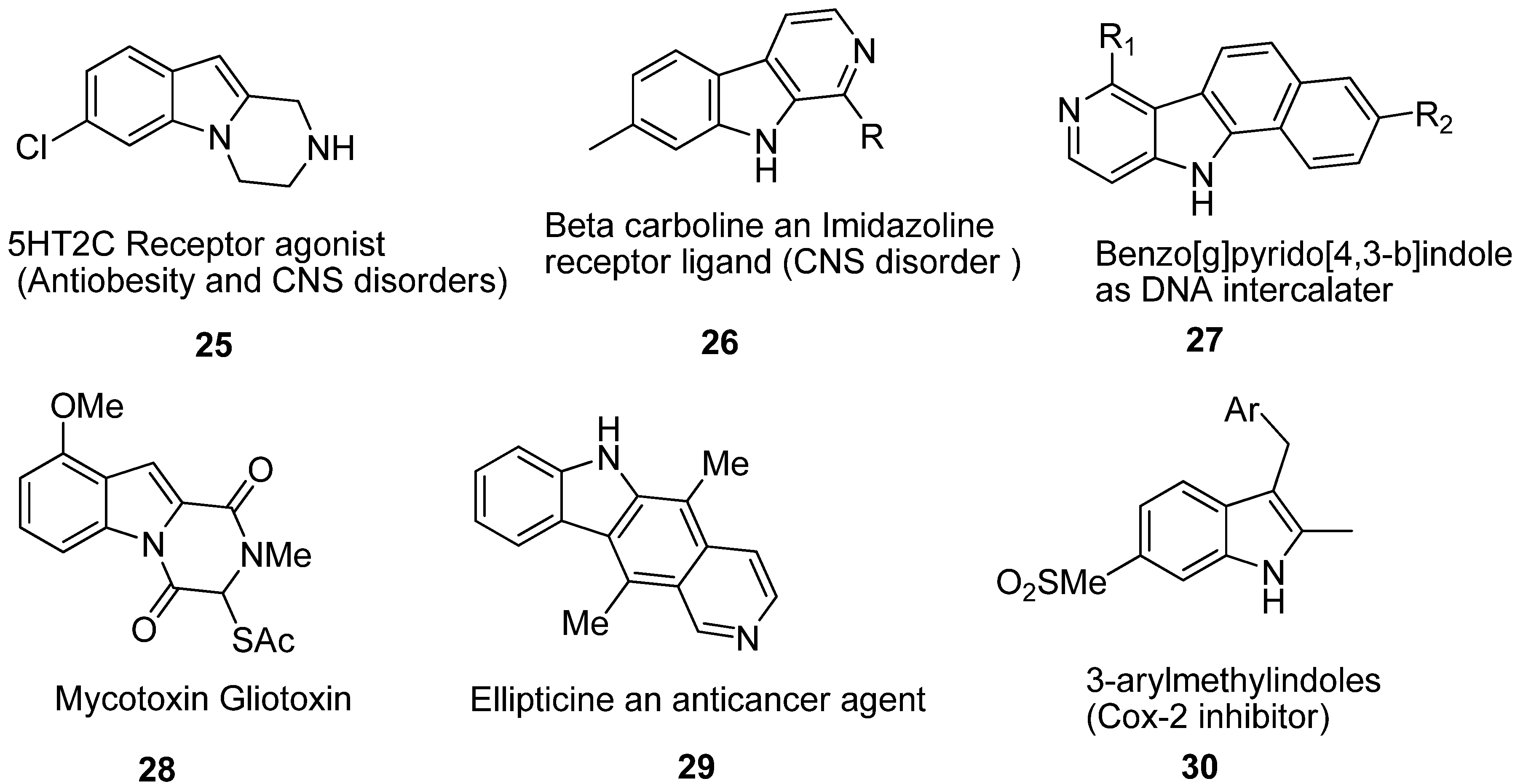
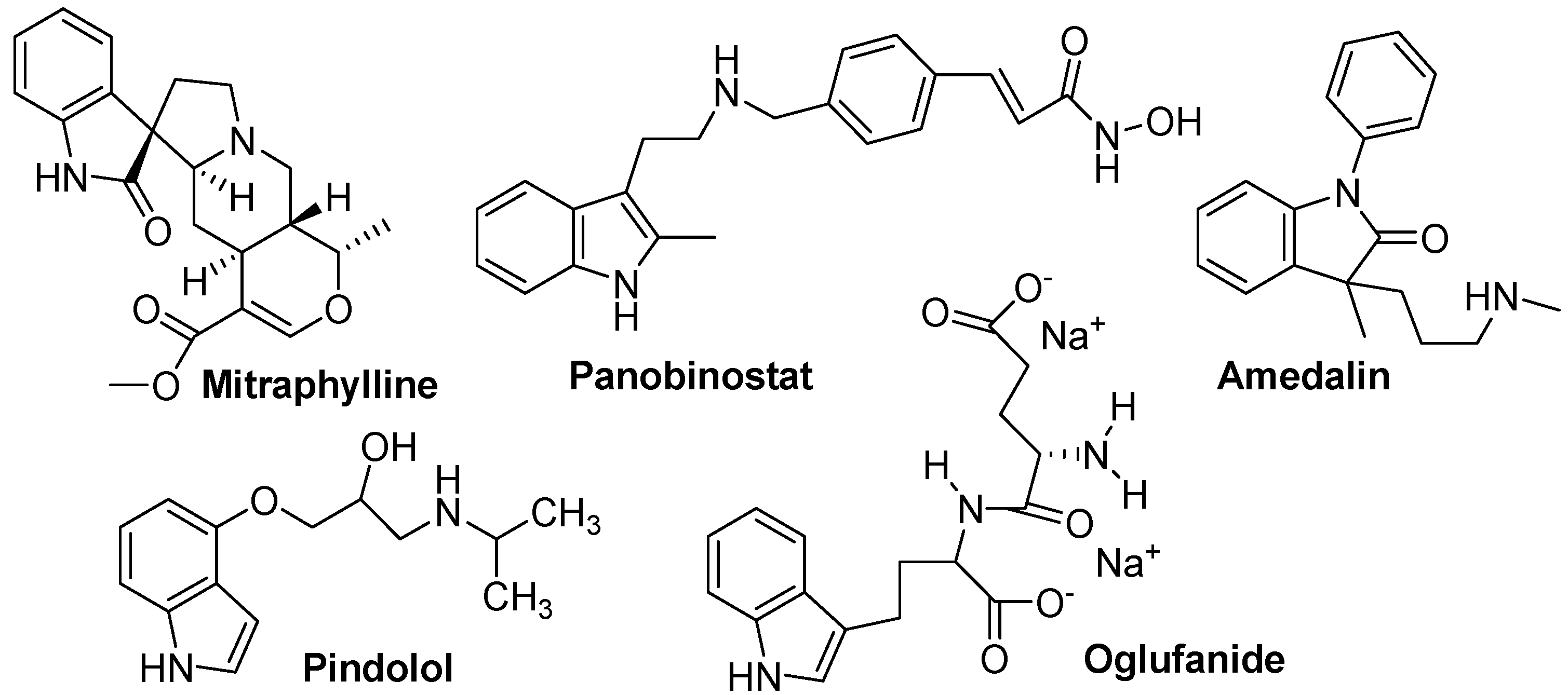
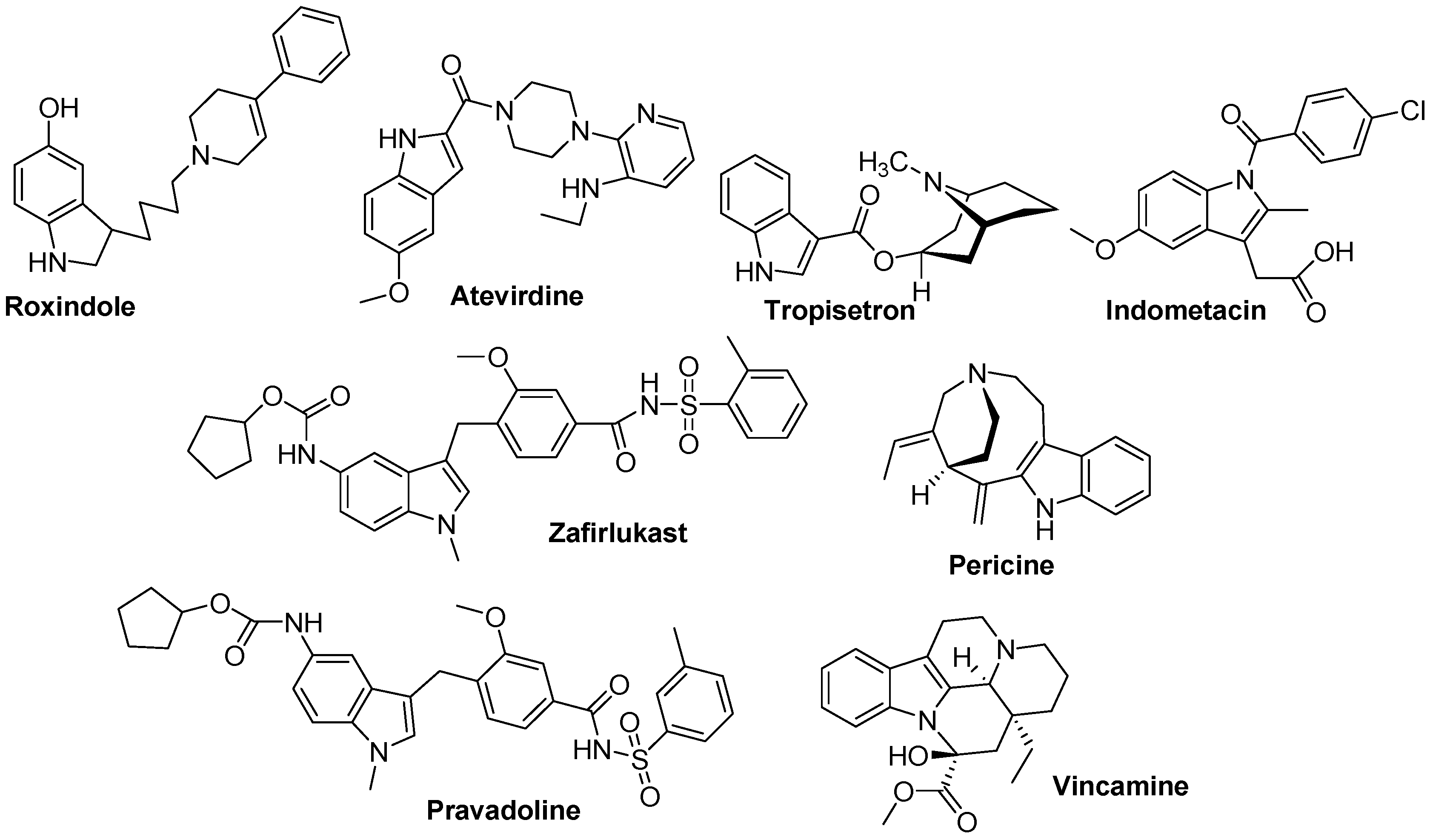
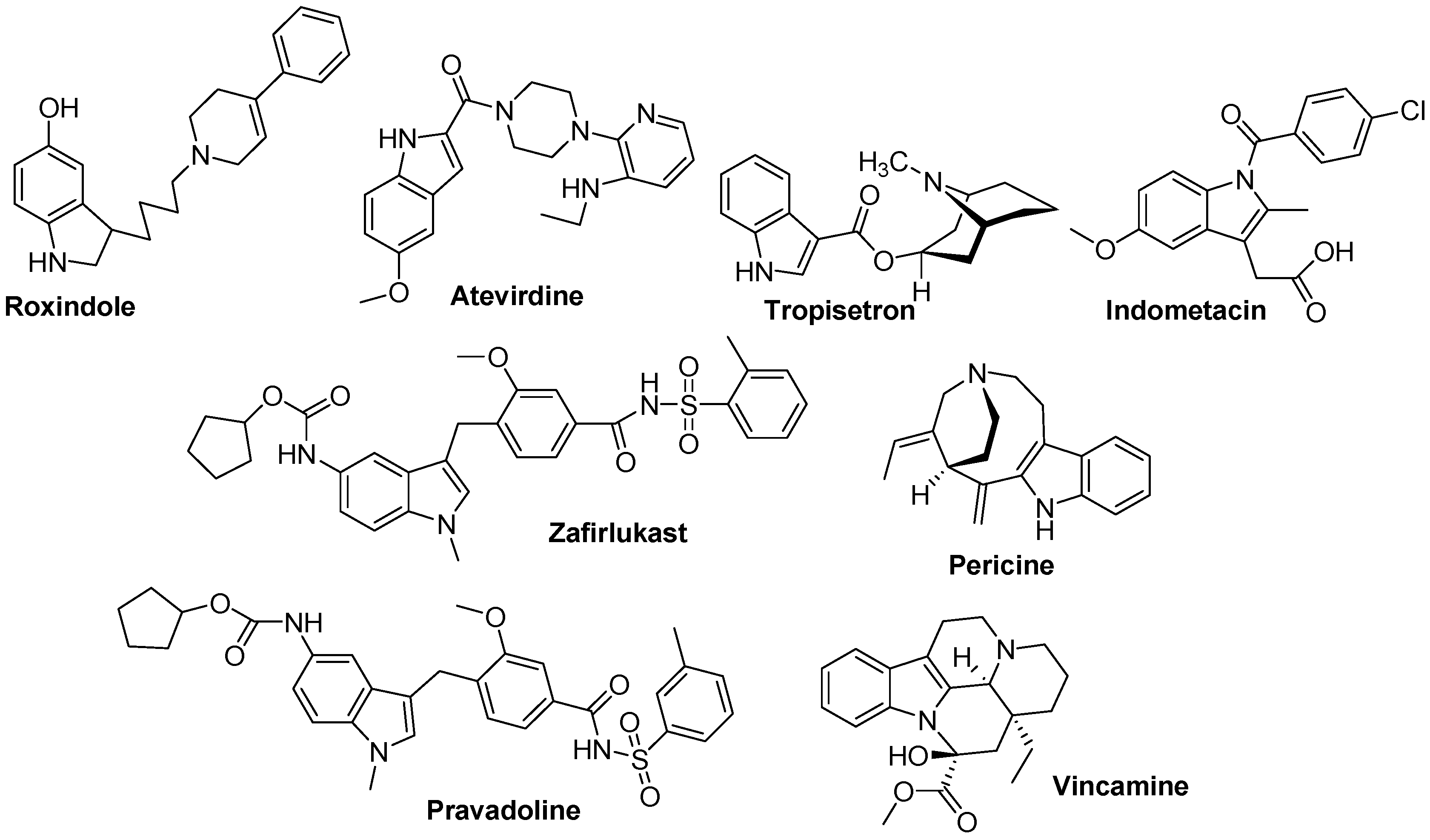
3. Indole Ring Containing Important Marketed Drug Molecules

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
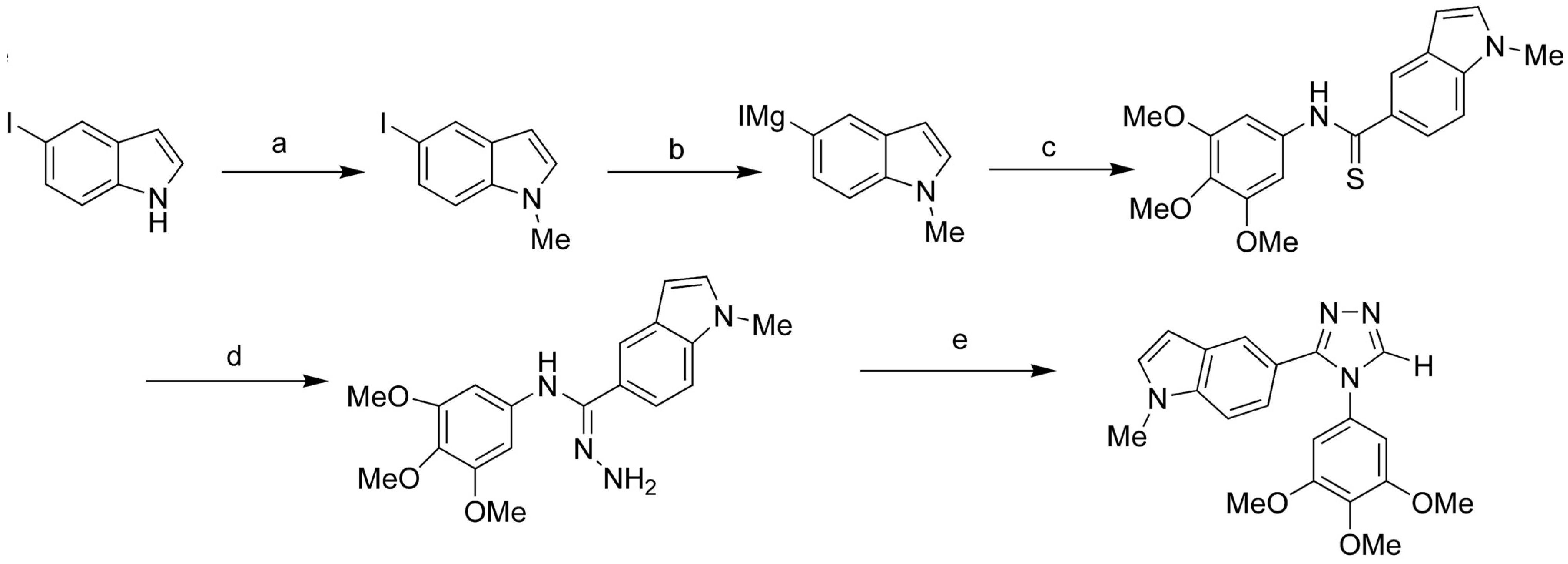{kind=link}
{kind=link}
{kind=link}
{kind=link}
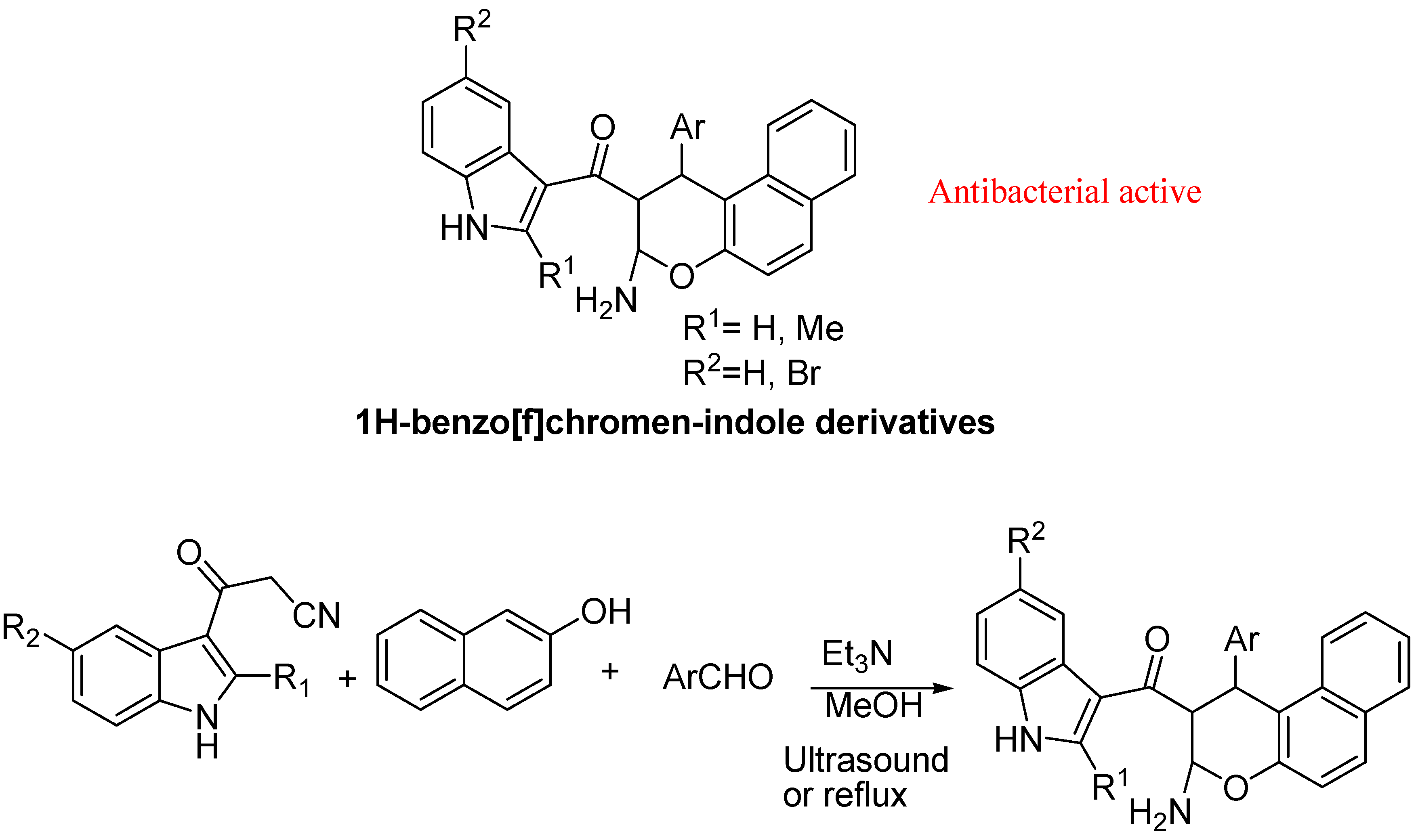{kind=link}
{kind=link}
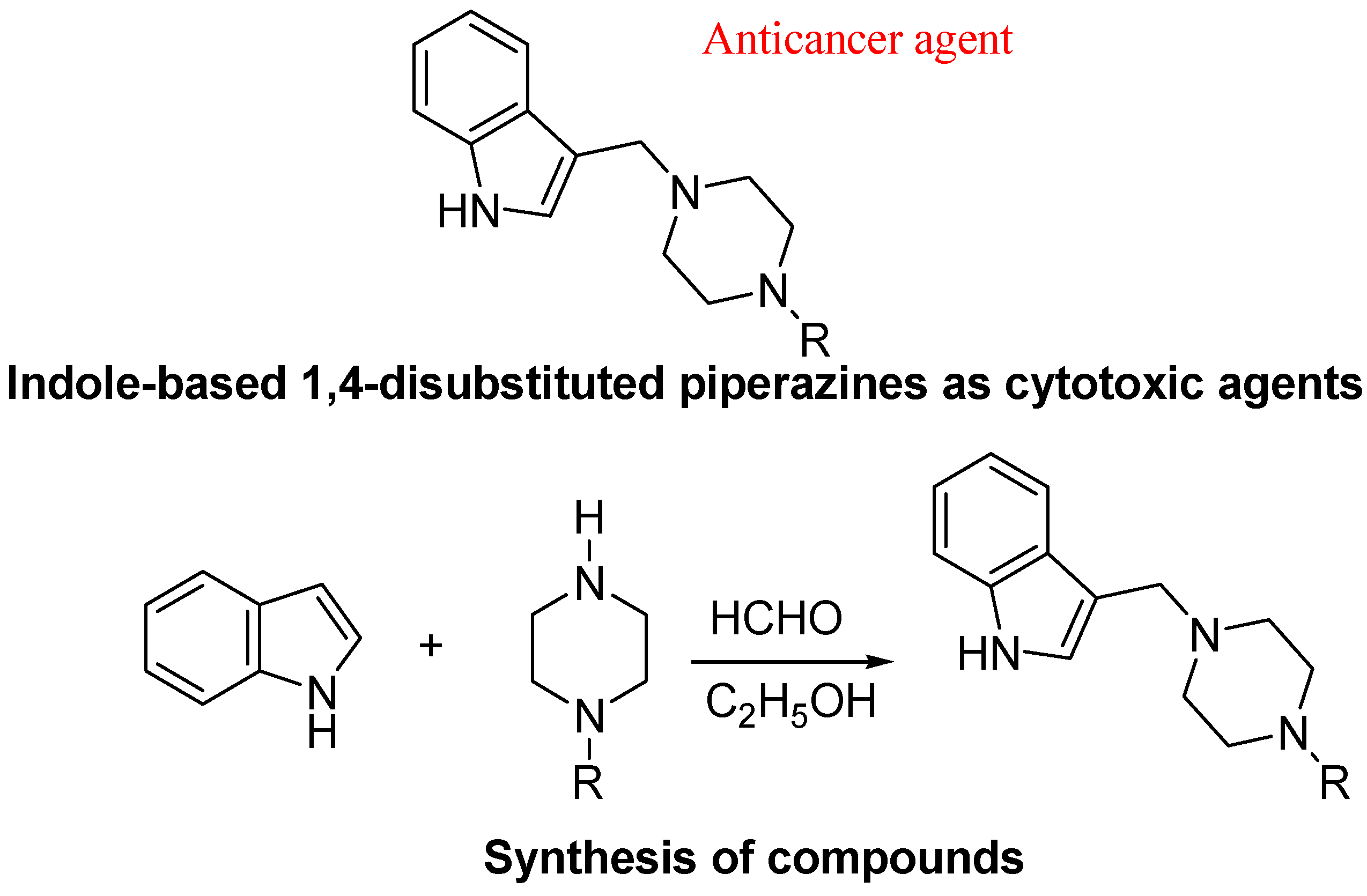{kind=link}
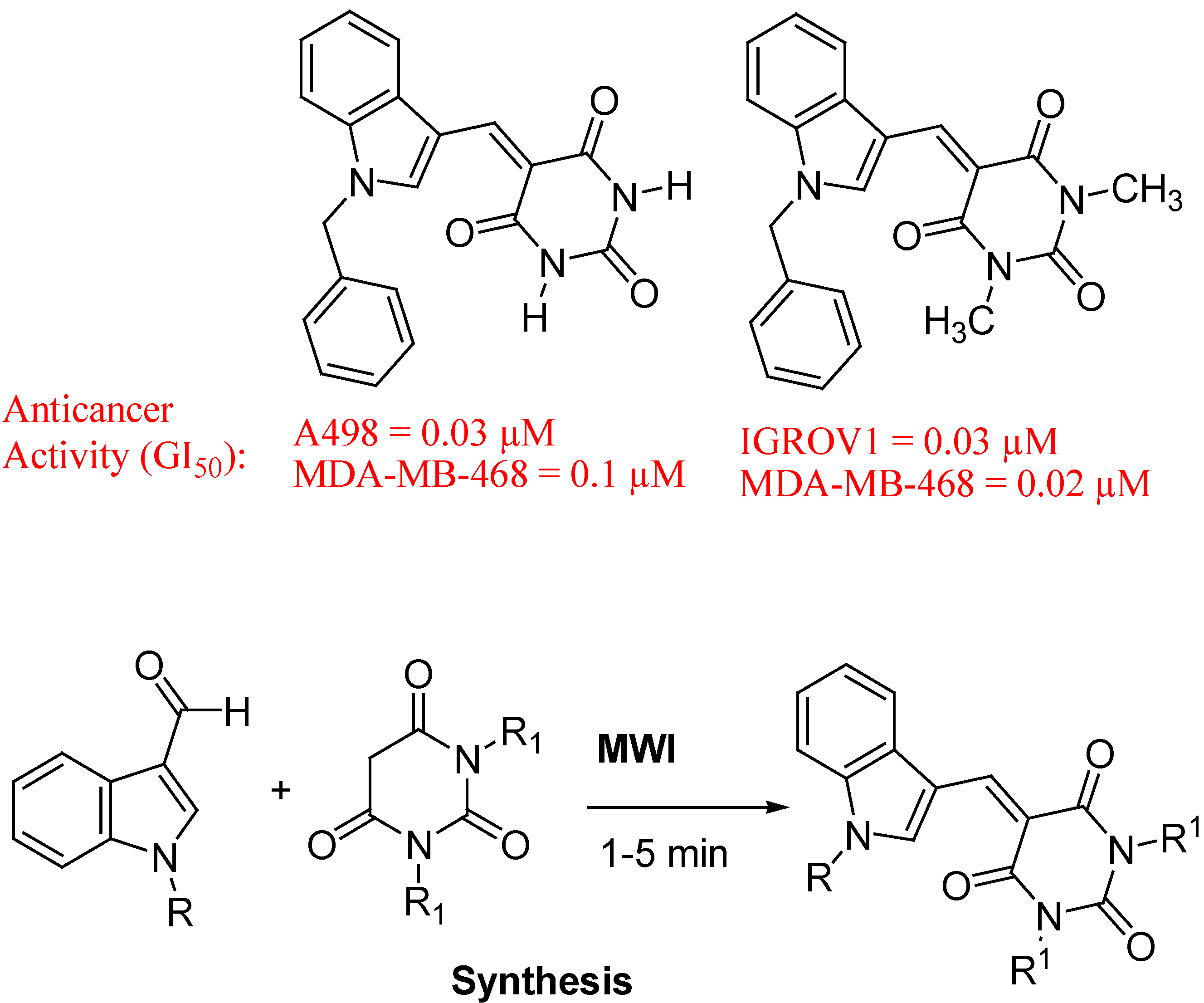{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Application | Drug | Application | Drug | Application |
|---|---|---|---|---|---|
| Vincristine | Anticancer | Vincamine | Vasodilator | Roxindole | Schizophrenia |
| Vinblastine | Anticancer | Reserpine | Antihypertensive | Delavirdine | Anti-HIV |
| Vinorelbine | Anticancer | Peridopril | Antihypertensive | Atevirdine | Anti-HIV |
| Vindesine | Anticancer | Pindolol | Antihypertensive | Arbidol | Antiviral |
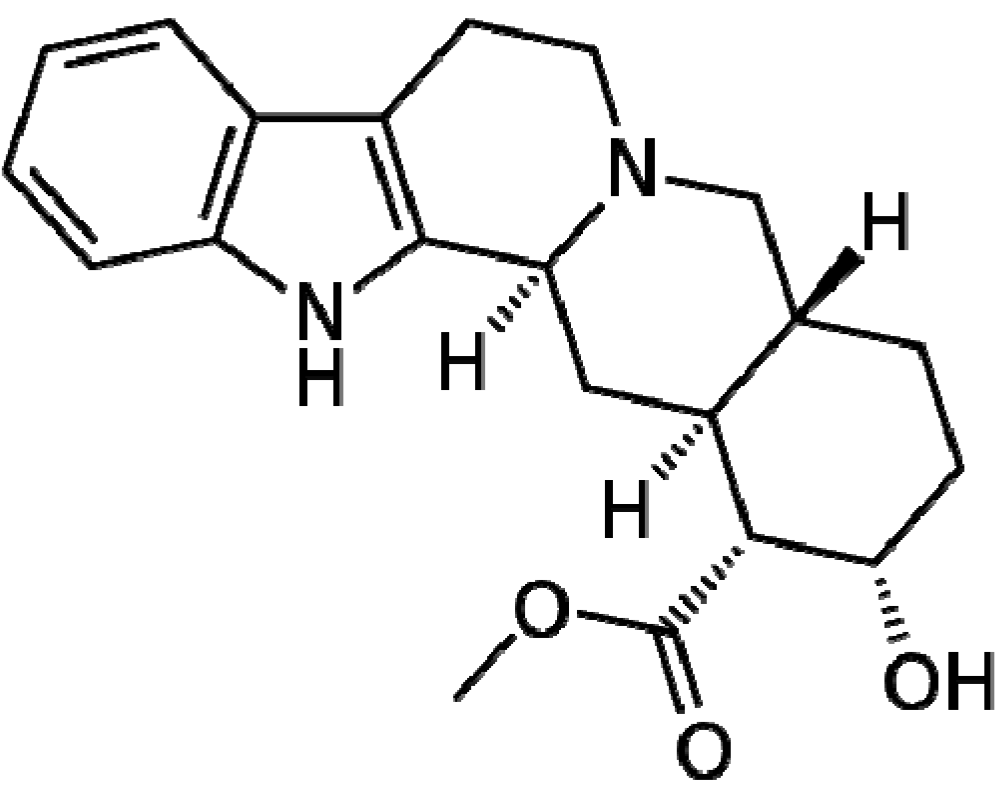
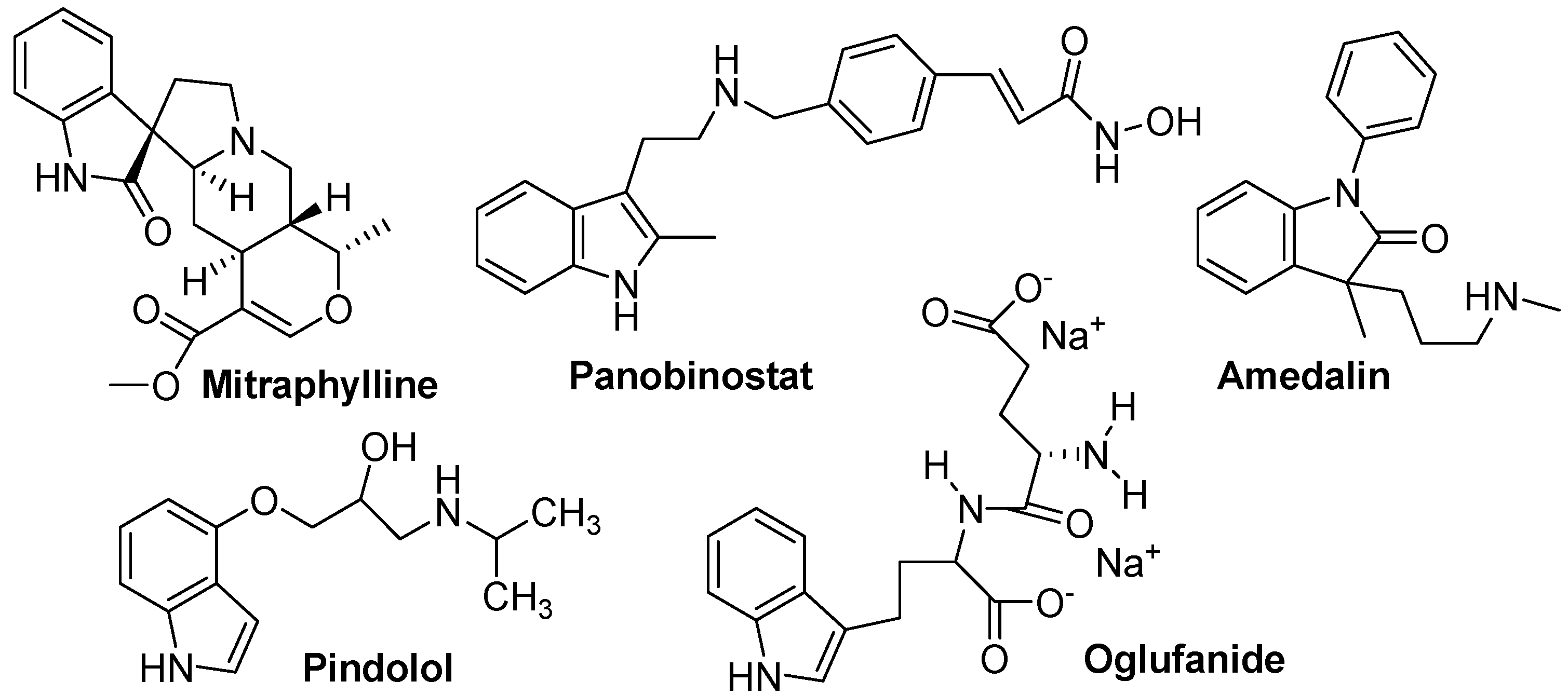
| Mitraphylline | Anticancer | Binedaline | Antidepressant | Zafirlukast | Anti-Asthmatic |
| Cediranib | Anticancer | Amedalin | Antidepressant | Bucindolol | β-Blockers |
| Panobinostat | Anti-leukamic | Oxypertine | Antipsychotic | Pericine | Opioid agonist |
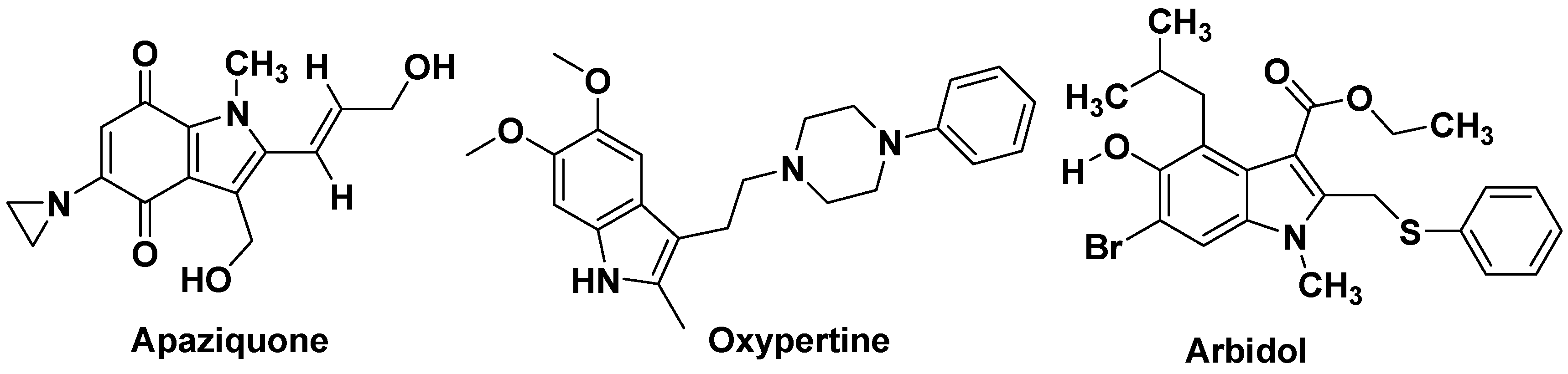
| Apaziquone | Anticancer | Siramesine | Antidepressant | Mitragynine | Opioid agonist |
| Tropisetron | Antiemetic | Indalpine | Antidepressant | Pravadoline | Analgesic |
| Doleasetron | Antiemetic | Yohimbine | Sexual Disorder | Bufotenidine | Toxin |
| Oglufanide | Immunomodulatory | Indomethacin | Anti-inflammatory | Proamanullin | Toxin |

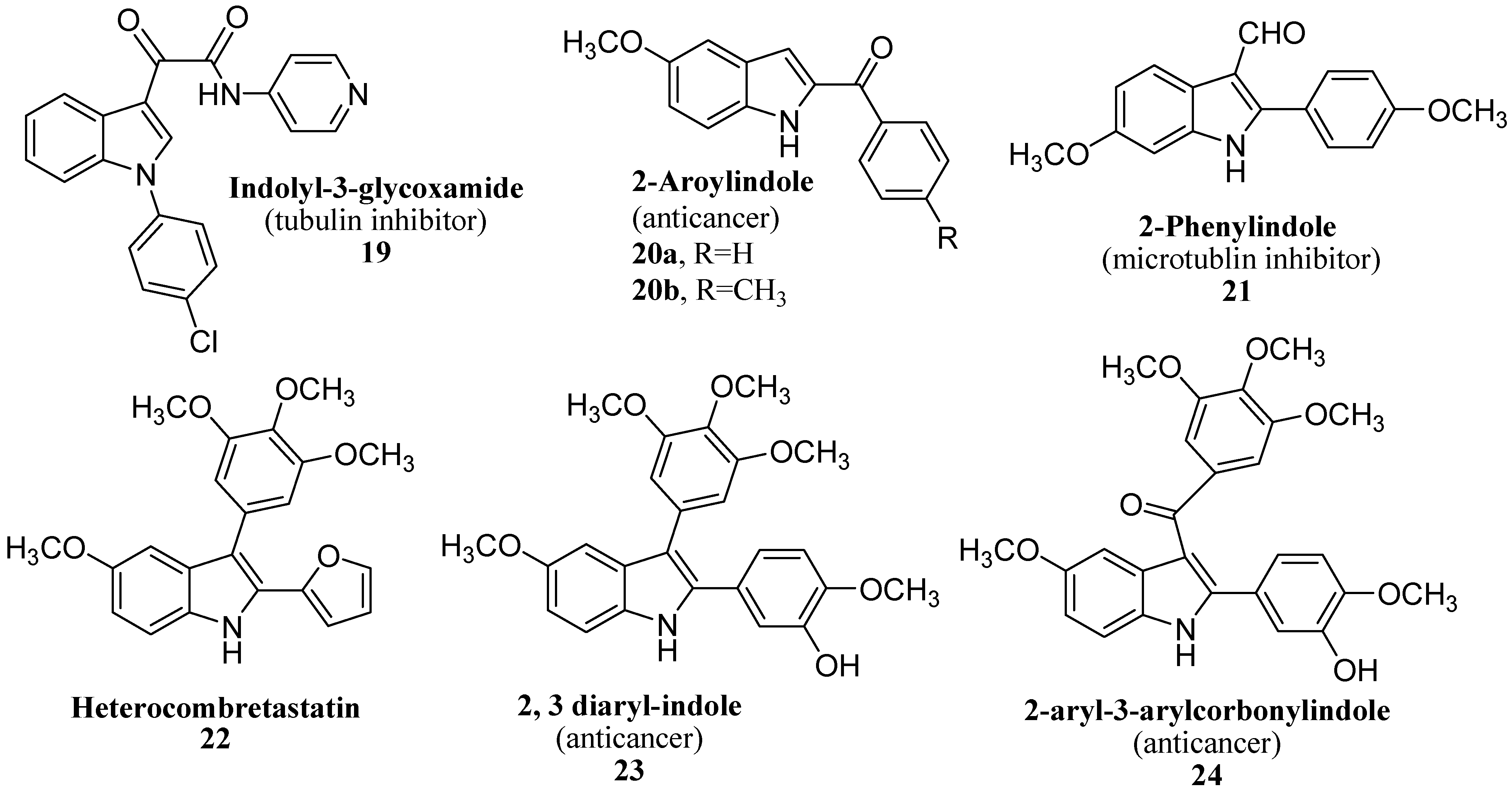
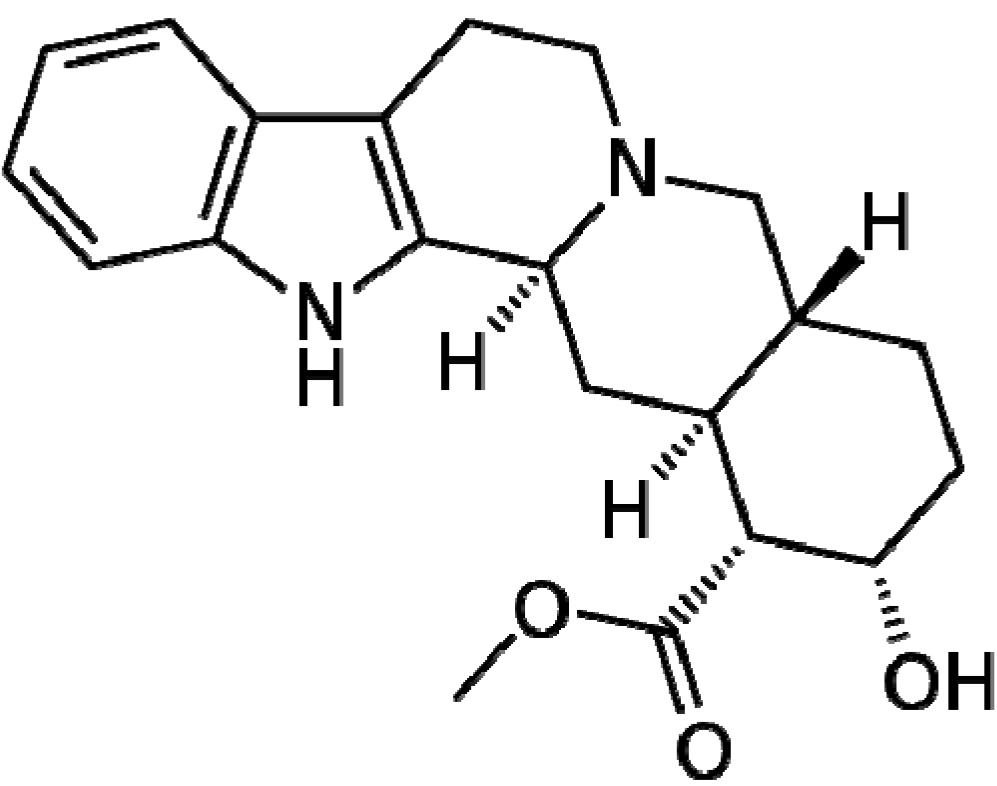
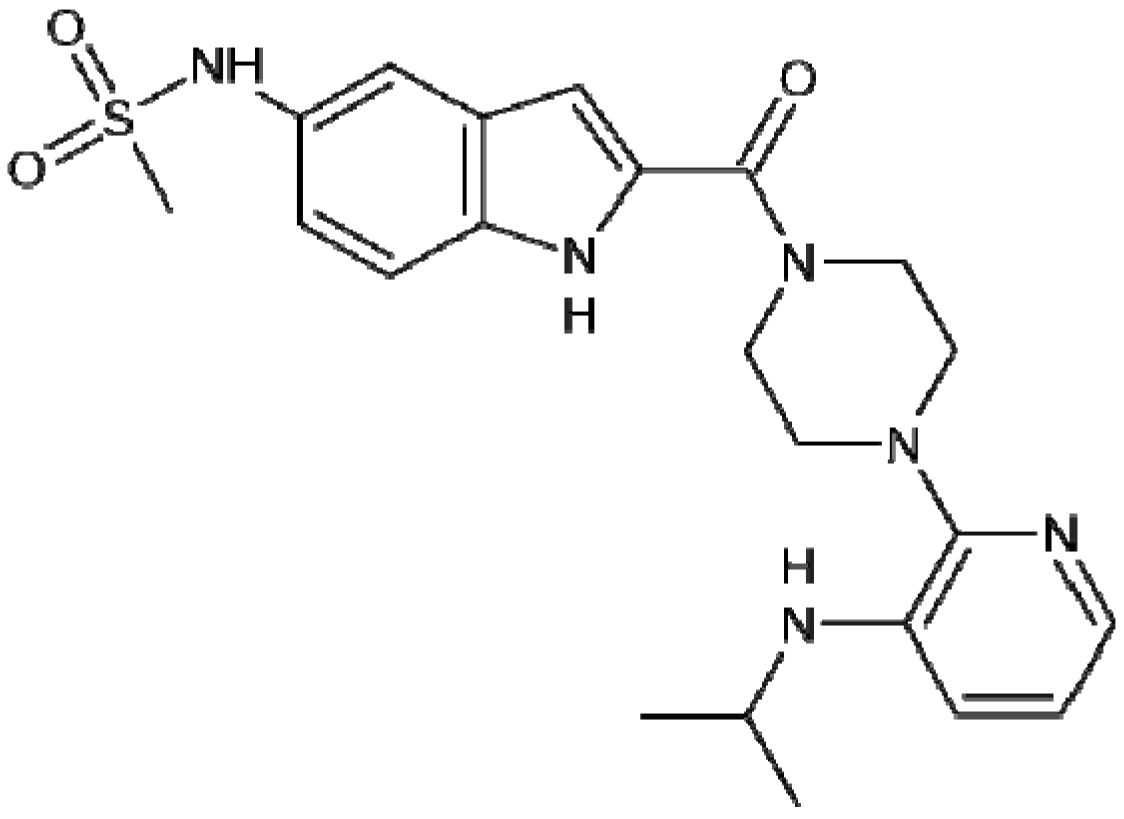
4. Focus on Bioactive Indoles Developed over the Past Few Years
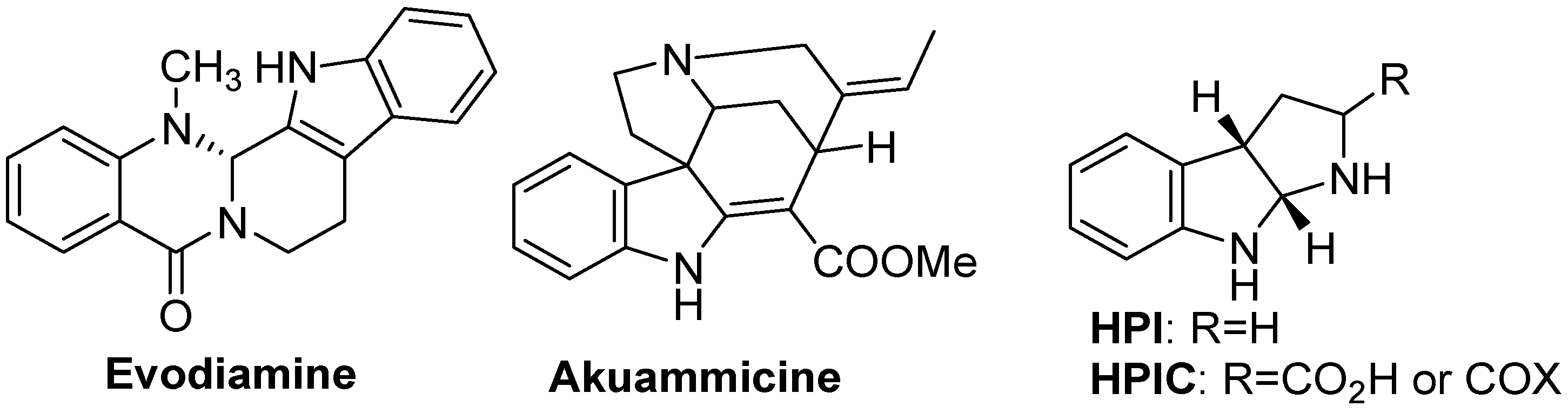
4.1. Natural Products Containing an Indole Core Nucleus
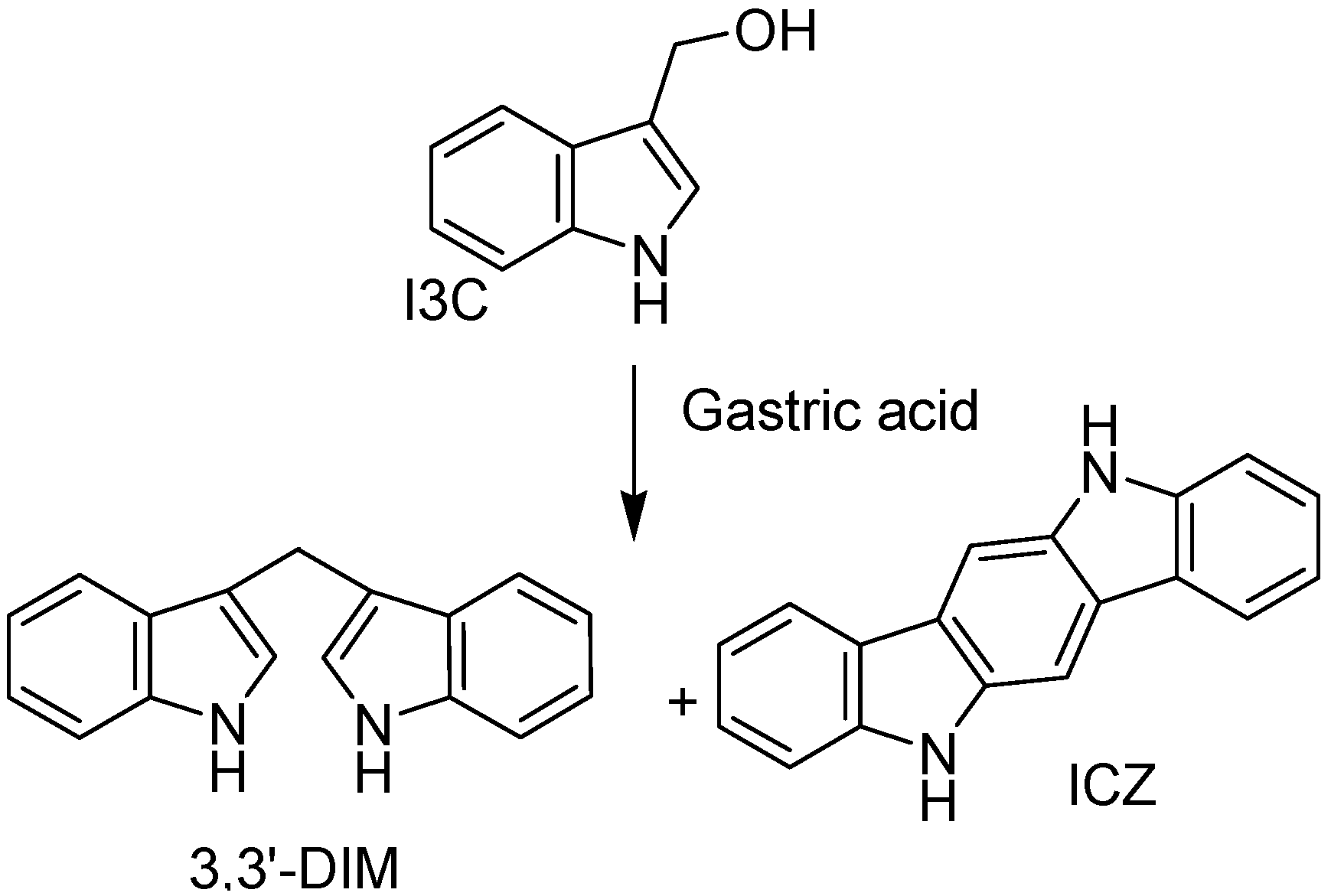
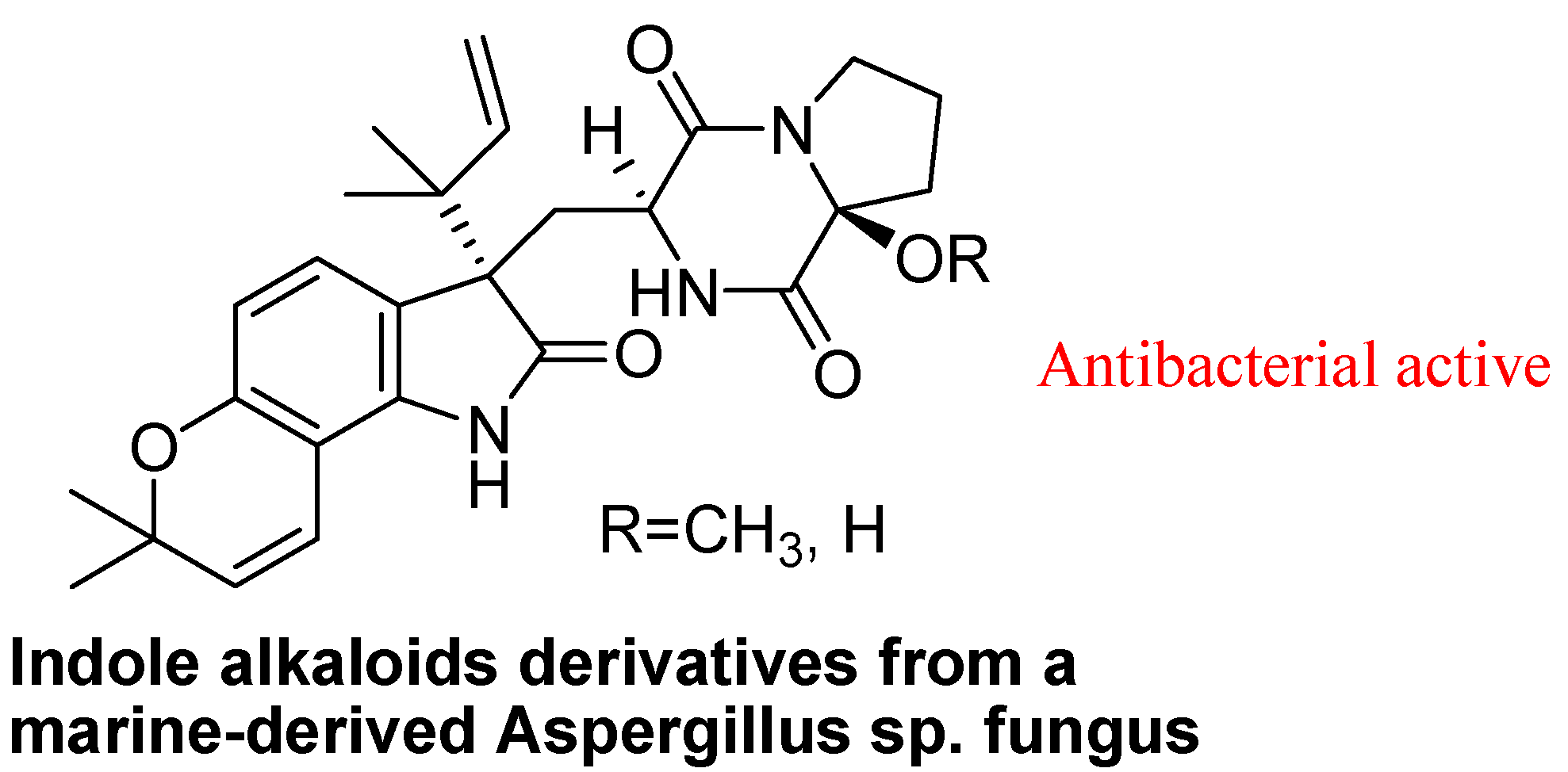
4.2. Marine Product Containing Indole Core Nucleus
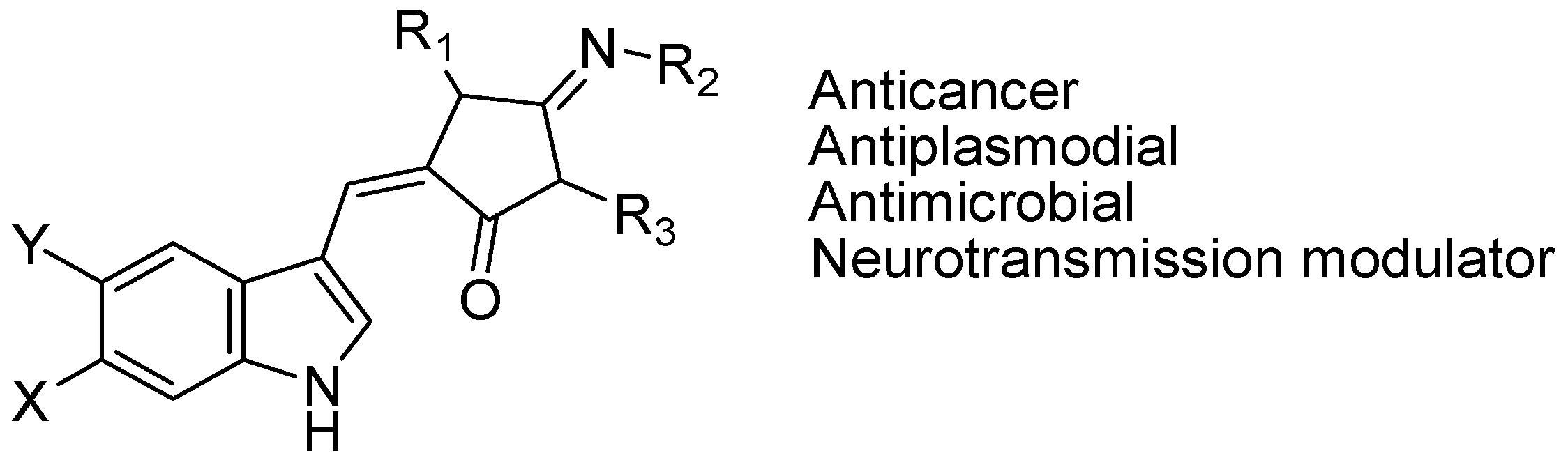
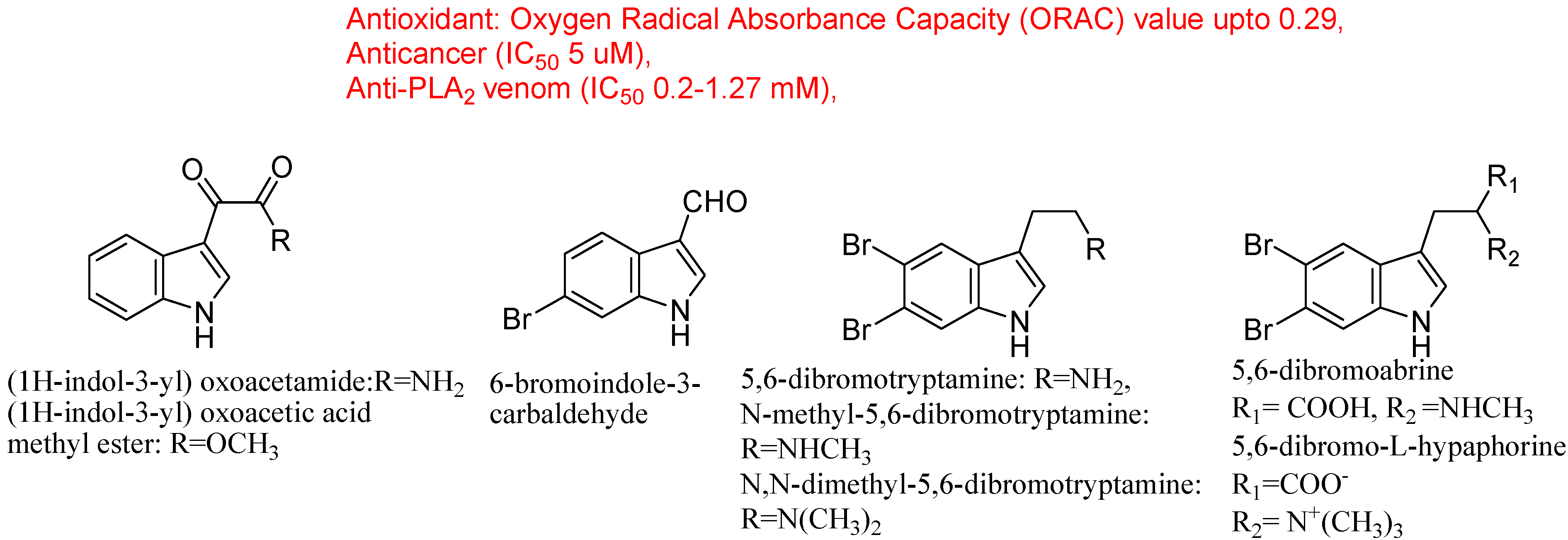
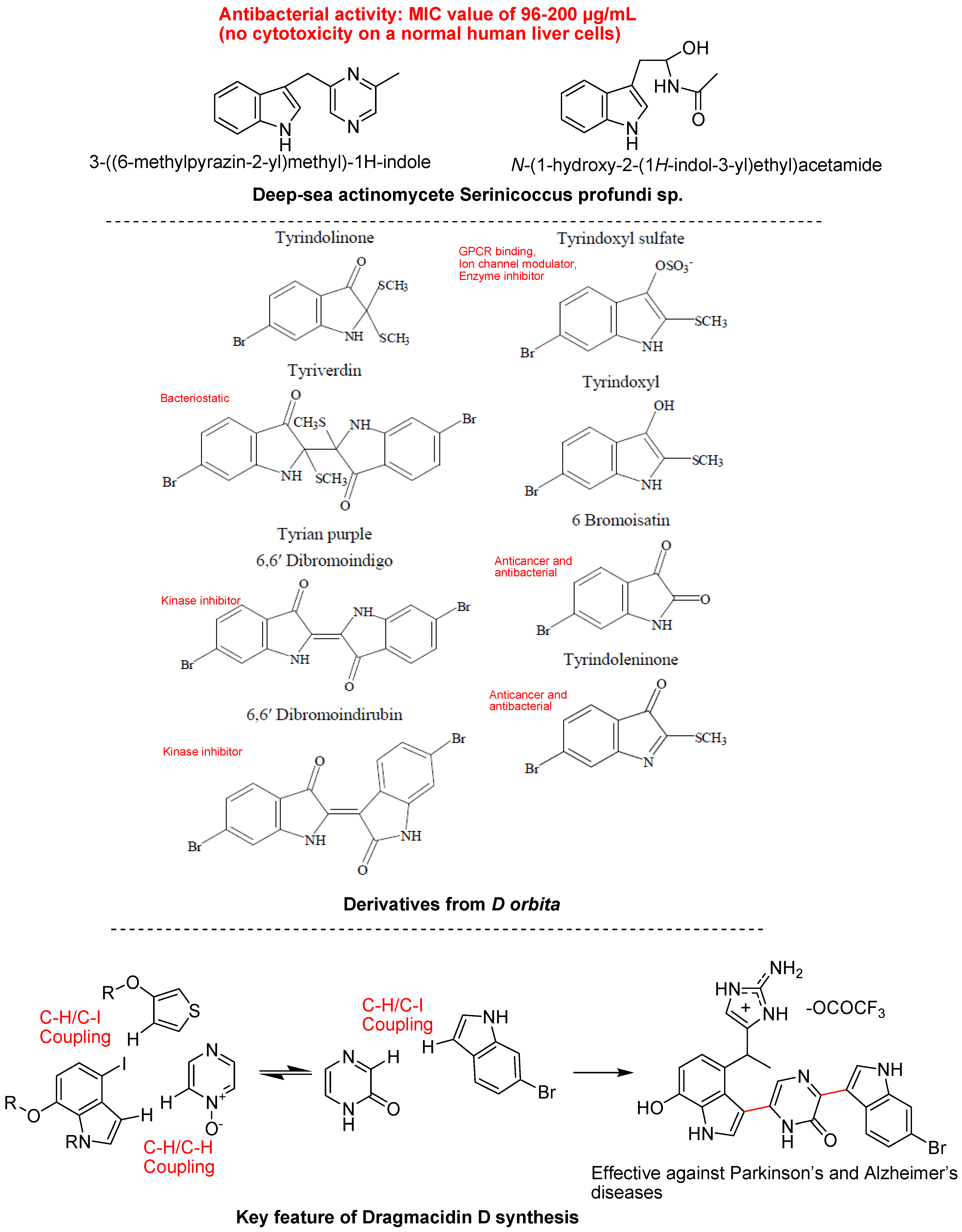
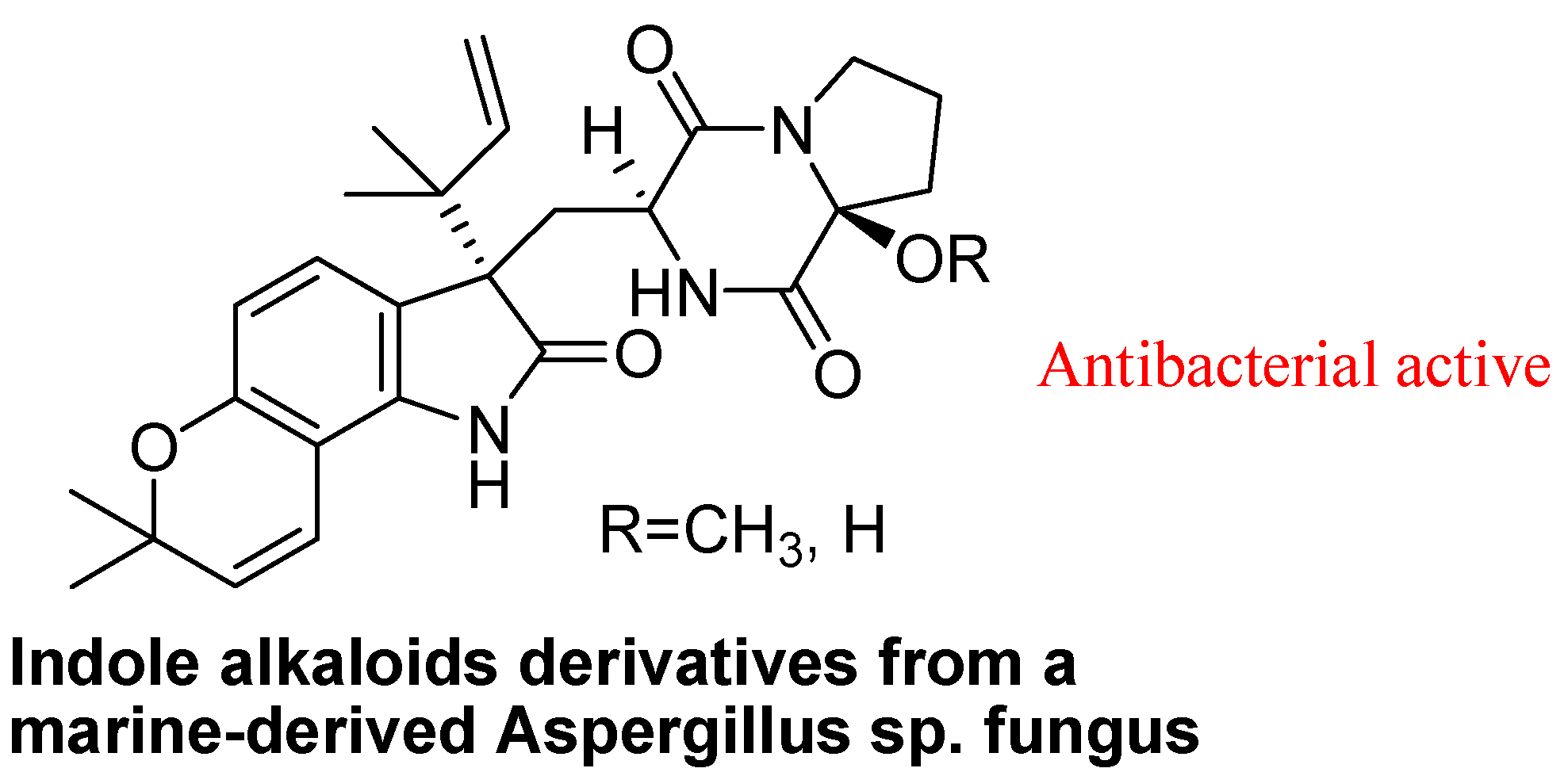
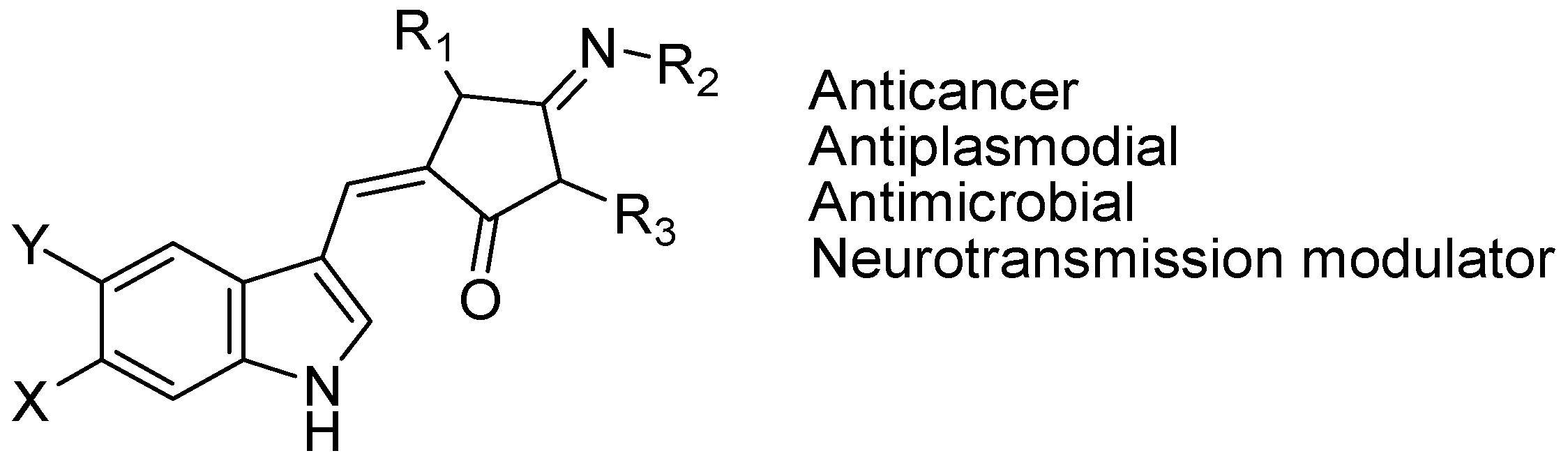
4.3. Synthetic Molecules Containing an Indole Nucleus and Having Medicinal Importance
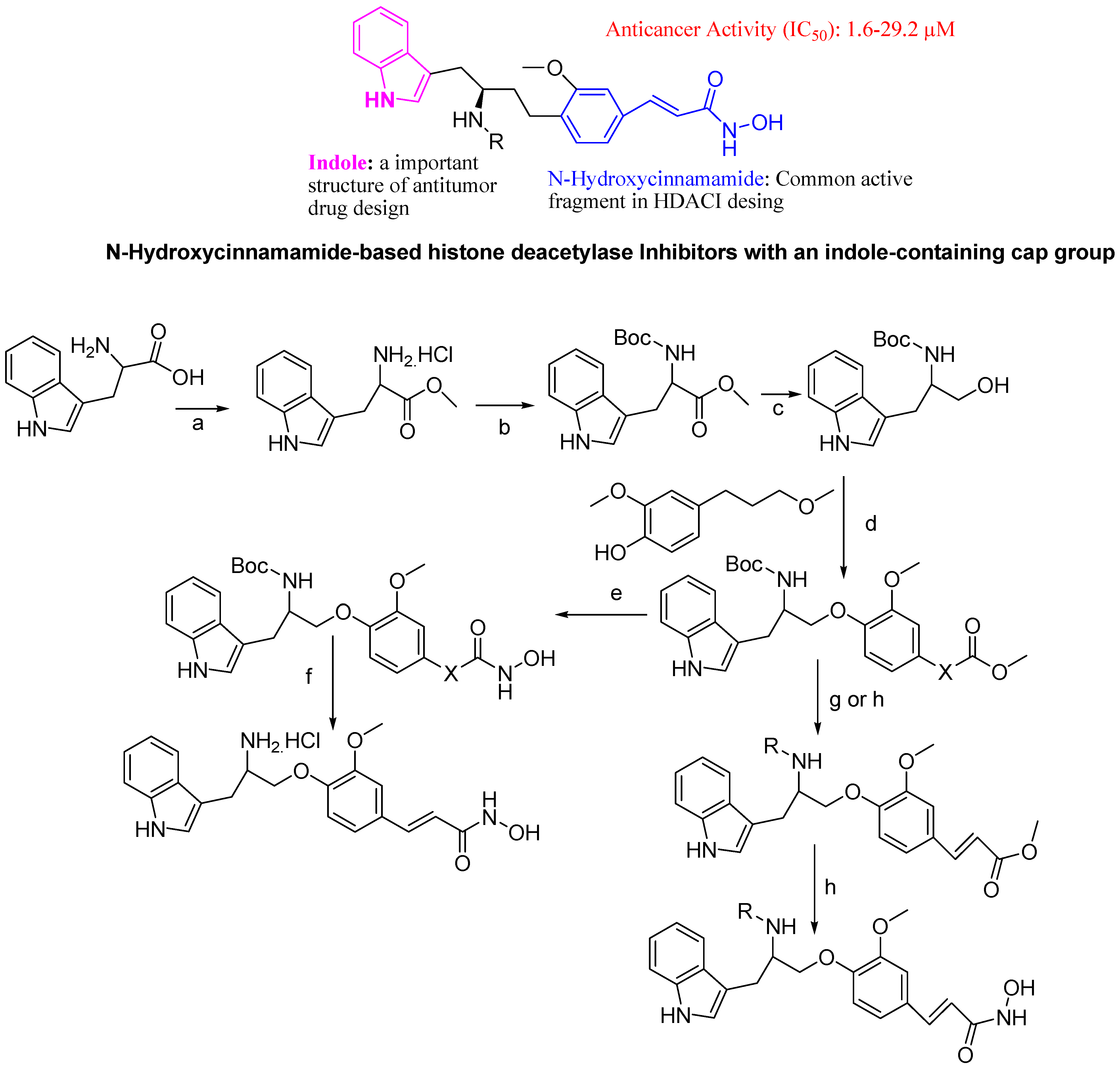
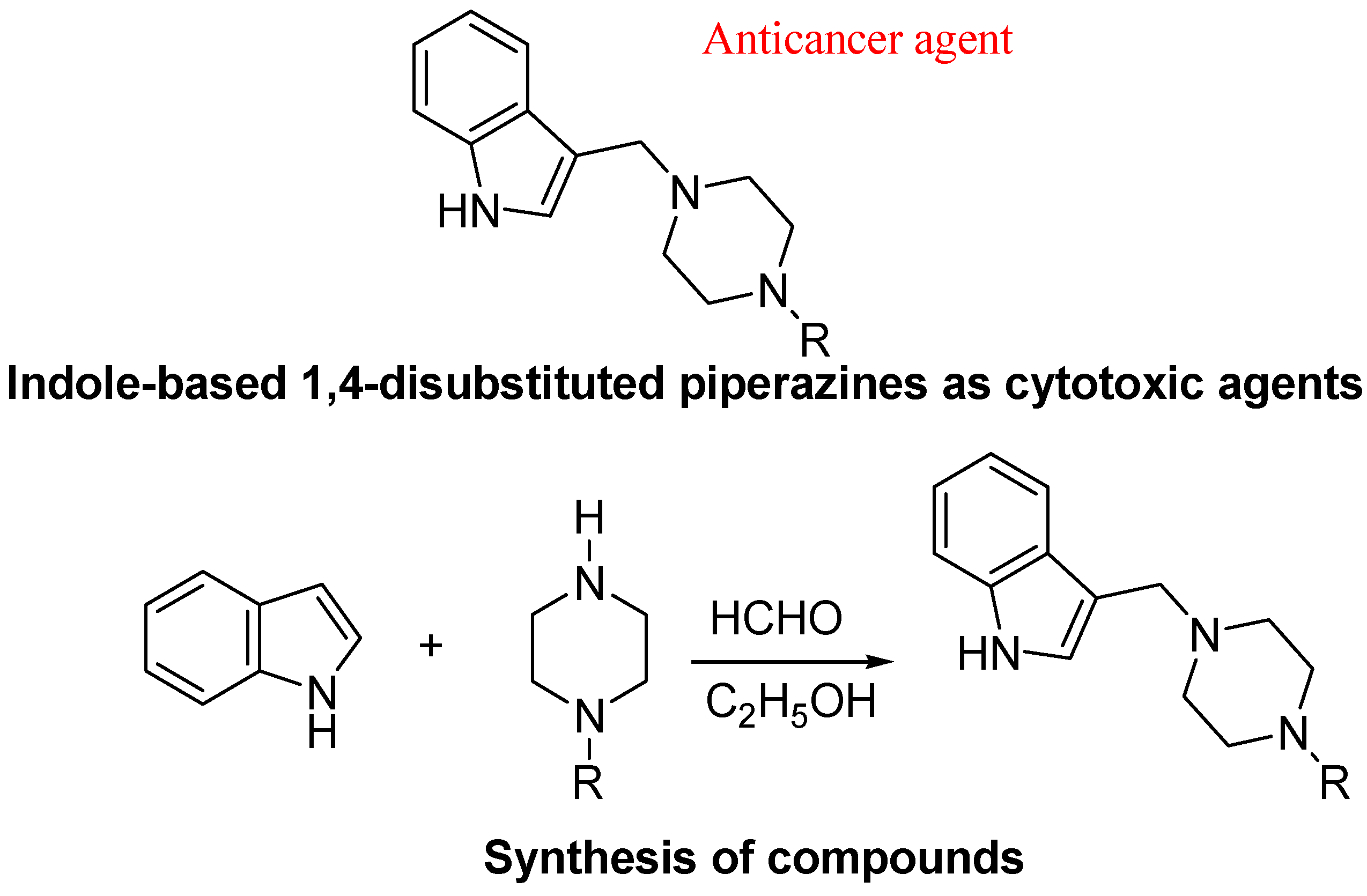
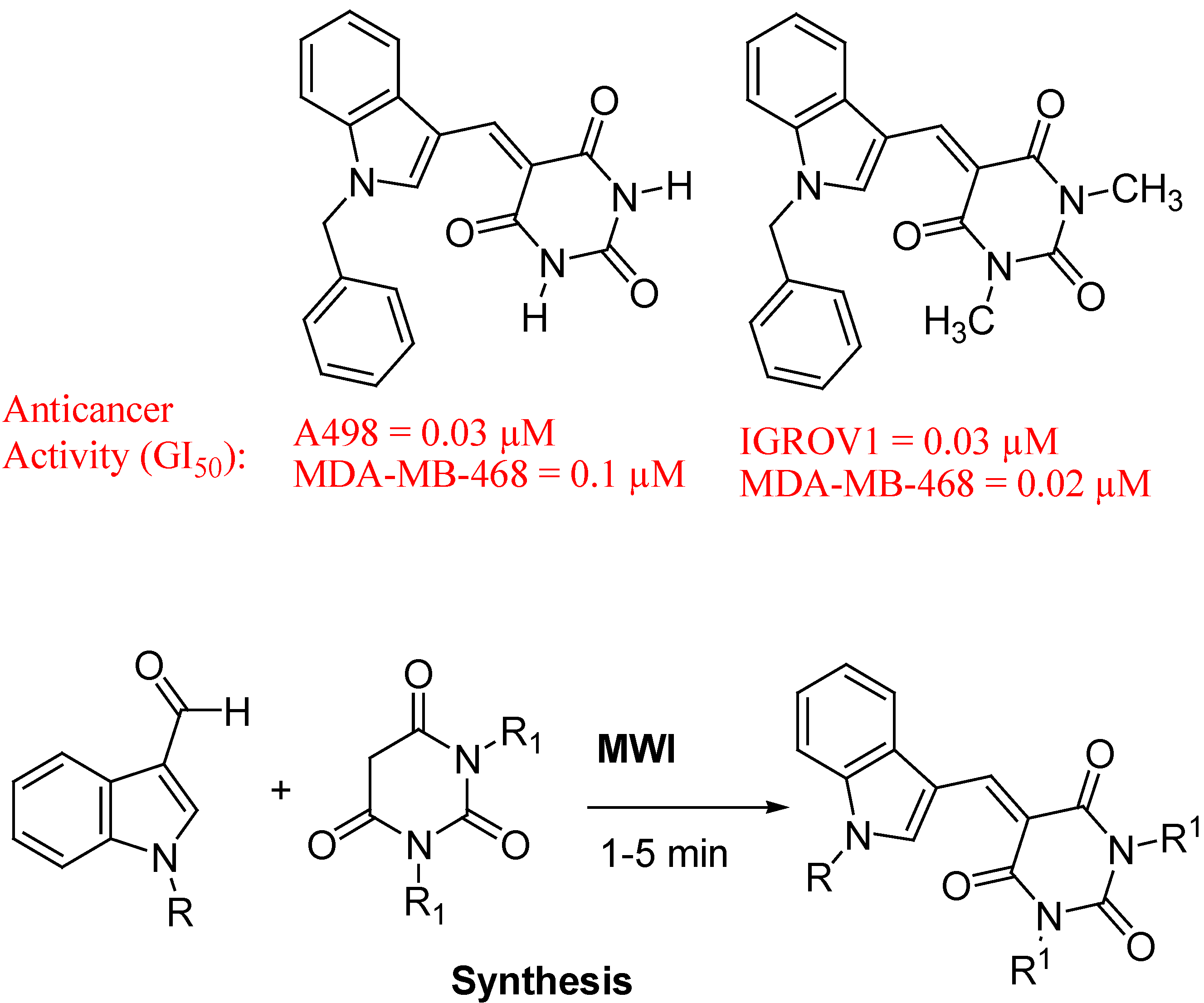


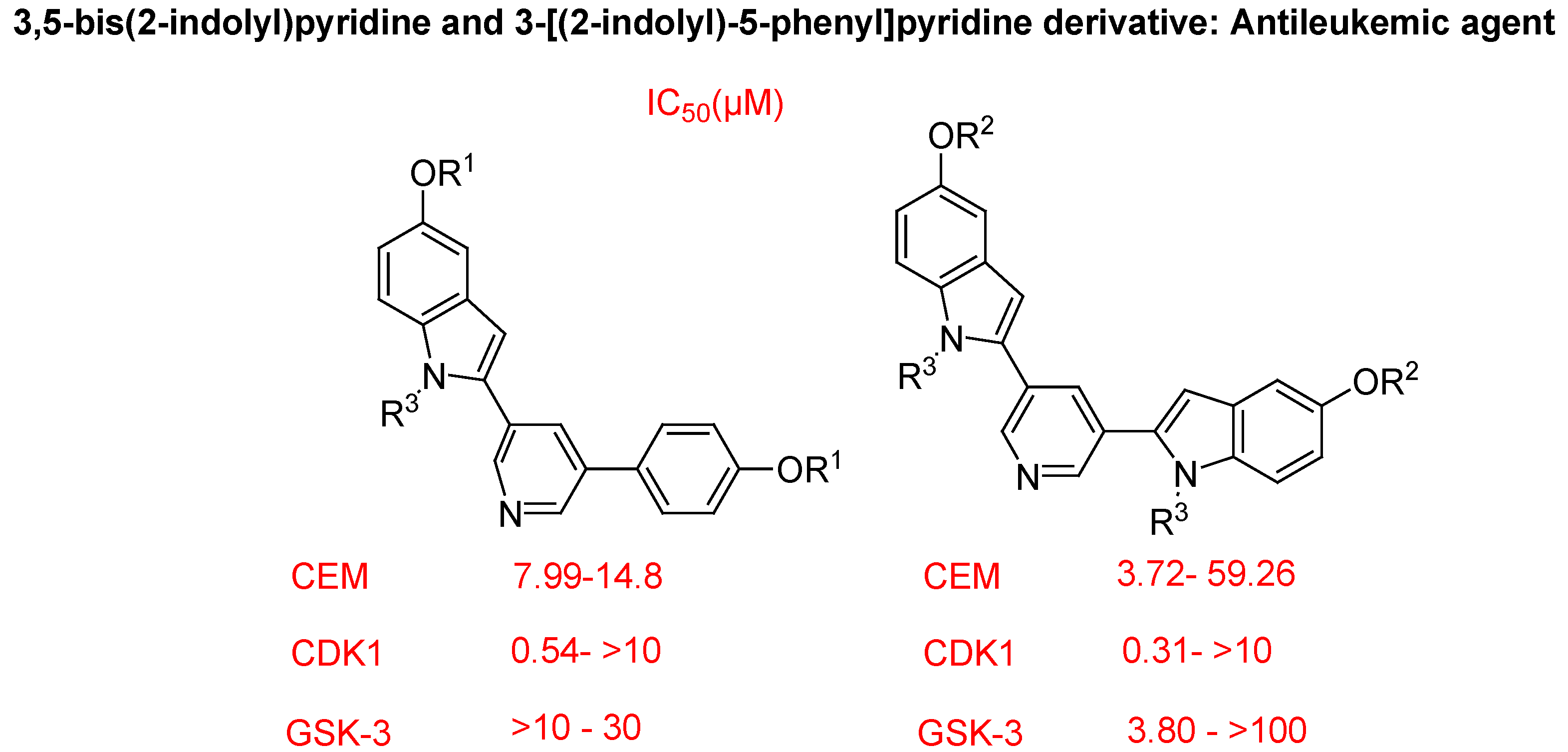
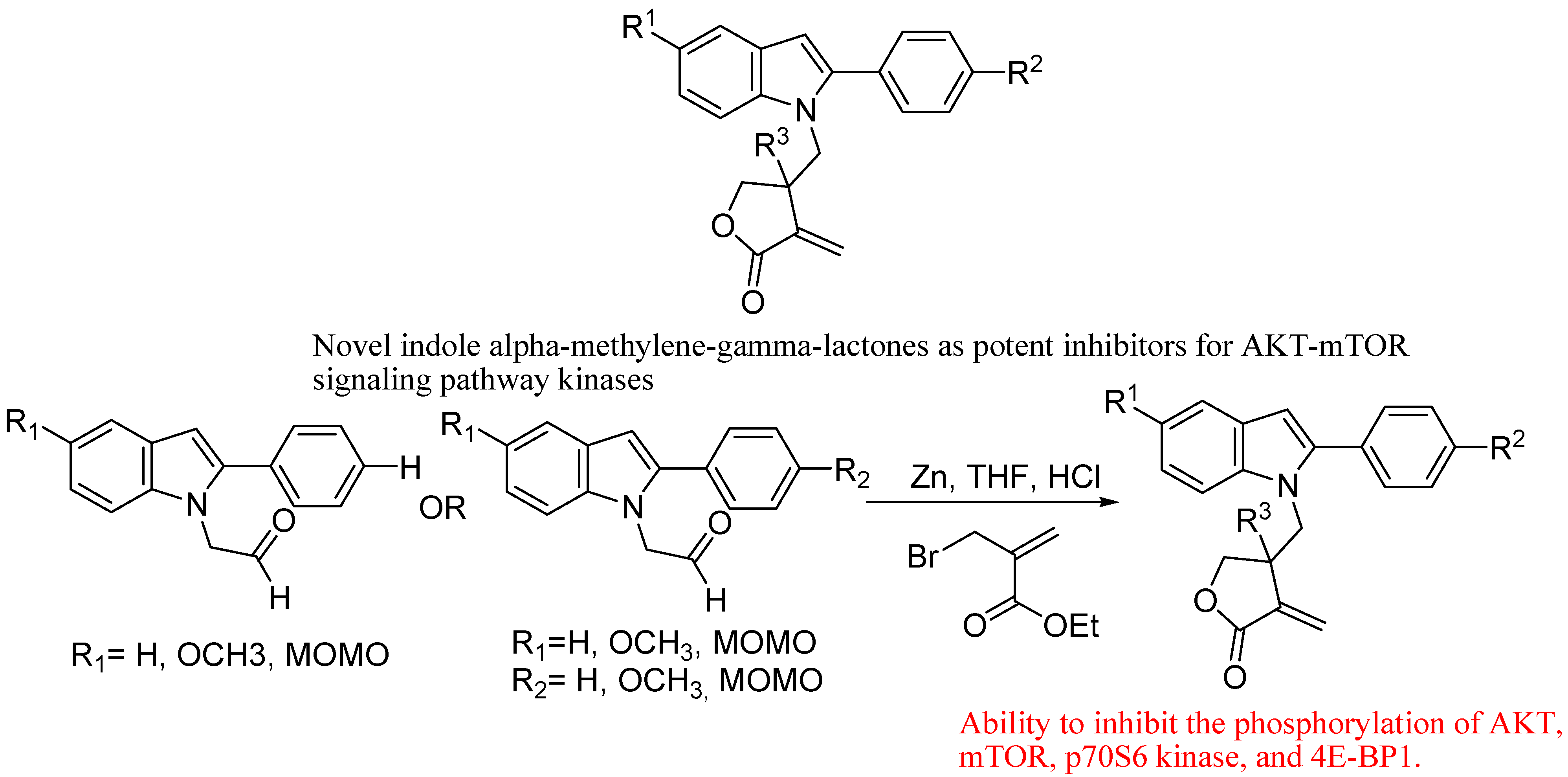
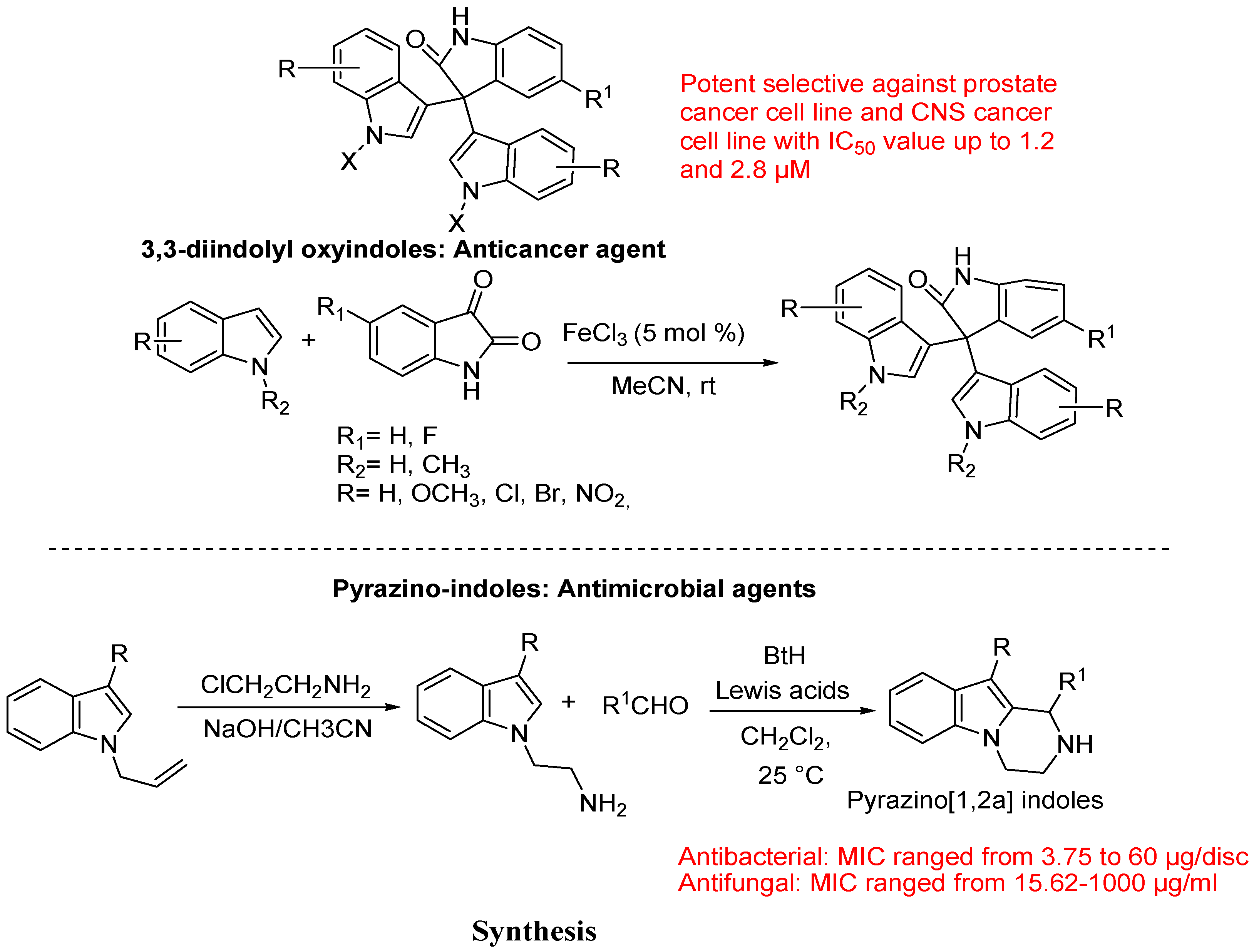
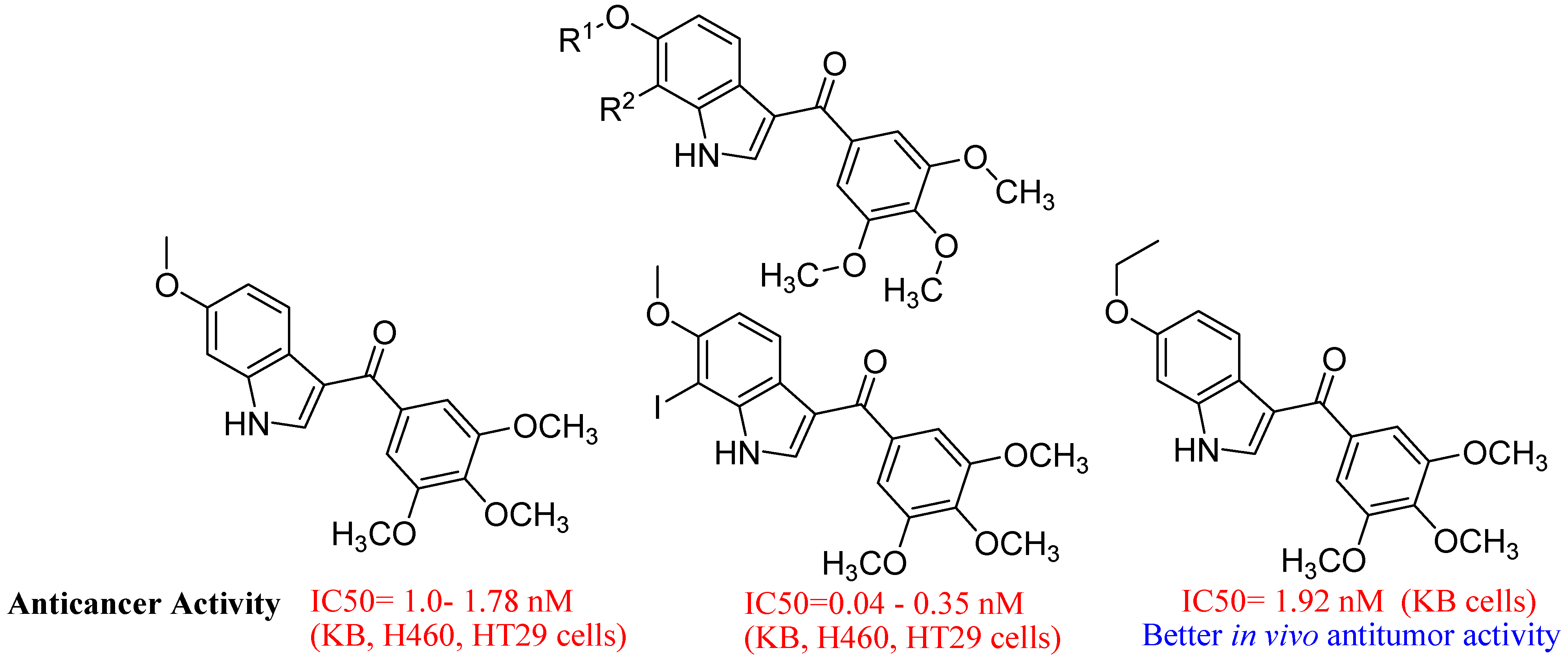
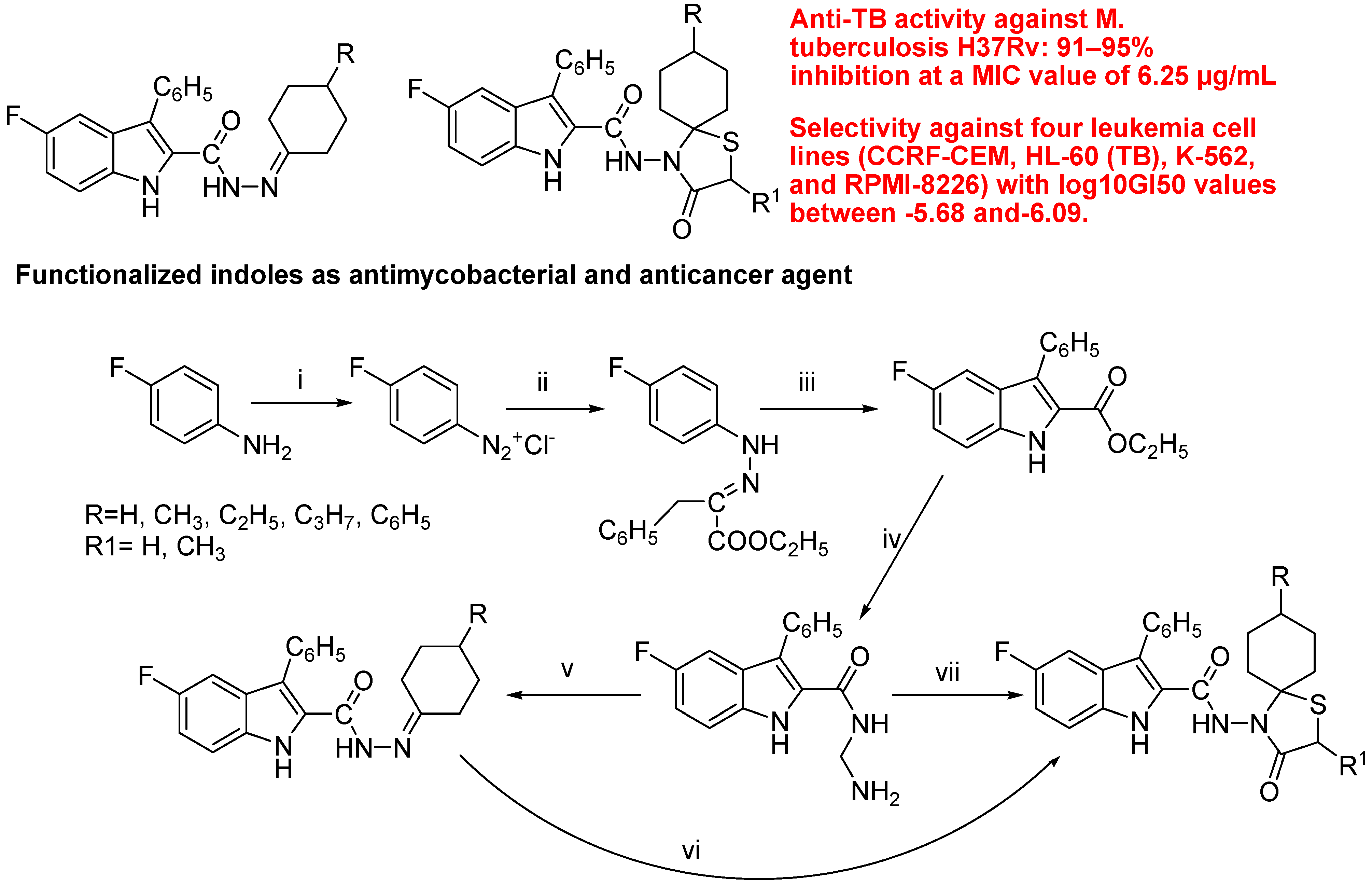
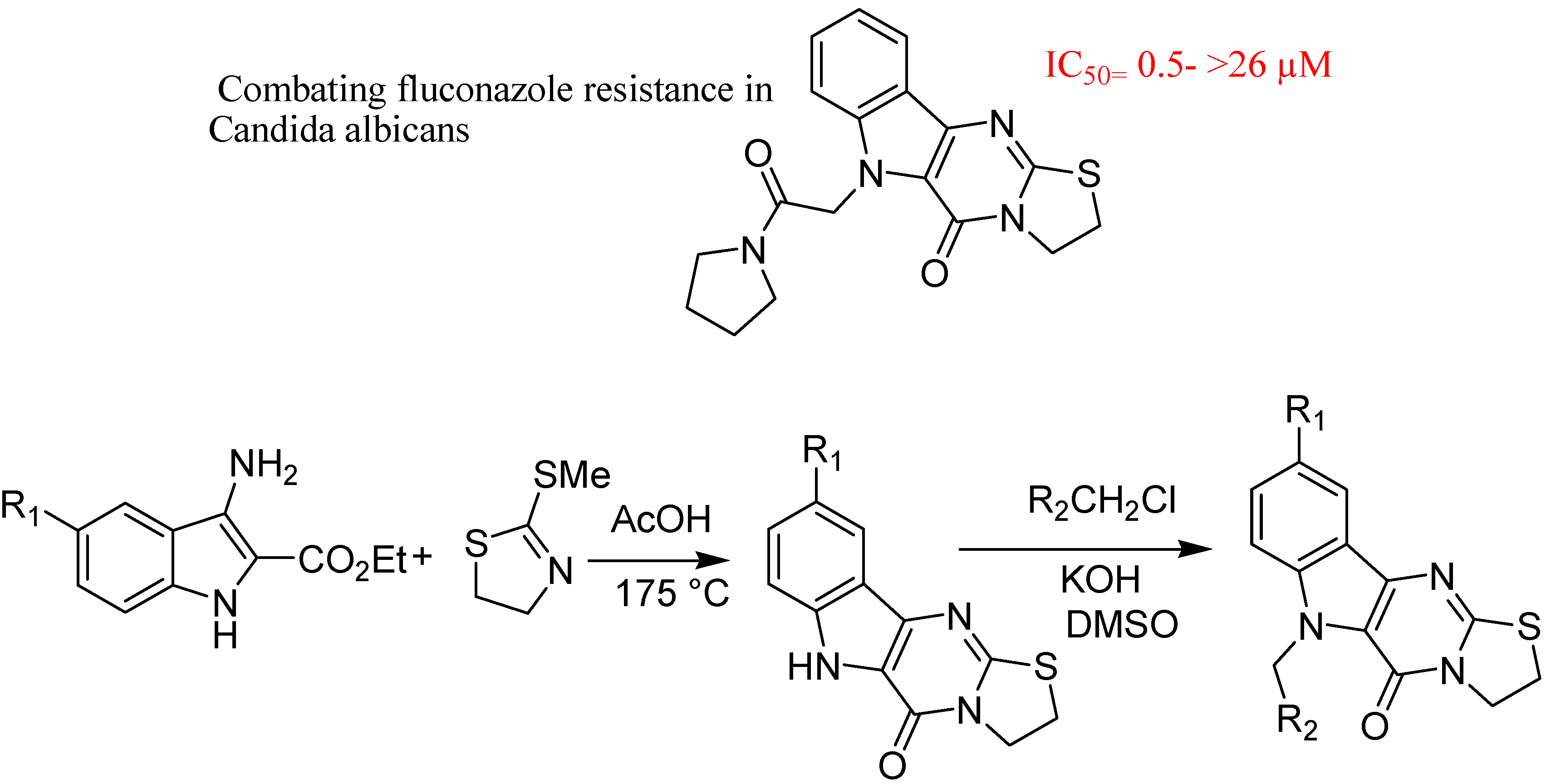
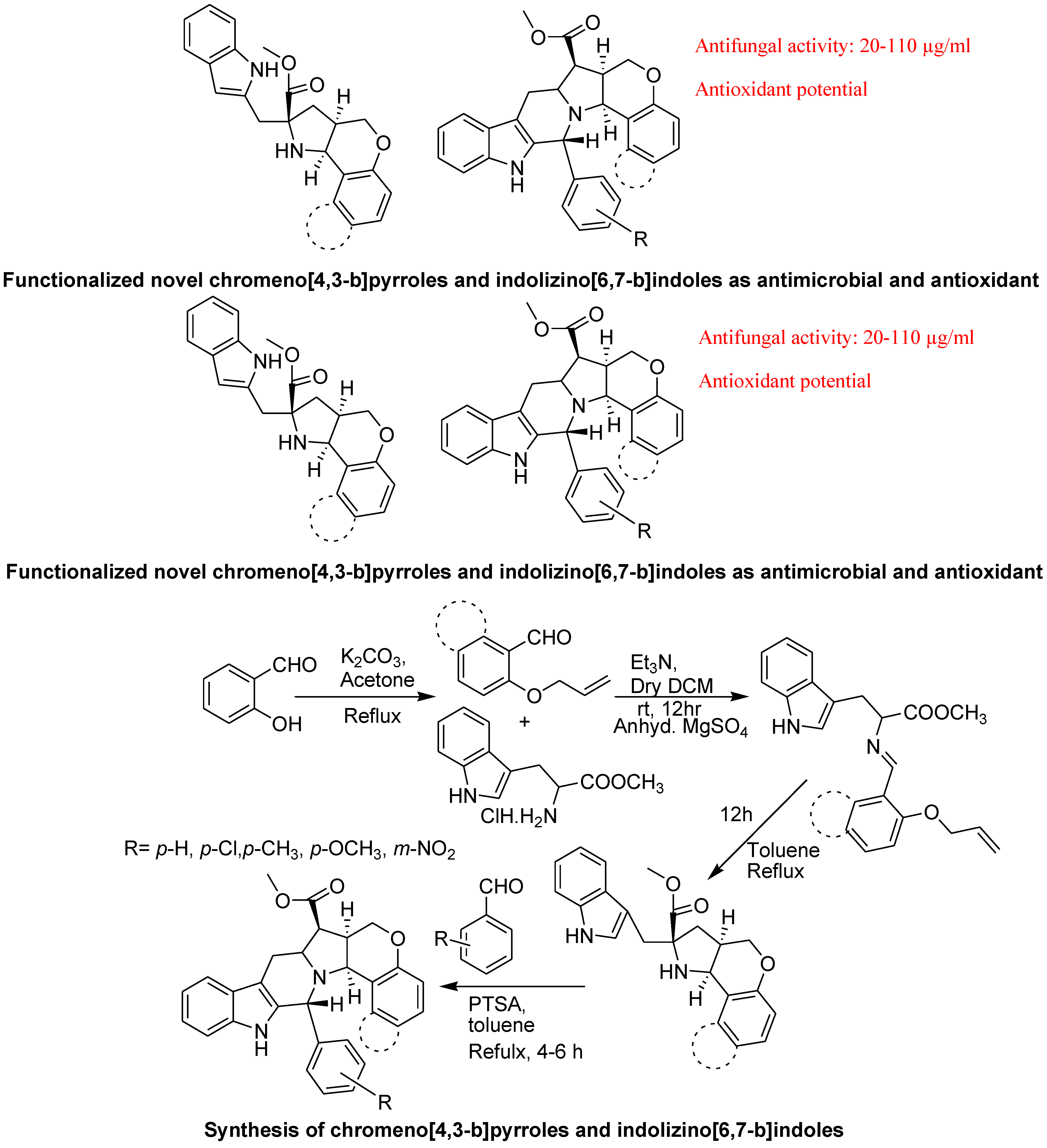
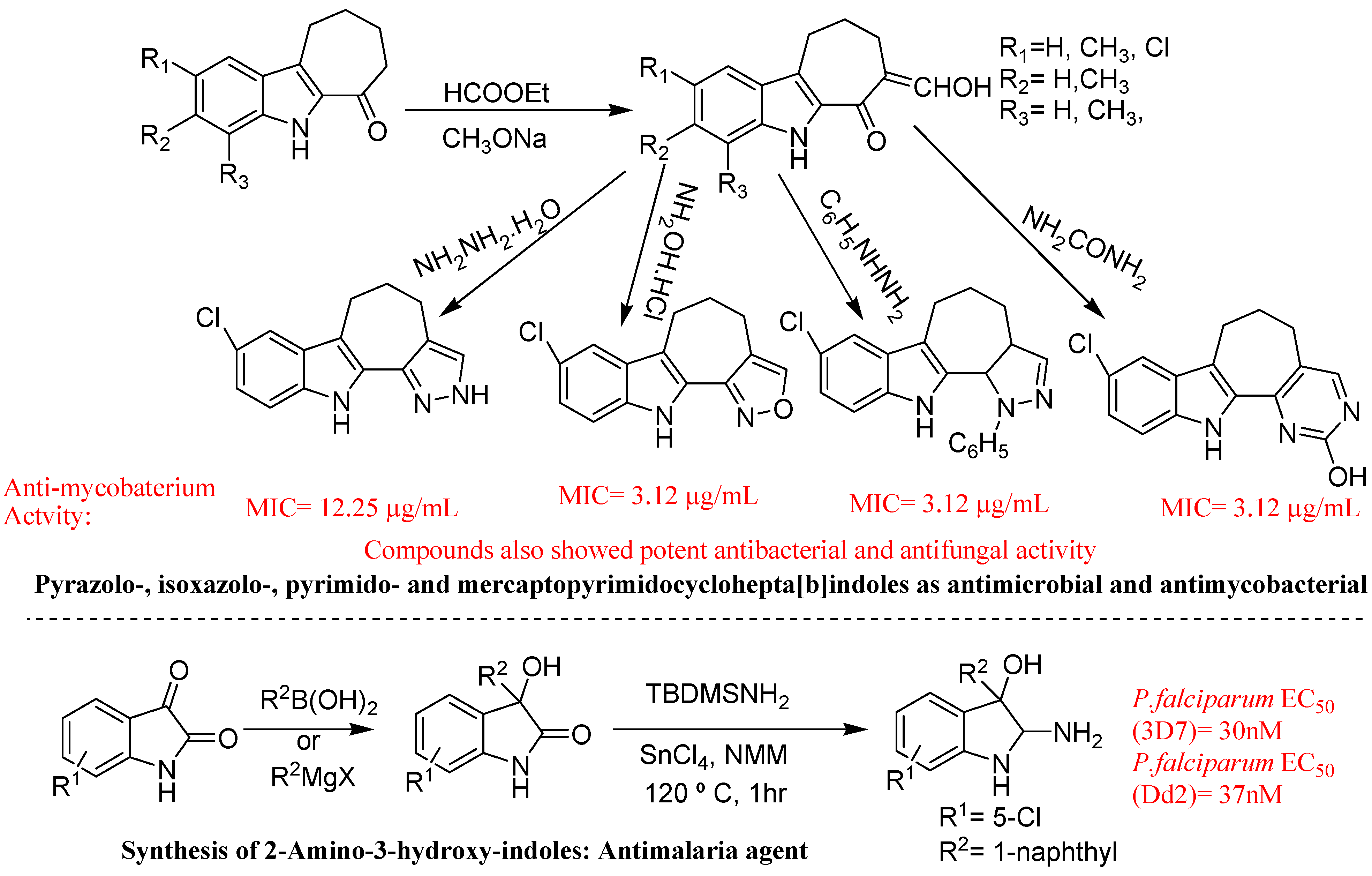


5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dolle, R.E.; Nelson, K.H. Comprehensive survey of combinatorial library synthesis: 1998. J. Comb. Chem. 1999, 1, 235–282. [Google Scholar]
- Franzen, R.G. Recent advances in the preparation of heterocycles on solid support: A review of the literature. J. Comb. Chem. 2000, 2, 195–214. [Google Scholar] [CrossRef]
- Dolle, R.E. Comprehensive survey of combinatorial library synthesis: 2000. J. Comb. Chem. 2001, 3, 477–517. [Google Scholar] [CrossRef]
- Hanessian, S.; McNaughton-Smith, G.; Lombart, H.G.; Lubell, W.D. Design and synthesis of conformationally constrained amino acids as versatile scaffolds and peptide mimetics. Tetrahedron 1997, 53, 12789–12854. [Google Scholar] [CrossRef]
- Radwanski, E.R; Last, R.L. Tryptophan biosynthesis and metabolism: Biochemical and molecular genetics. Plant Cell 1995, 7, 921–934. [Google Scholar]
- Jones, R.S. Tryptamine: A neuromodulator or neurotransmitter in mammalian brain? Progress Neurobiol. 1982, 19, 117–139. [Google Scholar] [CrossRef]
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu. Rev. Med. 2009, 60, 335–366. [Google Scholar]
- Chilton, W.S.; Bigwood, J.; Gensen, R.E. Psilocin, bufotenine and serotonin: Historical and biosynthetic observations. J. Psychedelic Drugs 1979, 11, 61–69. [Google Scholar] [CrossRef]
- Freidonk-Mueschenborn, E.; Fox, A. Resolution of concentration–response differences in onset of effect between subcutaneous and oral sumatriptan. Headache 2005, 45, 632–637. [Google Scholar] [CrossRef]
- Generali, J.A.; Cada, D.J. Off-label drug uses—Ondansetron: Postanesthetic shivering. Hospital Pharmacy 2009, 44, 670–671. [Google Scholar] [CrossRef]
- Horton, R. Lotronex and the FDA: A fatal erosion of integrity. Lancet 2001, 375, 1544–1545. [Google Scholar] [CrossRef]
- Saxton, J.E. Monoterpenoid indole alkaloids. In The Chemistry of Heterocyclic Compounds Part 4; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Mehta, R.G.; Liu, J.; Constantinou, A.; Thomas, C.F.; Hawthorne, M.; You, M.; Gerhüser, C.; Pezzuto, J.M.; Moon, R.C.; Moriarty, R.M. Cancer chemopreventive activity of brassinin, a phytoalexin from cabbage. Carcinogenesis 1995, 16, 399–404. [Google Scholar] [CrossRef]
- Cohen, S. The Beyond Within: The LSD Story; Atheneum: New York, NY, USA, 1964. [Google Scholar]
- Ferreira, S.; Moncada, S.; Vane, J. Indomethacin and aspirin abolish prostaglandin release from the spleen. Nat. New Biol. 1971, 231, 237–239. [Google Scholar] [CrossRef]
- Johnson, I.S.; Armstrong, J.G.; Gorman, M.; Burnett, J.P., Jr. The Vinca Alkaloids: A new class of oncolytic agents. Cancer Res. 1963, 23, 1390–1427. [Google Scholar]
- Duflos, A.; Kruczynski, A.; Barrat, J.M. Novel aspects of natural and modified vinca alkaloids. Curr. Med. Chem. Anticancer Agents 2002, 2, 55–70. [Google Scholar]
- Beckers, T.; Mahboobi, S. Natural, semisynthetic and synthetic microtubule inhibitors for cancer therapy. Drugs Future 2003, 28, 767–785. [Google Scholar] [CrossRef]
- Bacher, G.; Beckers, T.; Emig, P.; Klenner, T.; Kutshert, B.; Nickel, B. New small-molecule tubulin inhibitors. Pure Appl. Chem. 2001, 73, 1459. [Google Scholar] [CrossRef]
- Beckers, T.; Baasner, S.; Klenner, T.; Mahboobi, S.; Pongratz, H.; Frieser, M.; Hufsky, H.; Hockemeyer, J.; Fiebig, H.H.; Burger, A.; Bohmer, F.D. 2-Acyl indol derivatives and their use as antitumor agents. WO/2001/082909, 8 November 2001. [Google Scholar]
- Gastpar, R.; Goldbrunner, M.; Marko, D.; von Angere, E. Methoxy-substituted 3-formyl-2-phenylindoles inhibit tubulin polymerization. J. Med. Chem. 1998, 41, 4965–4972. [Google Scholar] [CrossRef]
- Medarde, M.; Ramos, A.; Caballero, E.; Pela’z-Lamamie’ de Clairac, R.; Lo’pez, J.L.; Garcia’ Gravalos, D.; San Felicia, A. Synthesis and antineoplastic activity of combretastatin analogues: Heterocombretastatins. Eur. J. Med. Chem. 1998, 33, 71–77. [Google Scholar] [CrossRef]
- Flynn, B.L.; Hamel, E.; Jung, M.K. One-pot synthesis of benzo[b]furan and indole inhibitors of tubulin polymerization. J. Med. Chem. 2002, 45, 2670–2673. [Google Scholar] [CrossRef]
- Yamamoto, K.; Noda, K.; Yoshimura, A.; Kukuoka, M.; Furuse, K.; Niitani, H. Phase-I Study of E7010. Cancer Chem. Pharmacol. 1998, 42, 127–134. [Google Scholar] [CrossRef]
- Flynn, B.L.; Flynn, G.P.; Hamel, E.; Jung, M.K. The synthesis and tubulin binding activity of thiophene-based analogues A-4 combretastatin. Bioorg. Med. Chem. Lett. 2001, 11, 2341–2343. [Google Scholar] [CrossRef]
- Bos, M.; Jenck, F.; Martin, J.R.; Moreau, J.L.; Mutel, V.; Sleight, A.J.; Widmer, U. Synthesis, pharmacology and therapeutic potential of 10-methoxypyrazino[1,2-a]indoles, partial agonists at the 5HT2C receptor. Eur. J. Med. Chem. 1997, 32, 253–261. [Google Scholar] [CrossRef]
- Hudson, A.L.; Price, R.; Tyacke, R.J.; Lalies, M.D.; Parker, C.A.; Nutt, D.J. Harmane, norharmane and tetrahydro β-carboline have high affinity for rat imidazoline binding sites. Br. J. Pharmacol. 1999, 126, 2. [Google Scholar]
- Husbands, S.M.; Glennon, R.A.; Gorgerat, S.; Gough, R.; Tyacke, R.; Crosby, J.; Nutt, D.J.; Lewis, J.W.; Hudson, A.L. β-carboline binding to imidazoline receptors. Drug Alcohol. Depend. 2001, 64, 203–208. [Google Scholar] [CrossRef]
- Escude, C.; Nguyen, C-H.; Mergny, J-L.; Sun, J-S.; Bisagni, E.; Garestier, T.; Helene, C. Selective Stabilization of DNA Triple Helixes by Benzopyridoindole Derivatives. J. Am. Chem. Soc. 1995, 117, 10212–10219. [Google Scholar] [CrossRef]
- Johnson, J.R.; Bruce, W.F.; Dutcher, J.D. Gliotoxin, the antibiotic principle of gliocladium fimbriatum. I. production, physical and biological properties. J. Am. Chem. Soc. 1943, 65, 2005–2009. [Google Scholar] [CrossRef]
- Fridrichsons, J.; Mathieson, A.M. The crystal structure of gliotoxin. Acta. Crystallogr. 1967, 23, 439–448. [Google Scholar] [CrossRef]
- Hara, M.; Han, M. Ras farnesyltransferase inhibitors suppress the phenotype resulting from an activated ras mutation in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 3333–3337. [Google Scholar] [CrossRef]
- Nagase, T.; Kawata, S.; Tamura, S.; Matsuda, Y.; Inui, Y.; Yamasaki, E.; Ishiguro, H.; Ito, T.; Miyagawa, J.; Mitsui, H.; Yamamoto, K.; Kinoshita, M.; Matsuzawa, Y. Manumycin and gliotoxin derivative KT7595 block Ras farnesylation and cell growth but do not disturb lamin farnesylation and localization in human tumour cells. Br. J. Cancer 1997, 76, 1001–1010. [Google Scholar] [CrossRef]
- Van der Pyl, D.; Inokoshi, J.; Shiomi, K.; Yang, H.; Takeshima, H.; Omura, S. Inhibition of farnesyl-protein transferase by gliotoxin and acetylgliotoxin. J. Antibiot. 1992, 45, 1802–1805. [Google Scholar] [CrossRef]
- Stiborova, M.; Bieler, C.A.; Wiessler, M.; Frei, E. The anticancer agent ellipticine on activation by cytochrome P450 forms covalent DNA adducts. Biochem. Pharmacol. 2001, 62, 1675–1684. [Google Scholar] [CrossRef]
- Auclair, C. Multimodal action of antitumor agents on DNA: the ellipticine series. Arch. Biochem. Biophys. 1987, 259, 1–14. [Google Scholar] [CrossRef]
- Frei, E.; Bieler, C.A.; Arlt, V.M.; Wiessler, M.; Stiborova, M. Covalent binding of the anticancer drug ellipticine to DNA in V79 cells transfected with human cytochrome P450 enzymes. Biochem. Pharmacol. 2002, 64, 289–295. [Google Scholar] [CrossRef]
- Stiborova, M.; Stiborova-Rupertova, M.; Borek-Dohalska, L.; Wiessler, M.; Frei, E. Rat microsomes activating the anticancer drug ellipticine to species covalently binding to deoxyguanosine in DNA are a suitable model mimicking ellipticine bioactivation in humans. Chem. Res. Toxicol. 2003, 16, 38–47. [Google Scholar] [CrossRef]
- Stiborova, M.; Breuer, A.; Aimova, D.; Stiborová-Rupertová, M.; Wiessler, M.; Frei, E. DNA adduct formation by the anticancer drug ellipticine in rats determined by 32P postlabeling. Int. J. Cancer 2003, 107, 885–890. [Google Scholar] [CrossRef]
- Murray, G.I.; Weaver, R.J.; Paterson, P.J.; Ewen, S.W.B.; Melvin, W.T.; Danny, M.B. Expression of xenobiotic metabolizing enzymes in breast cancer. J. Pathol. 1993, 169, 347–353. [Google Scholar] [CrossRef]
- Borek-Dohalska, L.; Frei, E.; Stiborova, M. DNA adduct formation by the anticancer drug ellipticine and its hydroxy derivatives in human breast adenocarcinoma MCF-7 Cells. Collect Czech Chem. Commun. 2004, 69, 603–615. [Google Scholar] [CrossRef]
- Campbell, J.A. ; Bordunov, V.; Broke, C.A.; Dhankwardt, J.; Hendricks, R.T.; Kress, J.M.; Walker, K.A.M.; Wang, J.H. Preparation of 3-arylmethylindoles as selective COX-2 inhibitors. Tetrahedron Lett. 2004, 45, 3793–3796. [Google Scholar] [CrossRef]
- Andersson, K.E. Pharmacology of penile erection. Pharmacol. Rev. 2001, 53, 417–450. [Google Scholar]
- Rosengren, A.H.; Jokubka, R.; Tojjar, D.; Granhall, C.; Hansson, O.; Li, D.Q.; Nagaraj, V.; Reinbothe, T.M.; Tuncel, J.; Eliasson, L.; et al. Overexpression of Alpha2A-Adrenergic receptors contributes to type 2 diabetes. Science 2009, 327, 217–220. [Google Scholar]
- Scott, L.J.; Perry, C.M. Delavirdine: A review of its use in HIV infection. Drugs 2000, 60, 1411–1444. [Google Scholar] [CrossRef]
- Biswal, S.; Sahoo, U.; Sethy, S.; Kumar, H.K.S.; Banerjee, M. Indole: The molecule of diverse biological activities. Asian J. Pharm. Clin. Res. 2012, 5, 1. [Google Scholar]
- Yutkin, V.; Chin, J. Apaziquone as an intravesical therapeutic agent for urothelial non-muscle-invasive bladder cancer. Expert Opin. Investig. Drugs. 2012, 21, 251–260. [Google Scholar] [CrossRef]
- Hall; Chapman; Rhodes, P.H. Dictionary of Organic Compounds; Chapman & Hall: London, UK, 1996. [Google Scholar]
- Leneva, I.A.; Russell, R.J.; Boriskin, Y.S.; Hay, A.J. Characteristics of arbidol-resistant mutants of influenza virus: Implications for the mechanism of anti-influenza action of arbidol. Antiviral Res. 2009, 81, 132–140. [Google Scholar] [CrossRef]
- Napalkov, D.A. Therapy with perindopril: organoprotection, not just the antihypertensive effect. Kardiologiia 2012, 52, 80–83. [Google Scholar]
- Bacher, N.; Tiefenthaler, M.; Sturm, S.; Stuppner, H.; Ausserlechner, M.J.; Kofler, R.; Konwalinka, G. Oxindole alkaloids from Uncaria tomentosa induce apoptosis in proliferating, G0/G1-arrested and bcl-2-expressing acute lymphoblastic leukaemia cells. Br. J. Haematol. 2006, 132, 615–622. [Google Scholar] [CrossRef]
- García Giménez, D.; García Prado, E.; Sáenz Rodríguez, T.; Fernández Arche, A.; de la Puerta, R. Cytotoxic effect of the pentacyclic oxindole alkaloid mitraphylline isolated from Uncaria tomentosa bark on human Ewing's sarcoma and breast cancer cell lines. Planta Med. 2010, 76, 133–136. [Google Scholar] [CrossRef]
- Prince, H.M.; Bishton, M. Panobinostat (LBH589): A novel pan-deacetylase inhibitor with activity in T cell lymphoma. Hematol. Meet. Rep. 2009, 3, 33–38. [Google Scholar]
- Mack, G.S. To selectivity and beyond. Nat. Biotech. 2010, 28, 1259–1266. [Google Scholar] [CrossRef]
- Isaac, M.T. Review: Combining pindolol with an SSRI improves early outcomes in people with depression. Evid. Based Ment. Health 2004, 7, 107. [Google Scholar] [CrossRef]
- Lednicer, D. The Organic Chemistry of Drug Synthesis; A John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; Volume 7, pp. 141–154. [Google Scholar]
- Klimke, A.; Klieser, E. Antipsychotic efficacy of the dopaminergic autoreceptor agonist EMD 49980 (Roxindol). Results of an open clinical study. Pharmacopsychiatry 1991, 24, 107–112. [Google Scholar] [CrossRef]
- Muller, W.; Stratz, T. Local treatment of tendinopathies and myofascial pain syndromes with the 5-HT3 receptor antagonist tropisetron. Scand. J. Rheumatic Suppl. 2004, 119, 44–48. [Google Scholar] [CrossRef]
- Macor, J.E.; Gurley, D.; Lanthorn, T.; Loch, J.; Mack, R.A.; Mullen, G.; Tran, O.; Wright, N.; Gordon, J.C. The 5-HT3 antagonist tropisetron (ICS 205-930) is a potent and selective alpha7 nicotinic receptor partial agonist. Bioorg. Med. Chem. Lett. 2001, 11, 319–321. [Google Scholar] [CrossRef]
- Cui, R.; Suemaru, K.; Li, B.; Kohnomi, S.; Araki, H. Tropisetron attenuates naloxone-induced place aversion in single-dose morphine-treated rats: role of alpha7 nicotinic receptors. Eur. J. Pharma. 2009, 609, 74–77. [Google Scholar] [CrossRef]
- Morse, G.D.; Reichman, R.C.; Fischl, M.A.; Para, M.; Leedom, J.; Powderly, W.; Demeter, L.M.; Resnick, L.; Bassiakos, Y.; Timpone, J.; Cox, S.; Batts, D. Concentration-targeted phase I trials of atevirdine mesylate in patients with HIV infection: dosage requirements and pharmacokinetic studies. The ACTG 187 and 199 study teams. Antiviral Res. 2010, 45, 47–58. [Google Scholar]
- Hart, F.; Boardman, P. Indomethacin: A New non-steroid anti-inflammatory agent. Br. Med. J. 1963, 2, 965–970. [Google Scholar] [CrossRef]
- Smyth, C.J. Indomethacin—Its rightful place in treatment. Ann. Intern. Med. 1970, 72, 430–432. [Google Scholar] [CrossRef]
- DrugBank.ca. Available online: http://www.drugbank.ca/drugs/DB00328 (accessed on 15 April 2013).
- Arens, H.; Borbe, H.O.; Ulbrich, B.; Stöckigt, J. Detection of pericine, a new cns-active indole alkaloid from picralima nitida cell suspension culture by opiate receptor binding studies. Planta Med. 1982, 46, 210–214. [Google Scholar] [CrossRef]
- Roberts, M.F.; Wink, M. Alkaloids: Biochemistry, Ecology, and Medicinal Applications; Plenum Press: New York, NY, USA, 1998. [Google Scholar]
- Haubrich, D.R.; Ward, S.J.; Baizman, E.; Bell, M.R.; Bradford, J.; Ferrari, R.; Miller, M.; Perrone, M.; Pierson, A.K.; Saelens, J.K.; et al. Pharmacology of pravadoline: A new analgesic agent. J. Pharmacol. Exp. Ther. 1990, 255, 511–522. [Google Scholar]
- Bell, M.R.; D'Ambra, T.E.; Kumar, V.; Eissenstat, M.A.; Herrmann, J.L.; Wetzel, J.R., Jr; Rosi, D.; Philion, R.E.; Daum, S.J.; Hlasta, D.J.; et al. Antinociceptive (aminoalkyl) indoles. J. Med. Chem. 1991, 34, 1099–1110. [Google Scholar] [CrossRef]
- Cook, P.; James, I. Cerebral Vasodilators. N. Engl. J. Med. 1981, 305, 1560–1564. [Google Scholar] [CrossRef]
- Arpe, H.-J.; Ullmann, F. Indole Alkaloids. In Ullmann's Encyclopedia of Industrial Chemistry, 5th ed.; Wiley-VCH: Weinheim, Germany, 1985. [Google Scholar]
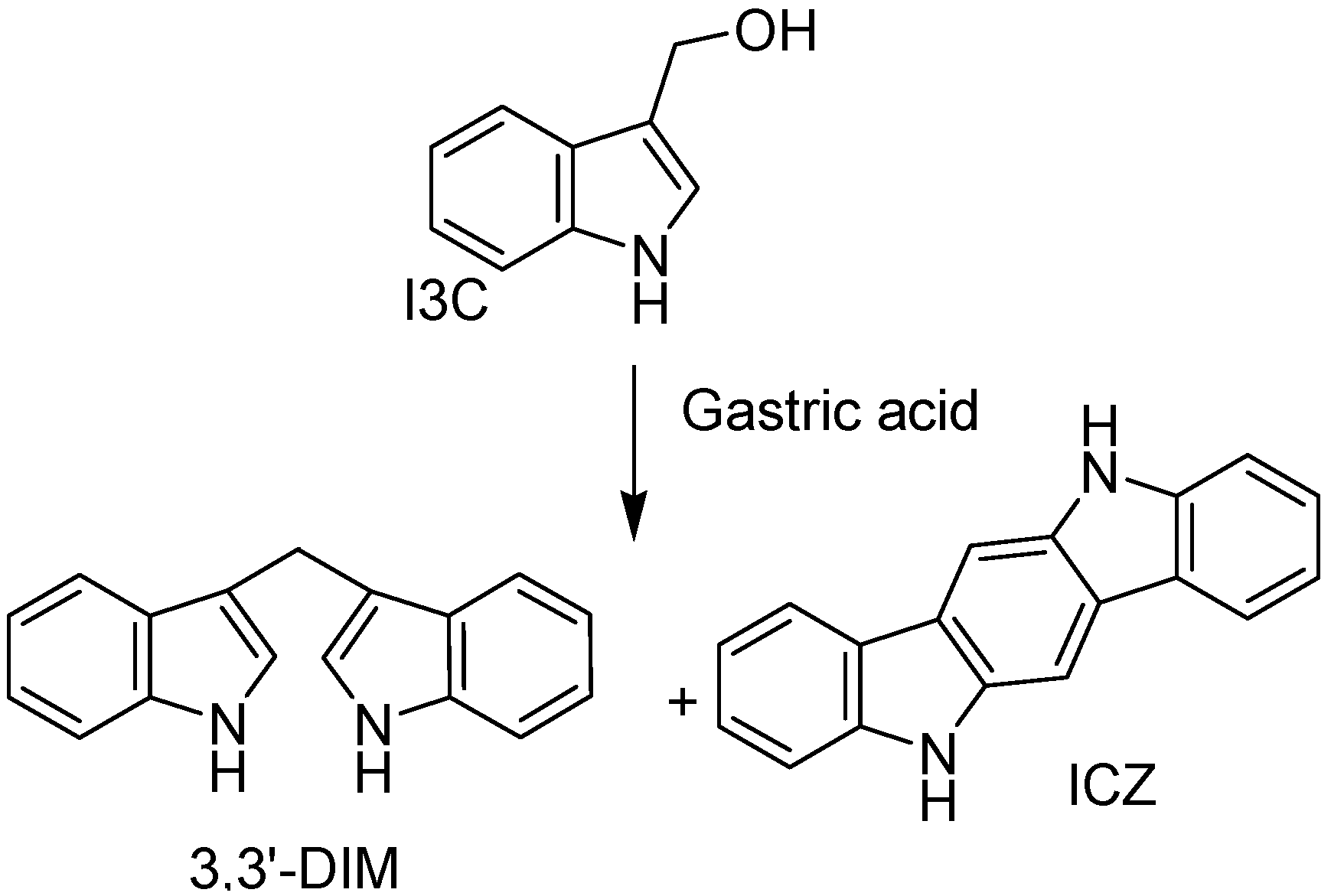
- Lynn, A.; Collins, A.; Fuller, Z.; Hillman, K; Ratcliffe, B. Cruciferous vegetables and colo-rectal cancer. Proc. Nutr. Soc. 2006, 65, 135–144. [Google Scholar] [CrossRef]
- Dashwood, R.H.; Arbogast, D.N.; Fong, A.T.; Pereira, C.; Hendricks, J.D.; Bailey, G.S. Quantitative inter-relationships between aflatoxin B1 carcinogen dose, indole-3-carbinol anti-carcinogen dose, target organ DNA adduction and final tumor response. Carcinogenesis 1989, 10, 175–181. [Google Scholar] [CrossRef]
- Ho, J.N.; Jun, W.; Choue, R.; Lee, J. I3C and ICZ inhibit migration by suppressing the EMT process and FAK expression in breast cancer cells. Mol. Med. Rep. 2012, 7, 384–388. [Google Scholar]
- Chang, X.; Tou, J.C.; Hong, C.; Kim, H.A.; Riby, J.E.; Firestone, G.L.; Bjeldane, L.F. 3,3′-Diindolylmethane inhibits angiogenesis and the growth of transplantable human breast carcinoma in athymic mice. Carcinogenesis 2005, 26, 771–778. [Google Scholar]
- Rahman, K.M.W.; Li, Y.; Wang, Z.; Sarkar, S.H.; Sarkar, F.H. Gene expression profiling revealed survivin as a target of 3,3′-Diindolylmethane-induced cell growth inhibition and apoptosis in breast cancer cells. Cancer Res. 2006, 66, 4952–4960. [Google Scholar] [CrossRef]
- Benninghoff, A.D.; Williams, D.E. The role of estrogen receptor β in transplacental cancer prevention by indole-3-Carbinol. Cancer Prev. Res. 2013, 6, 339–348. [Google Scholar] [CrossRef]
- Liu, A.G.; Volker, S.E.; Jeffery, E.H.; Erdman, J.W., Jr. Feeding tomato and broccoli powders enriched with bioactives improves bioactivity markers in rats. J. Agric. Food Chem. 2009, 57, 7304–7310. [Google Scholar] [CrossRef]
- Kim, Y.S.; Milner, J.A. Targets for indole-3-carbinol in cancer prevention. J. Nutr. Biochem. 2005, 16, 65–73. [Google Scholar] [CrossRef]
- Shorey, L.E.; Hagman, A.M.; Williams, D.E.; Ho, E.; Dashwood, R.H.; Benninghoff, A.D. 3,3′-Diindolylmethane induces G1 arrest and apoptosis in human acute T-Cell lymphoblastic leukemia cells. PLoS One 2012, 7, e34975. [Google Scholar]
- Tsai, J.T.; Liu, H.C.; Chen, Y.H. Suppression of inflammatory mediators by cruciferous vegetable-derived indole-3-carbinol and phenylethyl isothiocyanate in lipopolysaccharide-activated macrophages. Mediat. Inflamm. 2010, 2010, 293642. [Google Scholar]
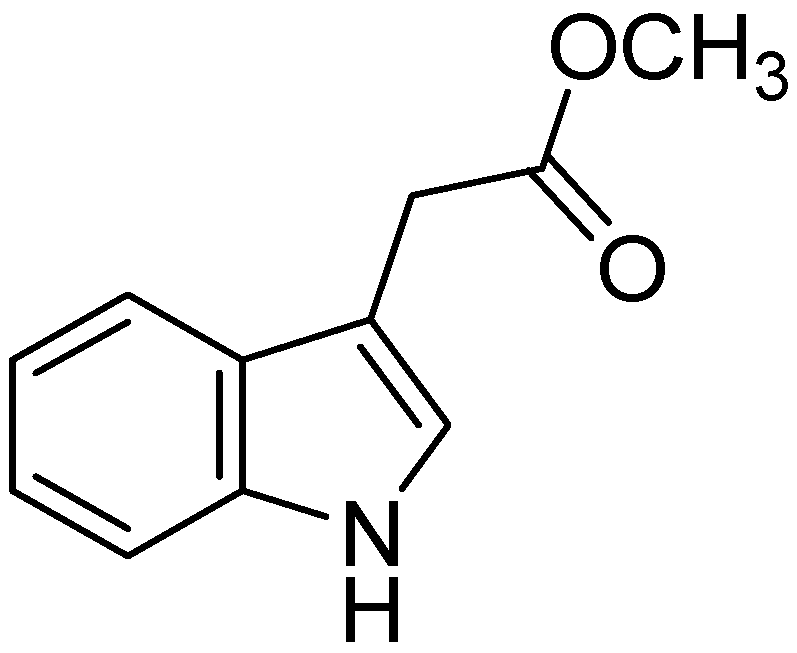
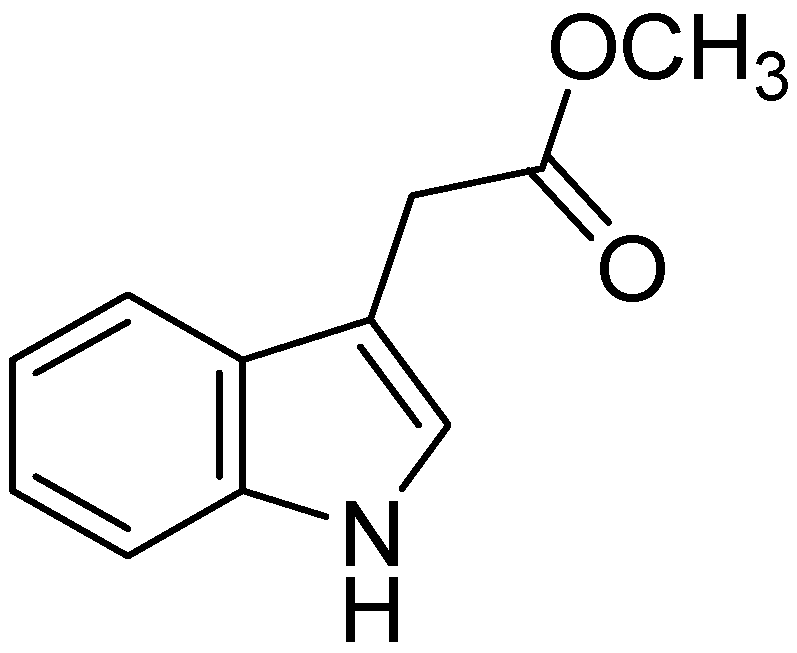
- Zhang, S.; Li, Z.; Wu, X.; Huang, Q.; Shen, H.M.; Ong, C.N. Methyl-3-indolylacetate inhibits cancer cell invasion by targeting the MEK1/2-ERK1/2 signaling pathway. Mol. Cancer Ther. 2006, 5, 3285–3293. [Google Scholar] [CrossRef]
- Jeffery, E.H.; Araya, M. Physiological effects of broccoli consumption. Phytochem. Rev. 2009, 8, 283–298. [Google Scholar] [CrossRef]
- Shin, J.S.; Yun, C.H.; Cho, Y.W.; Baek, N.I.; Choi, M.S.; Jeong, T.S.; Chung, H.G.; Lee, K.T. Indole-containing fractions of Brassica rapa inhibit inducible nitric oxide synthase and pro-inflammatory cytokine expression by inactivating nuclear factor-κB. J. Med. Food. 2011, 14, 1527–1537. [Google Scholar] [CrossRef]
- Fernández-Pérez, F.; Almagro, L.; Pedreño, M.A.; GómezRos, L.V. Synergistic and cytotoxic action of indole alkaloids produced from elicited cell cultures of catharanthus roseus. Pharm. Biol. 2013, 51, 304–310. [Google Scholar] [CrossRef]
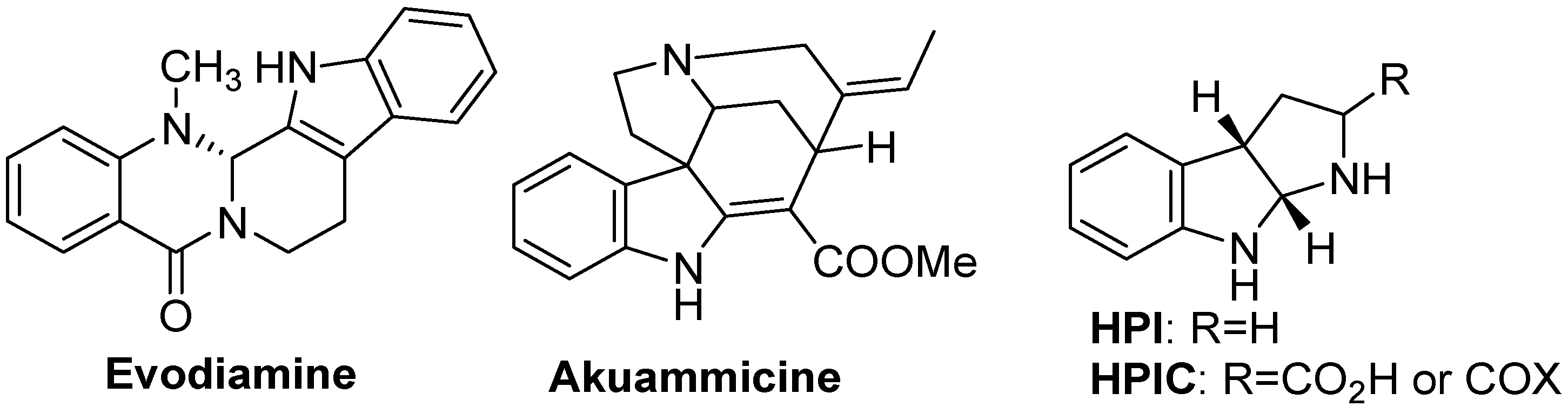
- Yu, H.; Jin, H.; Gong, W.; Wang, Z.; Liang, H. Pharmacological actions of multi-target-directed evodiamine. Molecules 2013, 18, 1826–1843. [Google Scholar] [CrossRef]
- Shittu, H.; Gray, A.; Furman, B.; Young, L. Glucose uptake stimulatory effect of akuammicine from Picralima nitida (Apocynaceae). Phytochem. Lett. 2010, 3, 53–55. [Google Scholar] [CrossRef]
- Ruiz-Sanchis, P.; Savina, S.A.; Albericio, F.; Álvarez, M. Structure, bioactivity and synthesis of natural products with hexahydropyrrolo[2,3-b]indole. Chem. Eur. J. 2011, 17, 1388–1408. [Google Scholar] [CrossRef]
- Bialonska, D.; Zjawiony, J.K. Aplysinopsins—Marine indole alkaloids: Chemistry, bioactivity and ecological significance. Mar. Drugs 2009, 7, 166–183. [Google Scholar] [CrossRef]
- Longeon, A.; Copp, B.R.; Quévrain, E.; Roué, M.; Kientz, B.; Cresteil, T.; Petek, S.; Debitus, C.; Bourguet-Kondracki, M.L. Bioactive indole derivatives from the south pacific marine sponges rhopaloeides odorabile and hyrtios sp. Mar. Drugs. 2011, 9, 879–888. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Mupparapu, N.; Battini, N.; Vishwakarma, R. A chemistry and biology of fascaplysin, a potent marine-derived CDK-4 inhibitor. Mini. Rev. Med. Chem. 2012, 12, 650–664. [Google Scholar] [CrossRef]
- Mollica, A.; Locatelli, M.; Stefanucci, A.; Pinnen, F. Synthesis and bioactivity of secondary metabolites from marine sponges containing dibrominated indolic systems. Molecules 2012, 17, 6083–6099. [Google Scholar] [CrossRef]
- Yang, X.W.; Zhang, G.Y.; Ying, J.X.; Yang, B.; Zhou, X.F.; Steinmetz, A.; Liu, Y.H.; Wang, N. Isolation, Characterization, and bioactivity evaluation of 3-((6-Methylpyrazin-2-yl)methyl)-1H-indole, a new alkaloid from a deep-sea-derived actinomycete Serinicoccus profundi sp. nov. Mar. Drugs 2013, 11, 33–39. [Google Scholar]
- Baker, J.T. Tyrian purple: An ancient dye, a modern problem. Endeavour 1974, 33, 11–17. [Google Scholar] [CrossRef]
- Baker, J.; Duke, C. Chemistry of the indoleninones. II. Isolation from the hypobranchial glands of marine molluscs of 6-bromo-2,2-dimethylthioindolin-3-one and 6-bromo-2-methylthioindoleninone as alternative precursors to Tyrian purple. Aust. J. Chem. 1973, 26, 2153–2157. [Google Scholar] [CrossRef]
- Westley, C.B.; Lewis, M.C.; Benkendorff, K. Histomorphology of the hypobranchial gland in Dicathais orbita (Gmelin, 1791) (Neogastropoda: Muricidae). J. Moll. Stud. 2010, 76, 186–195. [Google Scholar] [CrossRef]
- Benkendorff, K. Natural product research in the australian marine invertebrate dicathais orbita. Mar. Drugs 2013, 11, 1370–1398. [Google Scholar] [CrossRef]
- Devi, P.; Wahidullah, S.; Rodrigues, C.; Souza, L.D. The Sponge-associated bacterium bacillus licheniformis SAB1: A source of antimicrobial compounds. Mar. Drugs 2010, 8, 1203–1212. [Google Scholar] [CrossRef]
- Mandal, D.; Yamaguchi, A.D.; Yamaguchi, J.; Itami, K. Synthesis of dragmacidin D via direct C-H couplings. J. Am. Chem. Soc. 2011, 133, 19660–19663. [Google Scholar] [CrossRef]
- Chen, M.; Shao, C.L.; Fu, X.M.; Xu, R.F.; Zheng, J.J.; Zhao, D.L.; She, Z.G.; Wang, C.Y. Bioactive indole alkaloids and phenyl Ether derivatives from a marine-derived aspergillus sp. fungus. J. Nat. Prod. 2013, 76, 547–553. [Google Scholar] [CrossRef]
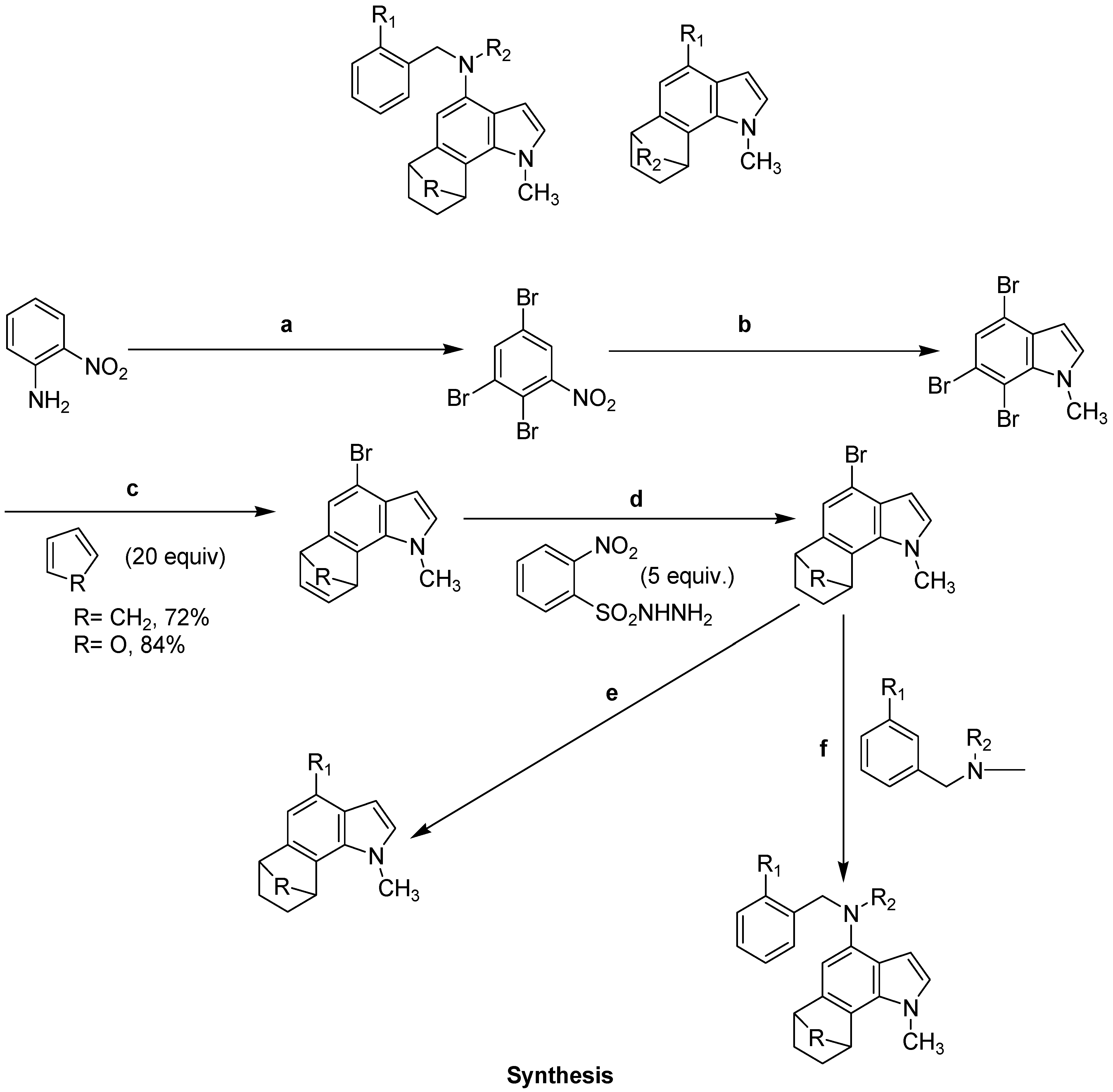
- Perchellet, J.P.; Waters, A.M.; Perchellet, E.M.; Thornton, P.D.; Brown, N.; Hill, D.; Neuenswander, B.; Lushington, G.H.; Santini, C.; Chandrasoma, N.; Buszek, K.R. Antitumor effects of synthetic 6,7-annulated-4-substituted indole compounds in L1210 leukemic cells in vitro. Anticancer. Res. 2012, 32, 4671–4684. [Google Scholar]
- Thornton, P.D.; Brown, N.; Hill, D.; Neuenswander, B.; Lushington, G.H.; Santini, C.; Buszek, K.R. Application of 6,7-indole aryne cycloaddition and Pd(0)-catalyzed Suzuki-Miyaura and Buchwald-Hartwig cross-coupling reactions for the preparation of annulated indole libraries. ACS Comb. Sci. 2011, 13, 443–448. [Google Scholar] [CrossRef]
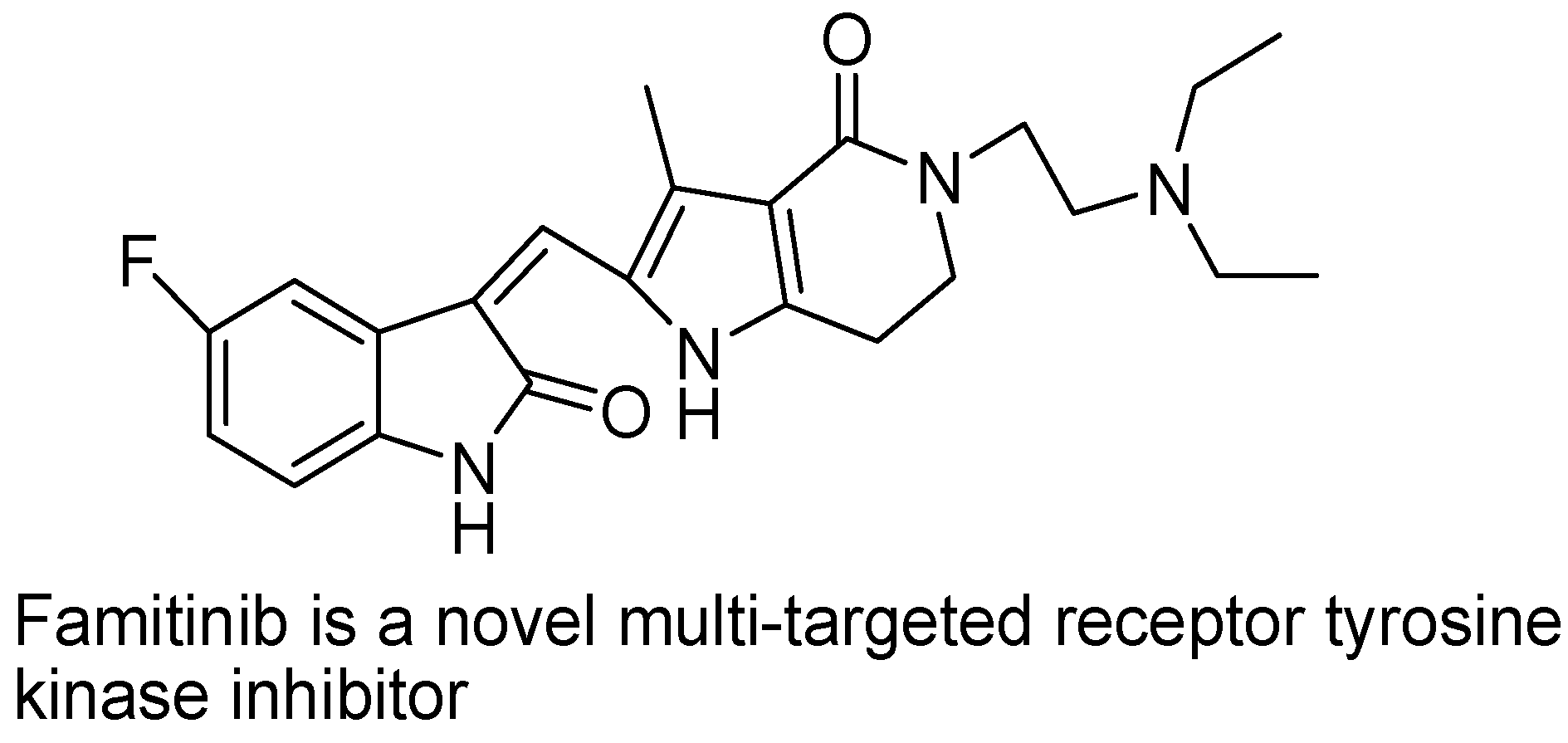
- Xie, C.; Zhou, J.; Guo, Z.; Diao, X.; Gao, Z.; Zhong, D.; Jiang, H.; Zhang, L.; Chen, X. Metabolism and bioactivation of famitinib, a novel inhibitor of receptor tyrosine kinase, in cancer patients. Br. J. Pharmacol. 2013, 168, 1687–1706. [Google Scholar] [CrossRef]
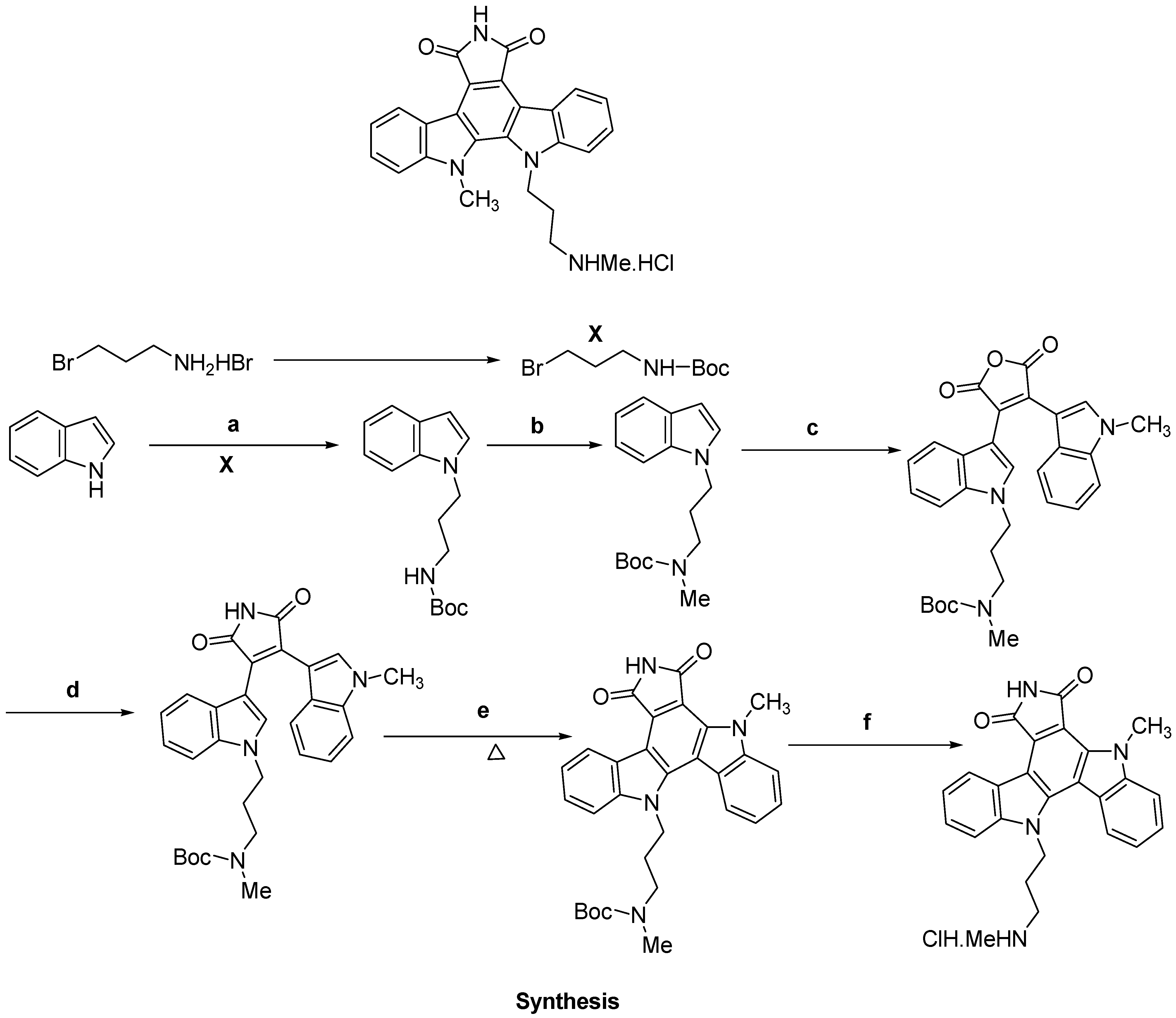
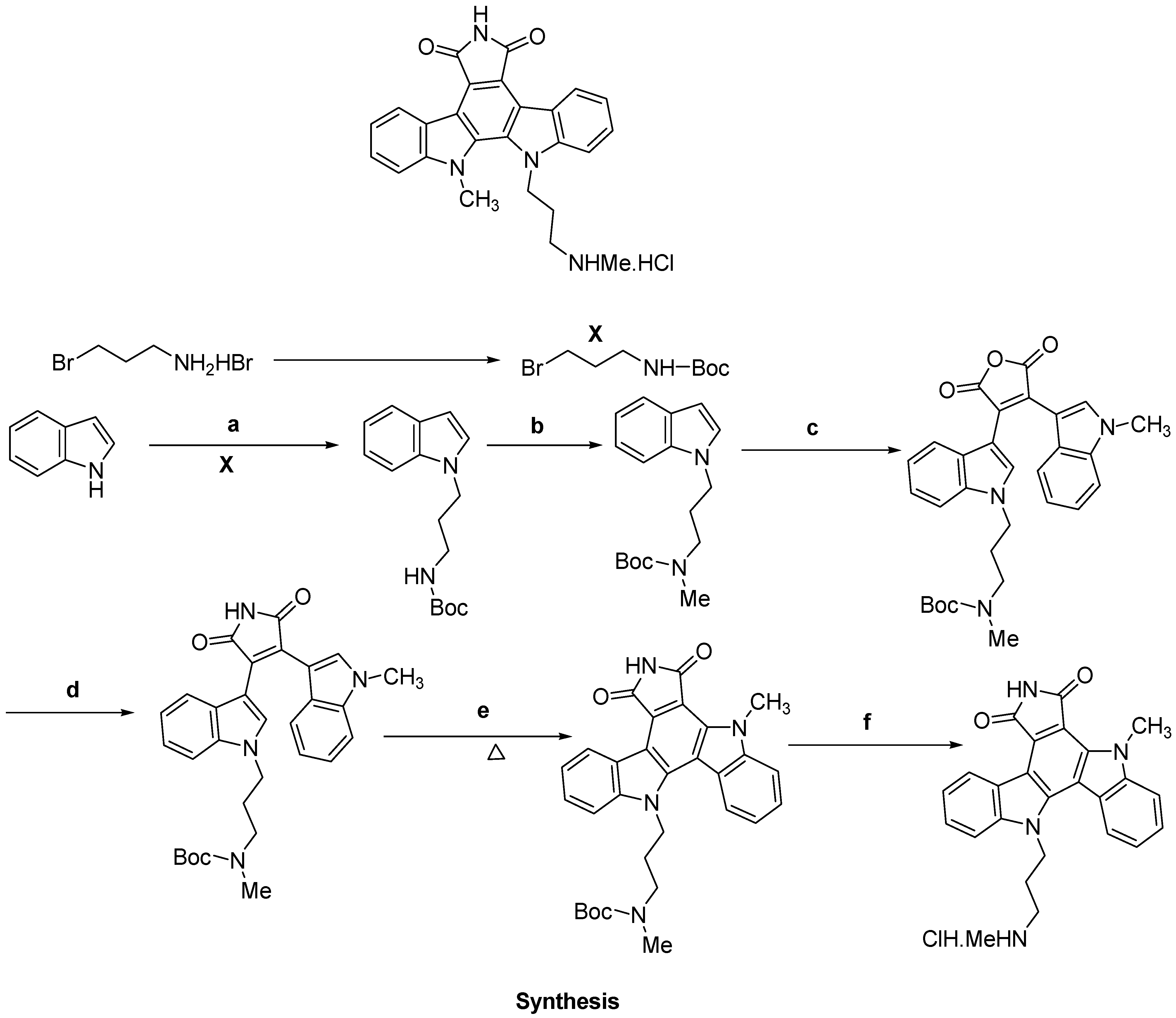
- Roy, S.; Eastman, A.; Gribble, G.W. Synthesis of bisindolylmaleimides related to GF109203x and their efficient conversion to the bioactive indolocarbazoles. Org. Biomol. Chem. 2006, 4, 3228–3234. [Google Scholar] [CrossRef]
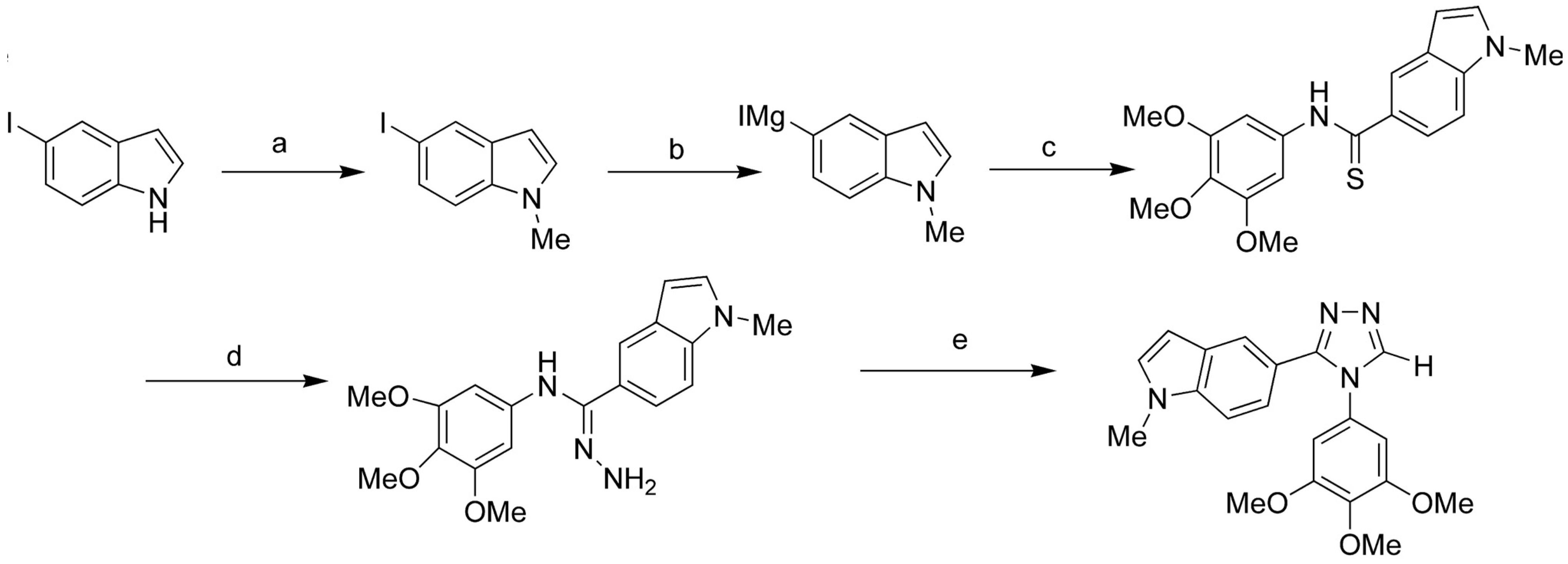
- Zhang, Q.; Peng, Y.; Wang, X.I.; Keenan, S.M.; Arora, S.; Welsh, W.J. Highly potent triazole-based tubulin polymerization inhibitors. J. Med. Chem. 2007, 50, 749–754. [Google Scholar] [CrossRef]
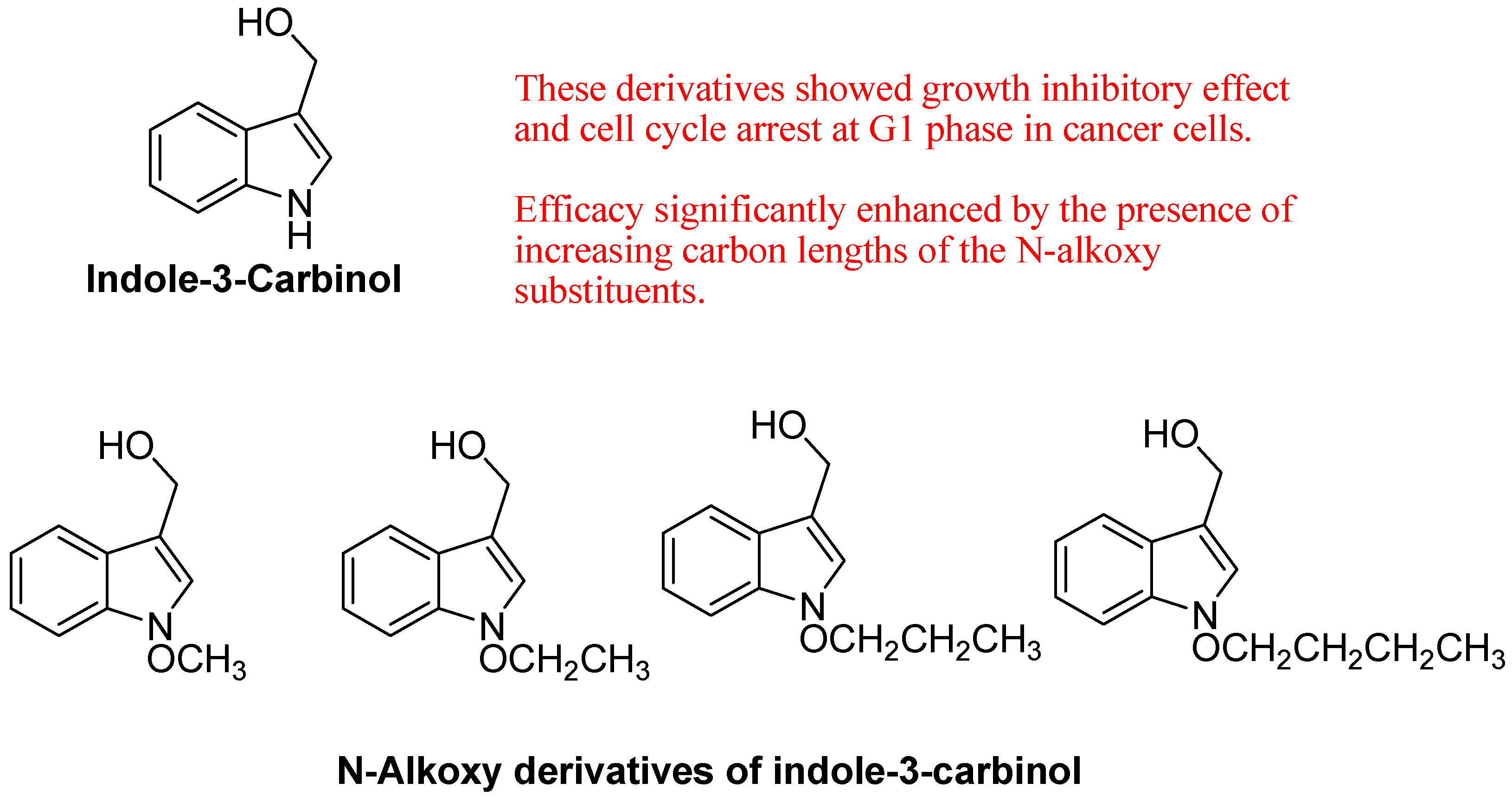
- Jump, S.M.; Kung, J.; Staub, R.; Kinseth, M.A.; Cram, E.J.; Yudina, L.N.; Preobrazhenskaya, M.N.; Bjeldanes, L.F.; Firestone, G.L. N-Alkoxy derivatization of indole-3-carbinol increases the efficacy of the G1 cell cycle arrest and of I3C-specific regulation of cell cycle gene transcription and activity in human breast cancer cells. Biochem. Pharmacol. 2008, 75, 713–724. [Google Scholar] [CrossRef]
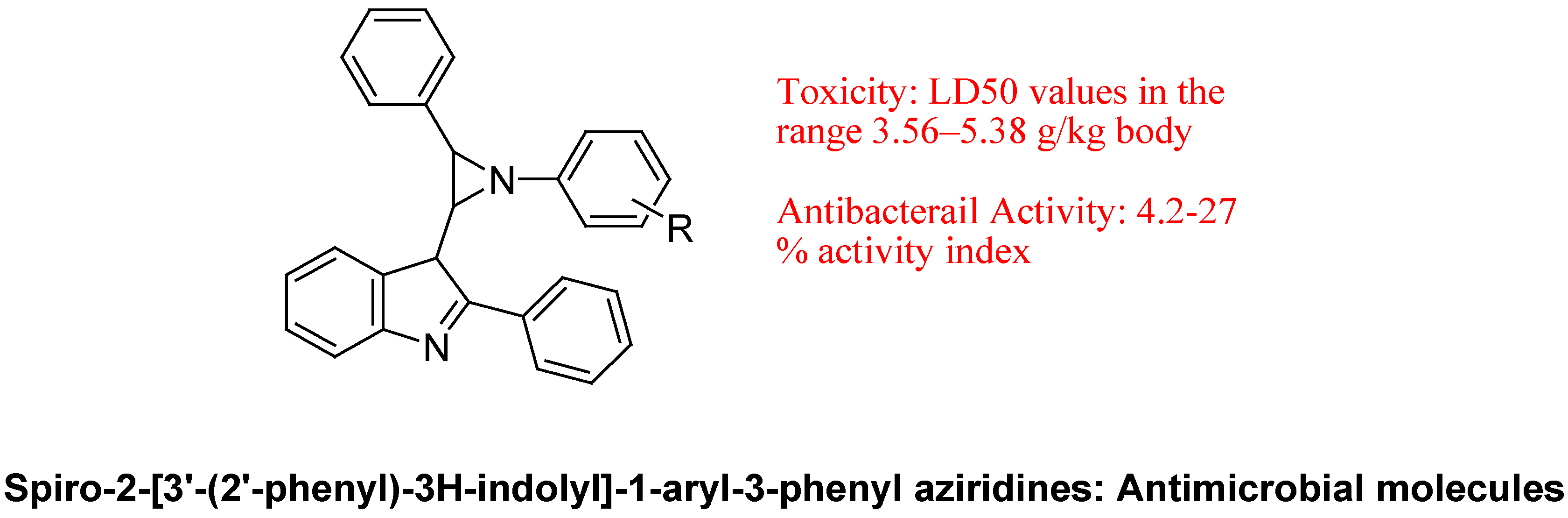
- Sharma, P.; Kumar, A.; Sahu, V.; Upadhyay, S.; Singh, J. Synthesis of bioactive spiro-2-[3′-(2′-phenyl)-3H-indolyl]-1-aryl-3-phenylaziridines and SAR studies on their antimicrobial behavior. Med. Chem. Res. 2009, 18, 383–395. [Google Scholar] [CrossRef]
- Chen, G.; Hao, X.J.; Sun, Q.Y.; Ding, J. Rapid synthesis and bioactivities of 3-(nitromethylene)indolin-2-one analogues. Chem. Pap. 2010, 64, 673–677. [Google Scholar] [CrossRef]
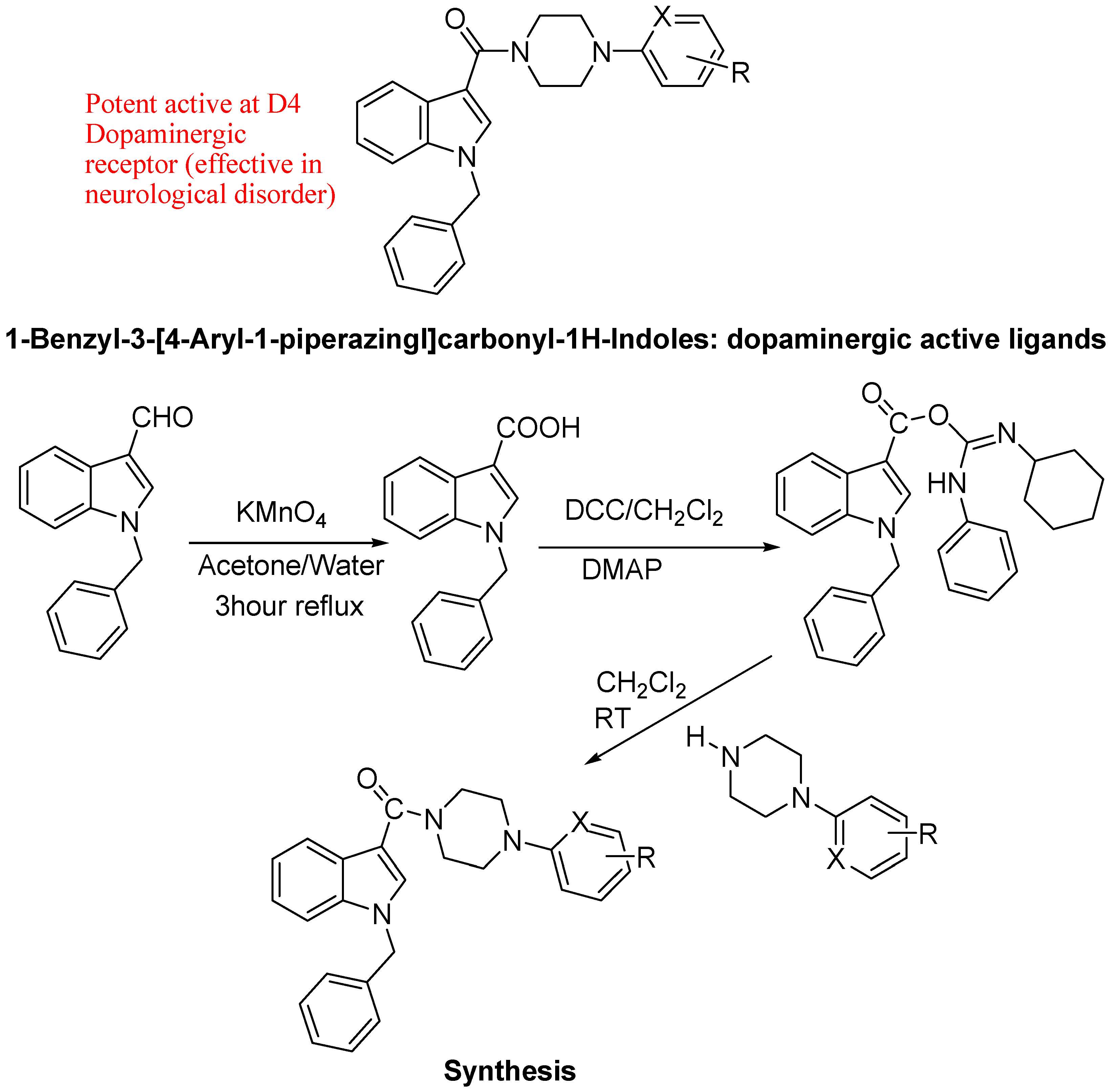
- Pessoa-Mahana, H.; Cuevas, M.I.; Pessoa-Mahana, C.D.; Araya-Maturana, R.; Fazardo, I.A.; Barria, C.S. Synthesis of 1-benzyl-3-[4-(aryl-1-piperazinyl) carbonyl]-1H-indoles.Novel ligands with potential D4 dopaminergic activity. J. Chil. Chem. Soc. 2011, 56, 866–869. [Google Scholar] [CrossRef]
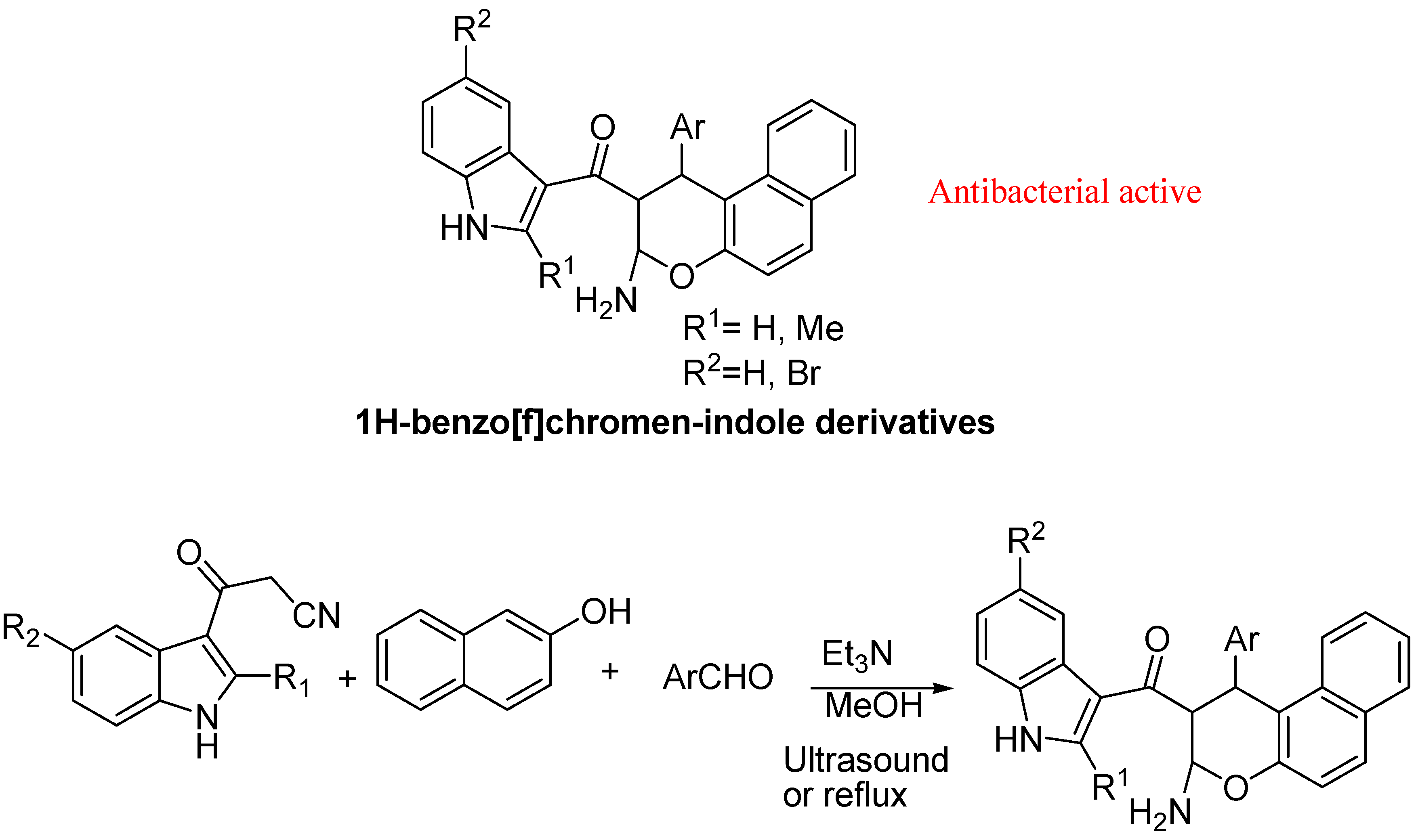
- Hosseinnia, R.; Mamaghani, M.; Tabatabaeian, K.; Shirini, F.; Rassa, M. An expeditious regioselective synthesis of novel bioactive indole-substituted chromene derivatives via one-pot three-component reaction. Bioorg. Med. Chem. Lett. 2012, 22, 5956–5960. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, P.; Chou, C.J.; Liu, C.; Wang, X.; Xu, W. Development of N-Hydroxycinnamamide-Based Histone Deacetylase Inhibitors with an Indole-Containing Cap Group. ACS Med. Chem. Lett. 2013, 4, 235–238. [Google Scholar] [CrossRef]
- Akkoc, M.K.; Yuksel, M.Y.; Durmaz, I.; Atalay, R.C. Design, synthesis, and biological evaluation of indole-based 1,4-disubstituted piperazines as cytotoxic agents. Turk. J. Chem. 2012, 36, 515–525. [Google Scholar]
- Singh, P.; Kaur, M.; Verma, P. Design, synthesis and anticancer activities of hybrids of indole and barbituric acids—Identification of highly promising leads. Bioorg. Med. Chem. Lett. 2009, 19, 3054–3058. [Google Scholar] [CrossRef]
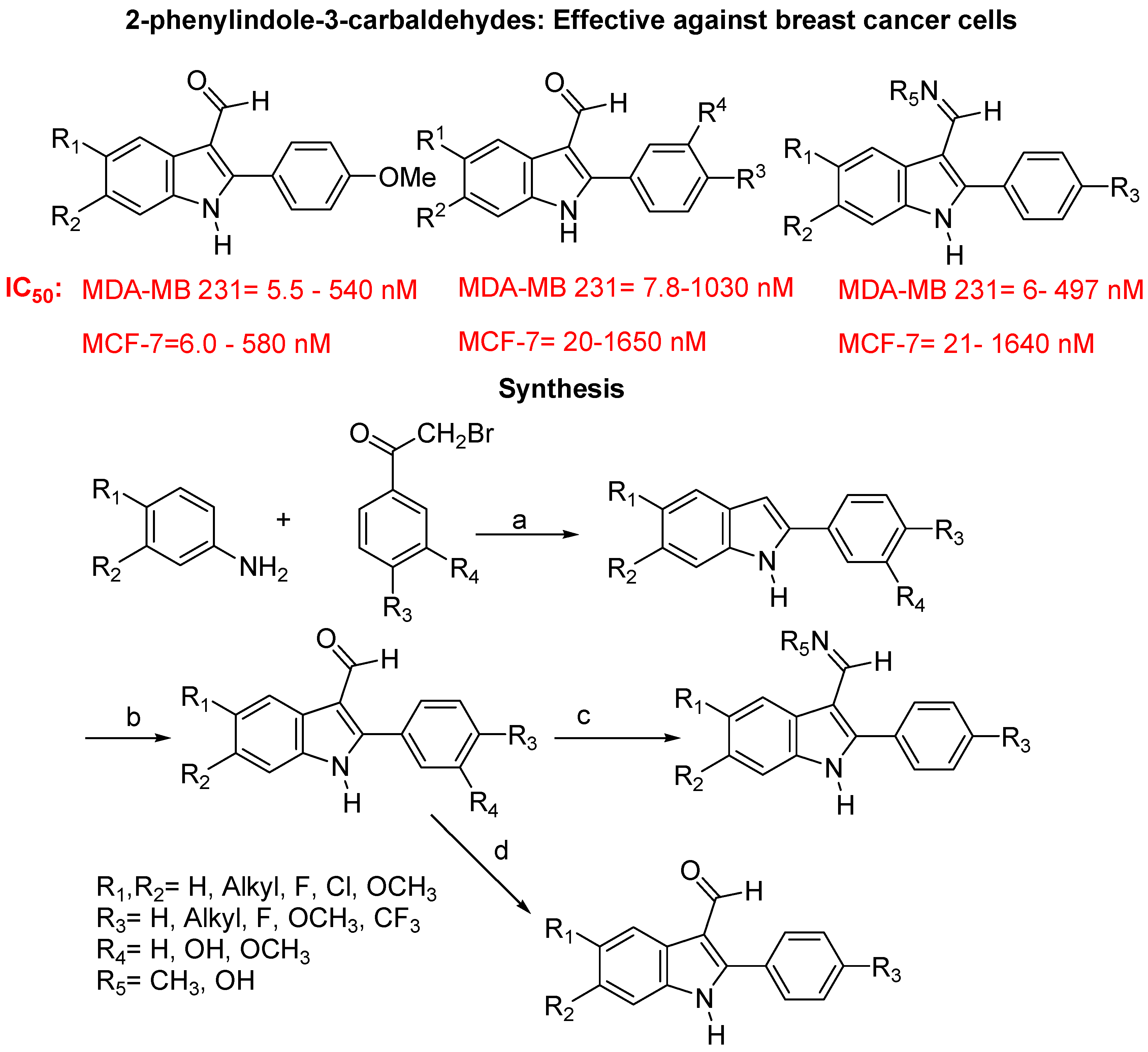
- Kaufmann, D.; Pojarová, M.; Vogel, S.; Liebl, R.; Gastpar, R.; Gross, D.; Nishino, T.; Pfaller, T.; Von, A.E. Antimitotic activities of 2-phenylindole-3-carbaldehydes in human breast cancer cells. Bioorg. Med. Chem. 2007, 15, 5122–5136. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, Y.; Sun, L.; Ding, L.; Gu, Y.; Gong, P. Synthesis and anti-tumor activity of 2-amino-3-cyano-6-(1H-indol-3-yl)-4-phenylpyridine derivatives in vitro. Eur. J. Med. Chem. 2011, 46, 3149–3157. [Google Scholar] [CrossRef]
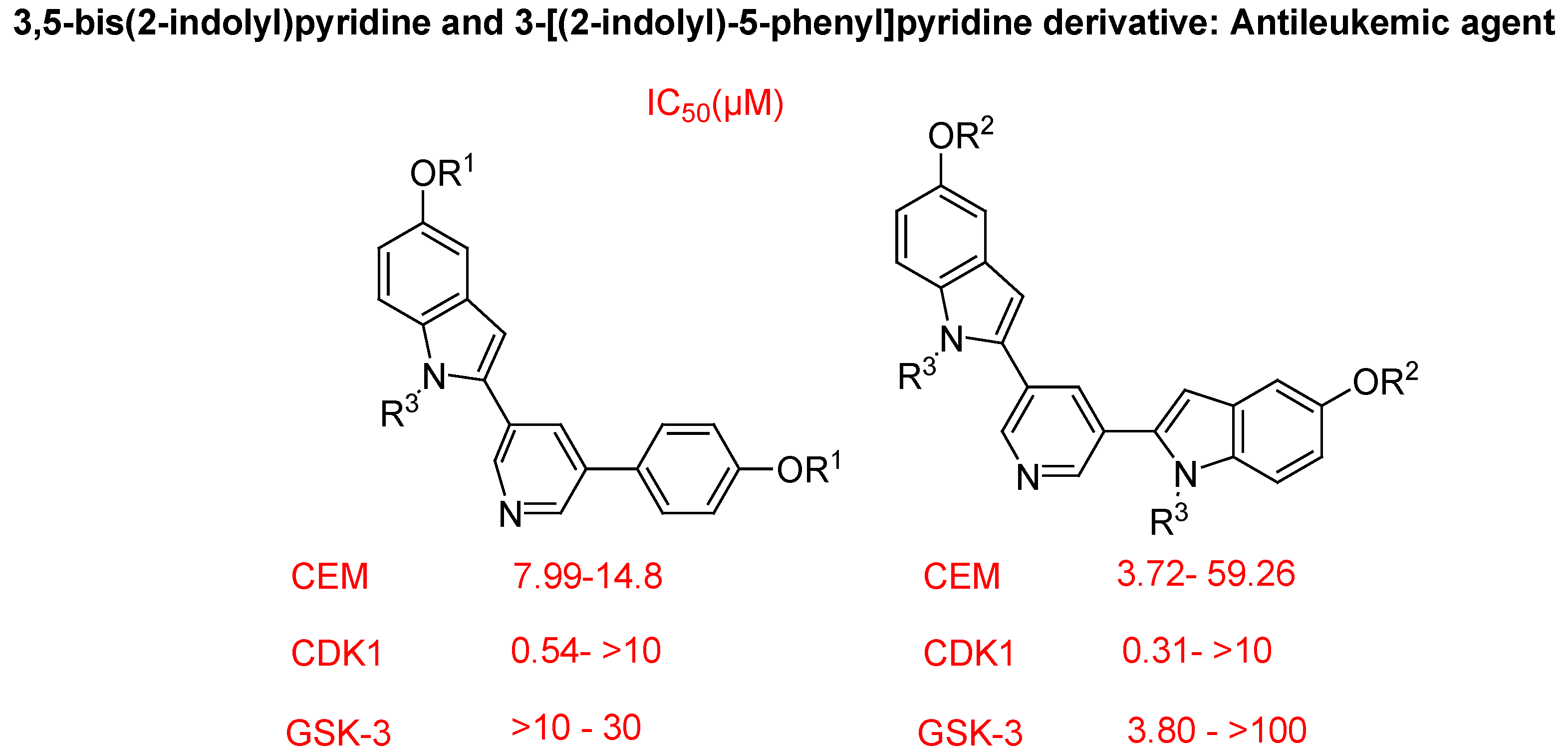
- Jacquemard, U.; Dias, N.; Lansiaux, A.; Bailly, C.; Logé, C.; Robert, J.M.; Lozach, O.; Meijer, L.; Mérour, J.Y.; Routier, S. Synthesis of 3,5-bis(2-indolyl)pyridine and 3-[(2-indolyl)-5-phenyl]pyridine derivatives as CDK inhibitors and cytotoxic agents. Bioorg. Med. Chem. 2008, 16, 4932–4953. [Google Scholar] [CrossRef]
- Nassar, E. Synthesis, (in vitro) antitumor and antimicrobial activity of some pyrazoline, pyridine, and pyrimidine derivatives linked to indole moiety. J. Am. Sci. 2010, 6, 463–471. [Google Scholar]
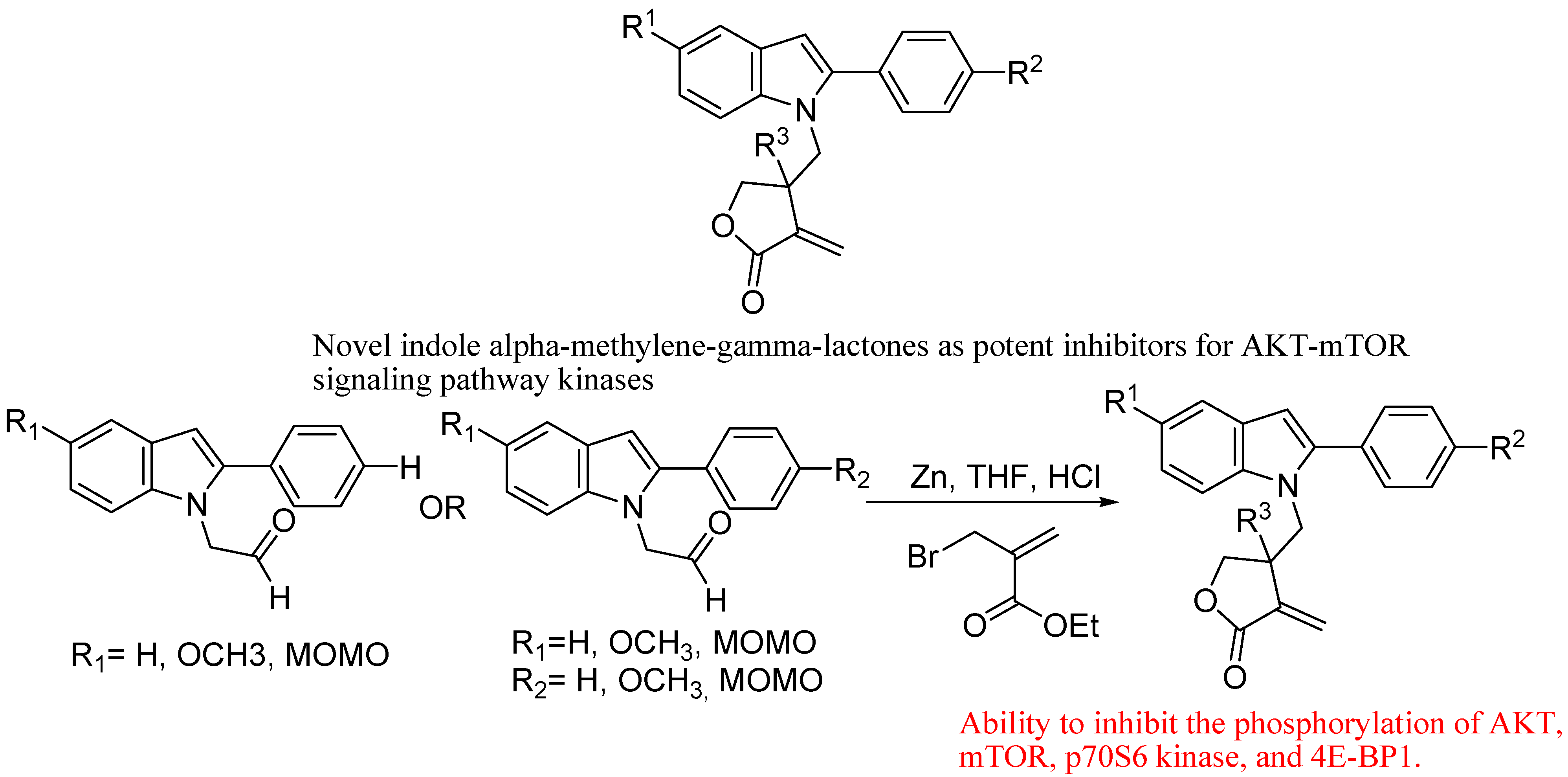
- Ding, H.; Zhang, C.; Wu, X.; Yang, C.; Zhang, X.; Ding, J.; Xie, Y. Novel indole α-methylene-γ-lactones as potent inhibitors for AKT-mTOR signaling pathway kinases. Bioorg. Med. Chem. Lett. 2005, 15, 4799–4802. [Google Scholar] [CrossRef]
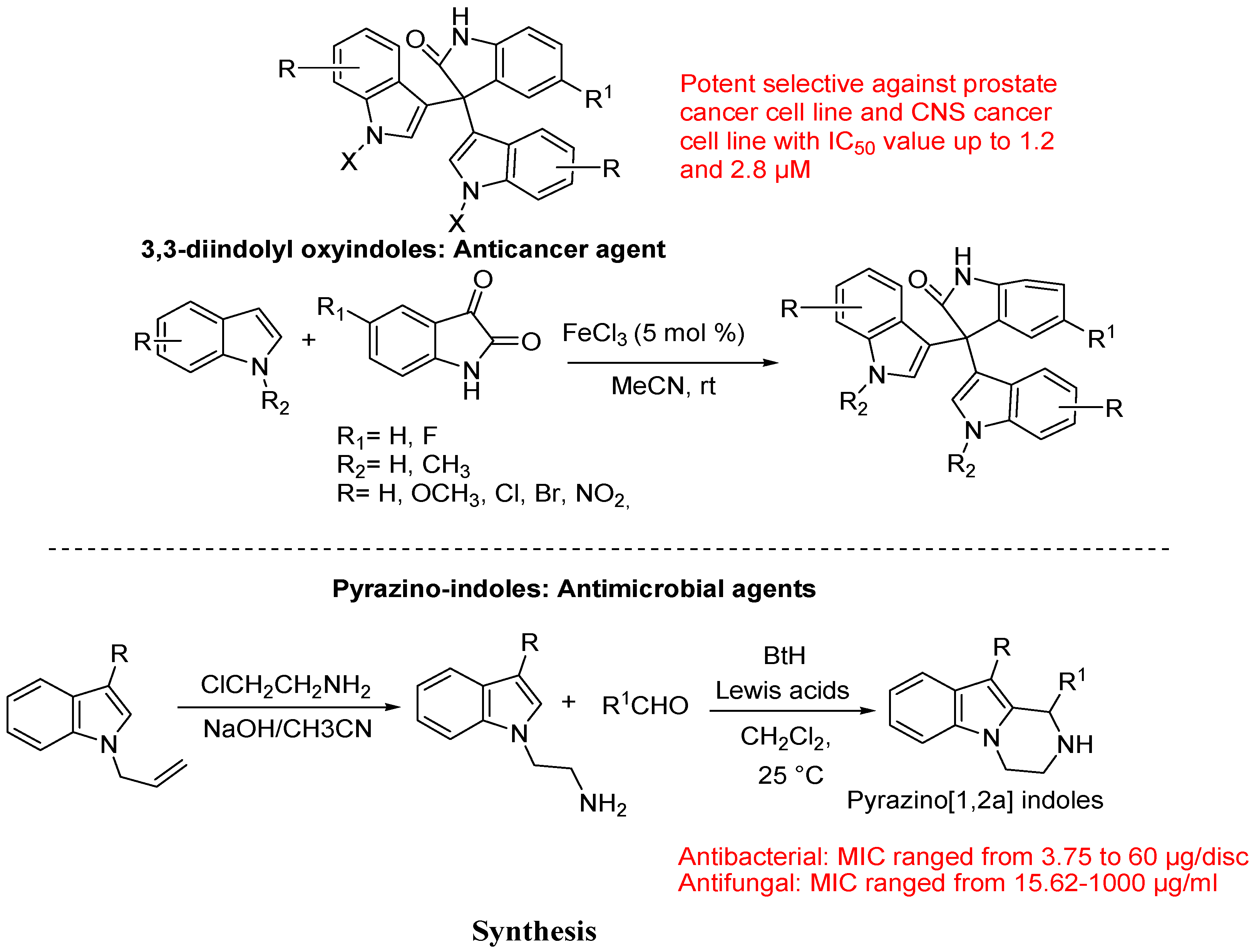
- Kamal, A.; Srikanth, Y.V.; Khan, M.N.; Shaik, T.B.; Ashraf, M. Synthesis of 3,3-diindolyl oxyindoles efficiently catalysed by FeCl3 and their in vitro evaluation for anticancer activity. Bioorg. Med. Chem. Lett. 2010, 20, 5229–5231. [Google Scholar] [CrossRef]
- Tiwari, R.K.; Singh, J.; Singh, D.; Verma, A.K.; Chandra, R. Highly efficient one-pot synthesis of 1-substituted-1,2,3,4-tetrahydropyrazino[1,2-a]indoles. Tetrahedron 2005, 61, 9513–9518. [Google Scholar] [CrossRef]
- Tiwari, R.K.; Singh, D.; Singh, J.; Yadav, V.; Pathak, A.K.; Dabur, R.; Chhillar, A.K.; Singh, R.; Sharma, G.L.; Chandra, R.; Verma, A.K. Synthesis and antibacterial activity of substituted 1,2,3,4-tetrahydropyrazino [1,2-a] indoles. Bioorg. Med. Chem. Lett. 2006, 16, 413–416. [Google Scholar] [CrossRef]
- Tiwari, R.K.; Verma, A.K.; Chhillar, A.K.; Singh, D.; Singh, J.; Sankar, V.K.; Yadav, V.; Sharma, G.L.; Chandra, R. Synthesis and antifungal activity of substituted 1,2,3,4-tetrahydropyrazino[1,2-a] indoles. Bioorg. Med. Chem. 2006, 14, 2747–2752. [Google Scholar] [CrossRef]
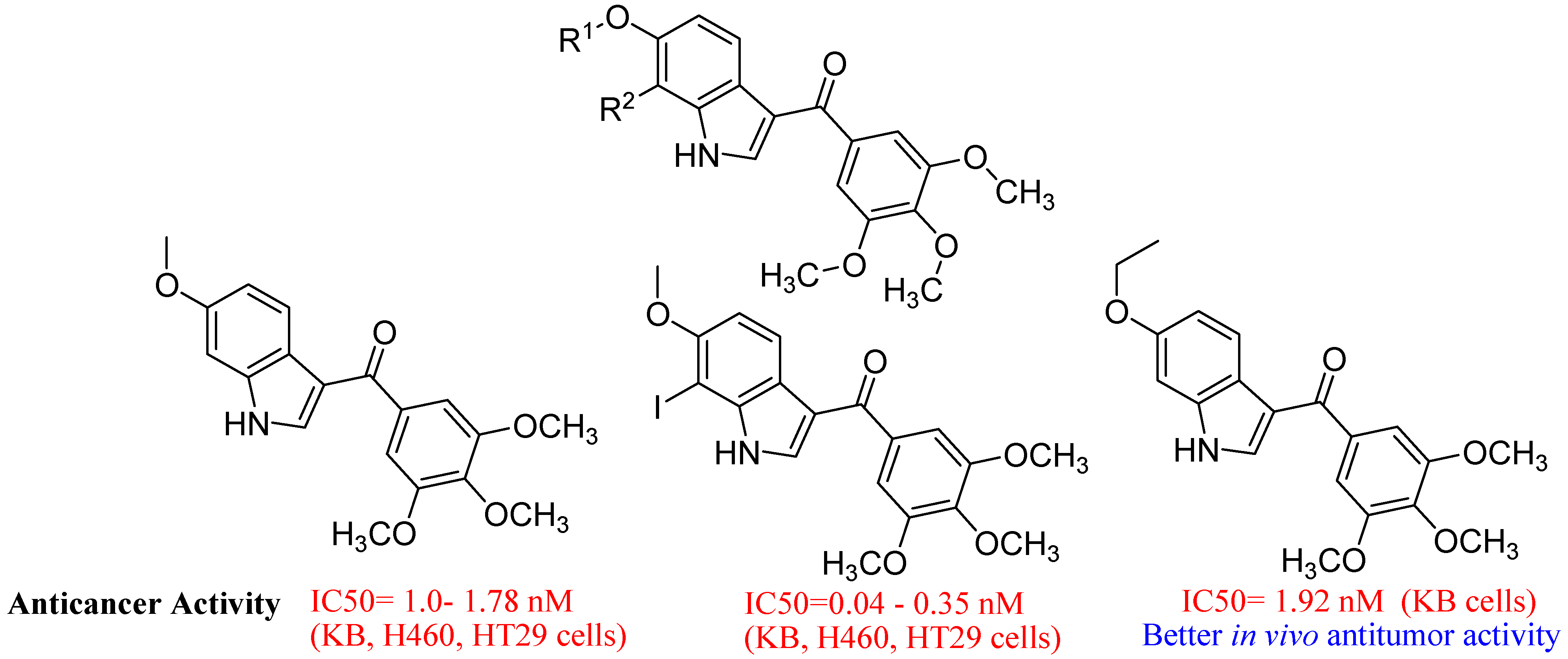
- Wu, Y.S.; Coumar, M.S.; Chang, J.Y.; Sun, H.Y.; Kuo, F.M.; Kuo, C.C.; Chen, Y.J.; Chang, C.Y.; Hsiao, C.L.; Liou, J.P.; et al. Synthesis and Evaluation of 3-Aroylindoles as Anticancer Agents: Metabolite Approach. J. Med. Chem. 2009, 52, 4941–4945. [Google Scholar] [CrossRef]
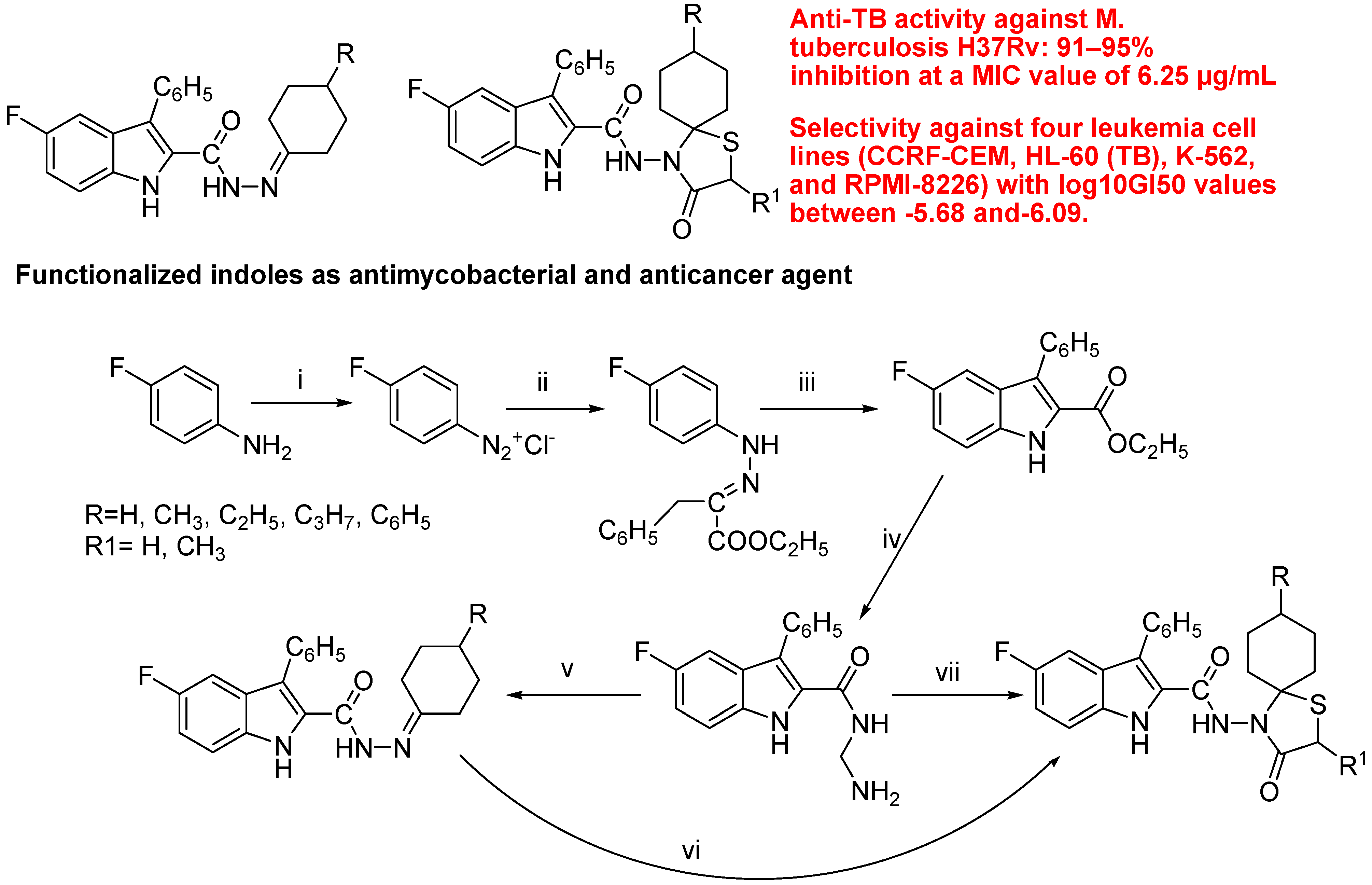
- Cihan-Üstündağ, G.; Capan, G. Synthesis and evaluation of functionalized indoles as antimycobacterial and anticancer agents. Mol. Divers. 2012, 16, 525–539. [Google Scholar] [CrossRef]
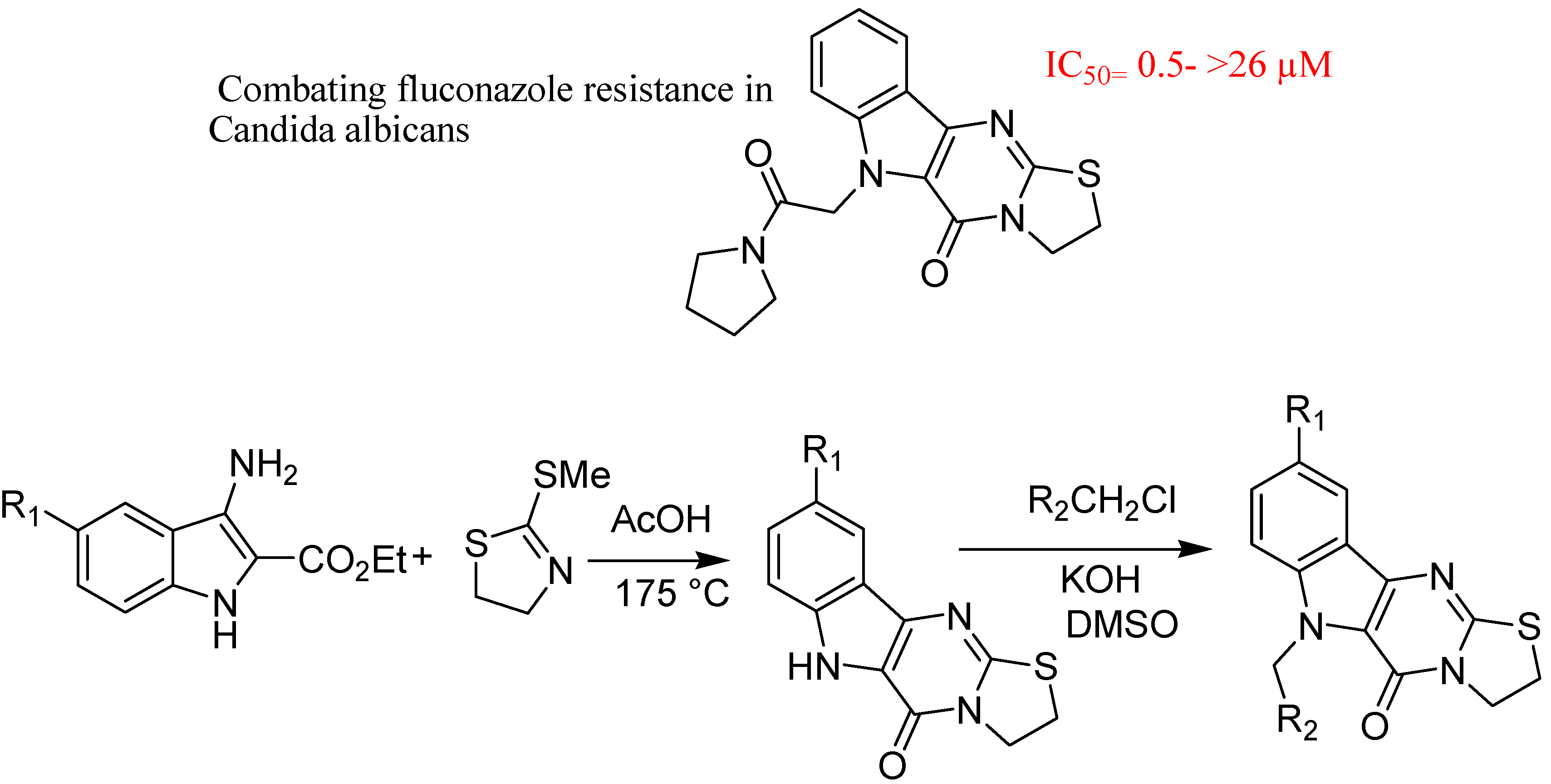
- Youngsaye, W.; Dockendorff, C.; Vincent, B.; Hartland, C.L.; Bittker, J.A.; Dandapani, S.; Palmer, M.; Whitesell, L.; Lindquist, S.; Schreiber, S.L.; et al. Overcoming fluconazole resistance in Candida albicans clinical isolates with tetracyclic indoles. Bioorg. Med. Chem. Lett. 2012, 22, 3362–3365. [Google Scholar] [CrossRef]
- Arumugam, N.; Raghunathan, R.; Almansour, A.I.; Karama, U. An efficient synthesis of highly functionalized novel chromeno[4,3-b]pyrroles and indolizino[6,7-b]indoles as potent antimicrobial and antioxidant agents. Bioorg. Med. Chem. Lett. 2012, 22, 1375–1379. [Google Scholar] [CrossRef]
- Yamuna, E.; Kumar, R.A.; Zeller, M.; Rajendra, P.K.J. Synthesis, antimicrobial, antimycobacterial and structure-activity relationship of substituted pyrazolo-, isoxazolo-, pyrimido- and mercaptopyrimidocyclohepta[b]indoles. Eur. J. Med. Chem. 2012, 47, 228–238. [Google Scholar] [CrossRef]
- Diana, P.; Carbone, A.; Barraja, P.; Montalbano, A.; Parrino, B.; Lopergolo, A.; Pennati, M.; Zaffaroni, N.; Cirrincione, G. Synthesis and Antitumor Activity of 3-(2-Phenyl-1,3-thiazol-4-yl)-1H-indoles and 3-(2-Phenyl-1,3-thiazol-4-yl)-1H-7-azaindoles. Chem. Med. Chem. 2011, 6, 1300–1309. [Google Scholar]
- Urgaonkar, S.; Cortese, J.F.; Barker, R.H.; Cromwell, M.; Serrano, A.E.; Wirth, D.F.; Clardy, J.; Mazitschek, R. A Concise Silylamine Approach to 2-Amino-3-hydroxy-indoles with Potent in vivo Antimalaria Activity. Org. Lett. 2010, 12, 3998–4001. [Google Scholar] [CrossRef]
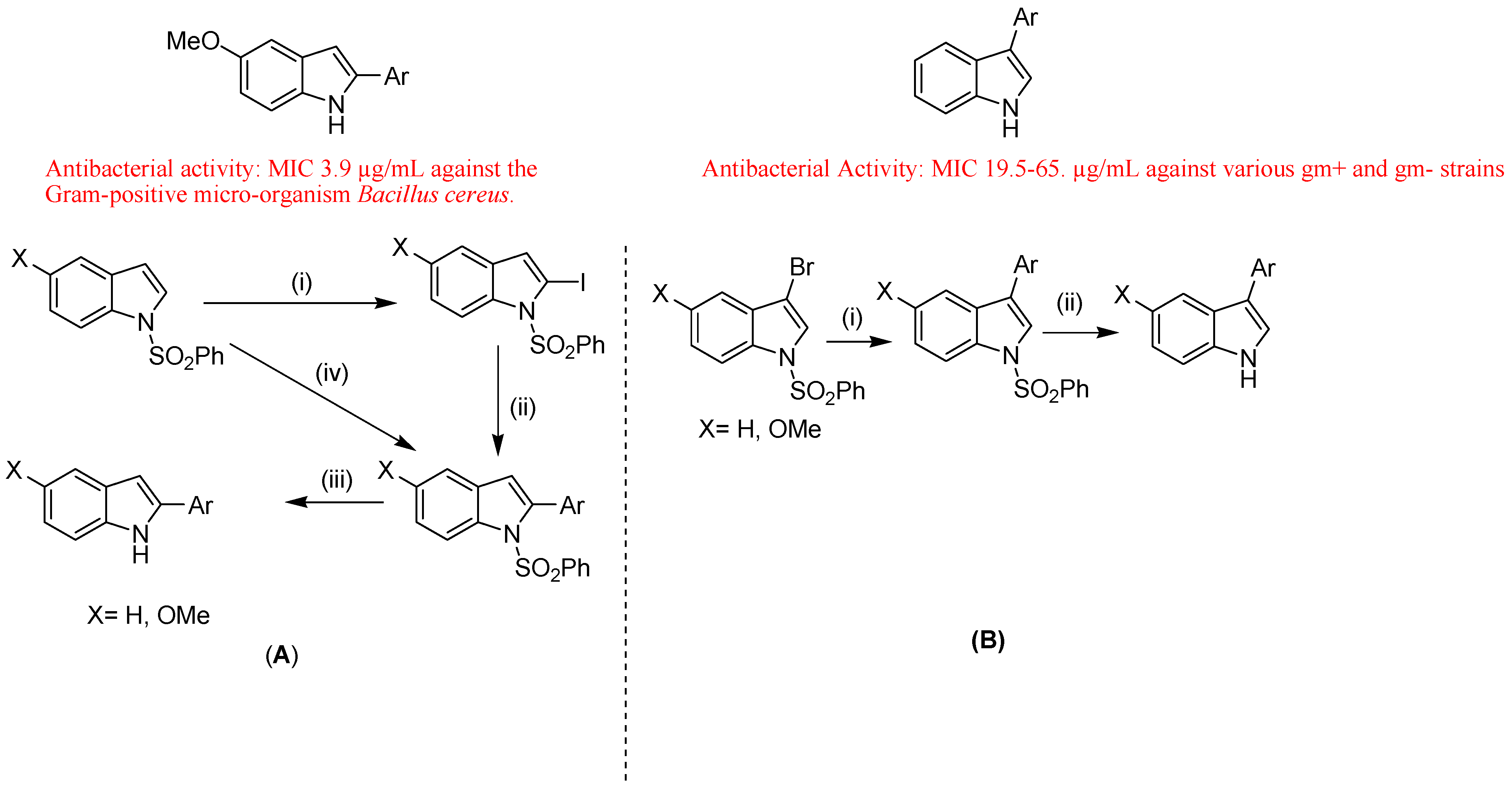
- Leboho, T.C.; Michael, J.P.; van Otterlo, W.A.; van Vuuren, S.F.; de Koning, C.B. The synthesis of 2- and 3-aryl indoles and 1,3,4,5-tetrahydropyrano[4,3-b]indoles and their antibacterial and antifungal activity. Bioorg. Med. Chem. Lett. 2009, 19, 4948–4951. [Google Scholar]
- Giraud, F.; Loge, C.; Pagniez, F.; Crepin, D.; Barres, S.; Picot, C.; Le Pape, P.; le Borgne, M. Design, synthesis and evaluation of 3-(imidazol-1-ylmethyl)indoles as antileishmanial agents. Part II. J. Enzyme Inhib. Med. Chem. 2009, 24, 1067–1075. [Google Scholar] [CrossRef]
- Xu, H.; Lv, M. Developments of Indoles as Anti-HIV-1 Inhibitors. Curr. Pharm. Des. 2009, 15, 2120–2148. [Google Scholar] [CrossRef]
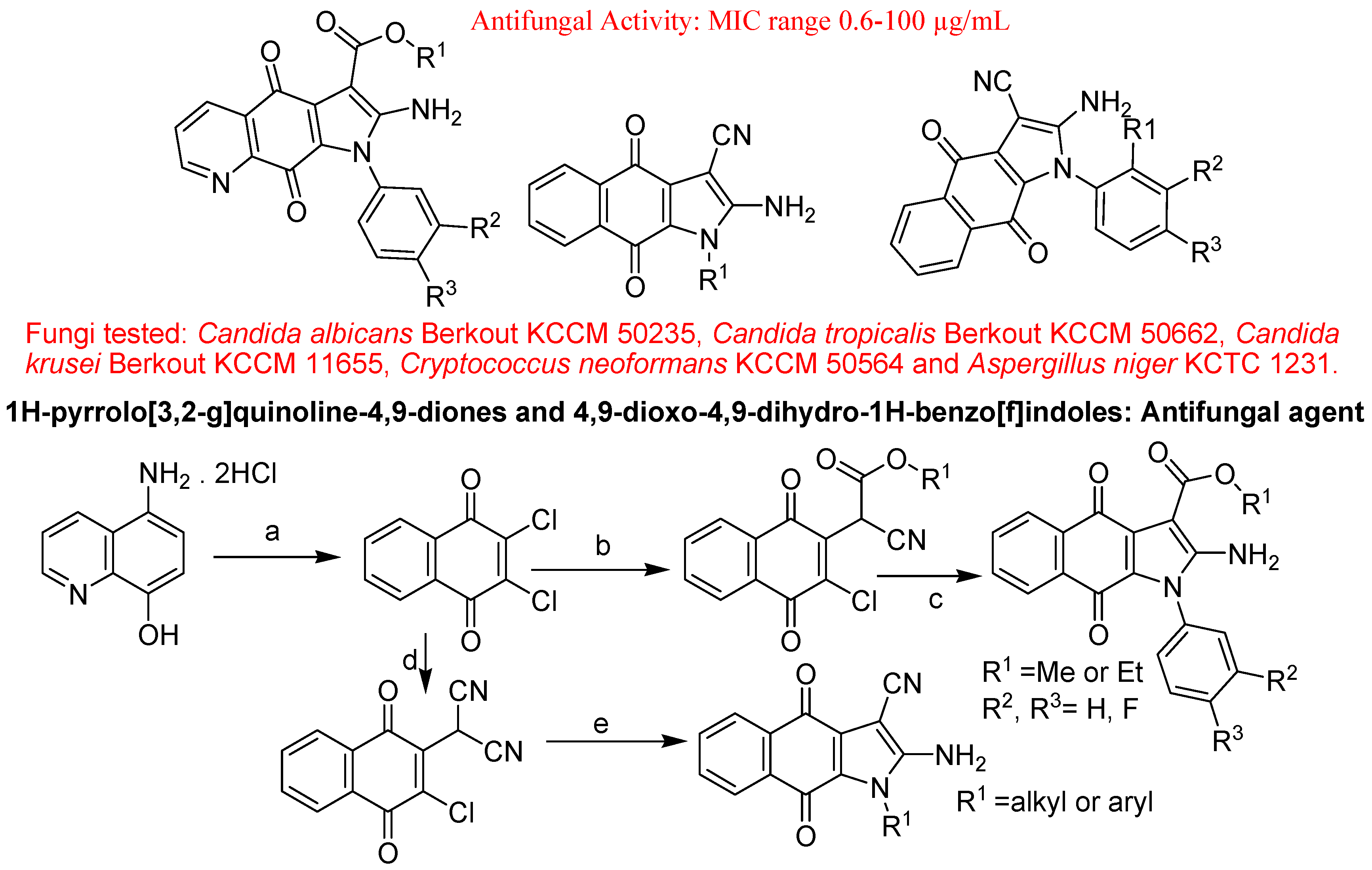
- Ryu, C.K.; Lee, J.Y.; Jeong, S.H.; Nho, J.H. Synthesis and antifungal activity of 1H-pyrrolo[3,2-g]quinoline-4,9-diones and 4,9-dioxo-4,9-dihydro-1H-benzo[f]indoles. Bioorg. Med. Chem. Lett. 2009, 19, 146–148. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical Importance of Indoles. Molecules 2013, 18, 6620-6662. https://doi.org/10.3390/molecules18066620
Kaushik NK, Kaushik N, Attri P, Kumar N, Kim CH, Verma AK, Choi EH. Biomedical Importance of Indoles. Molecules. 2013; 18(6):6620-6662. https://doi.org/10.3390/molecules18066620
Chicago/Turabian StyleKaushik, Nagendra Kumar, Neha Kaushik, Pankaj Attri, Naresh Kumar, Chung Hyeok Kim, Akhilesh Kumar Verma, and Eun Ha Choi. 2013. "Biomedical Importance of Indoles" Molecules 18, no. 6: 6620-6662. https://doi.org/10.3390/molecules18066620