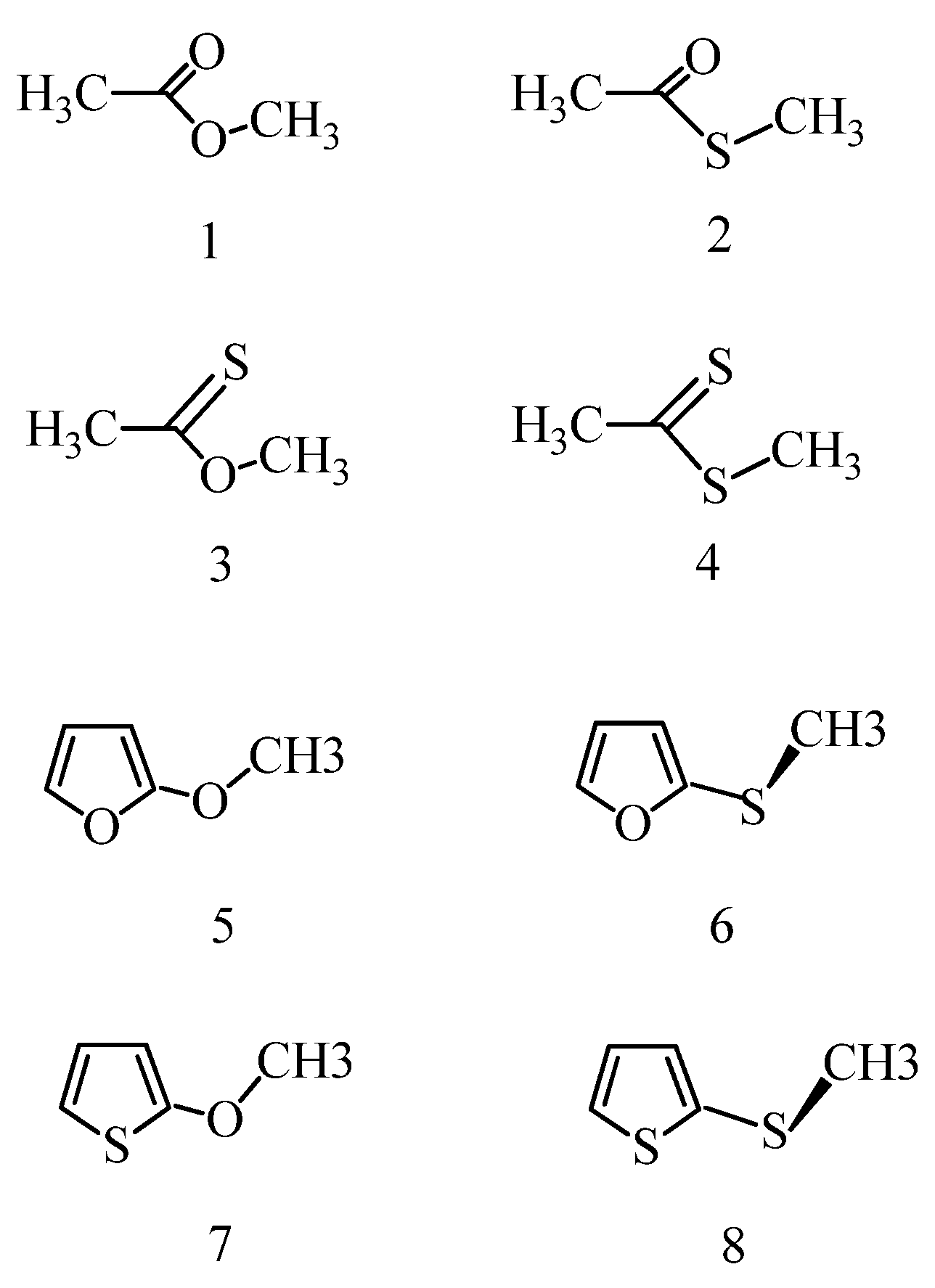
In cases, experimental information is available regarding the structure and the equilibrium composition. Experimental data are mostly available for gas-phase systems, and generally only for the structure of the predominant conformer. Even less is known for in-solution equilibria and related conformer structures. Theoretical calculations become of paramount interest in these cases. Since no experimental data have been found for the in-solution structure and equilibrium of the eight target molecules in the present study, the trends for their conformational preferences may provide useful information regarding their behavior through in-solution reactions and interactions with protein receptors in a binding cavity.
3.1. Geometries
Optimized molecular geometric parameters are compared with available experimental values for furan and thiophene (
Table 1) and for methyl acetate (structure 1) and thioacetic acid S-methyl ester (structure 2) (
Table 2). The calculated bond distances were estimated in remarkably better accord with the experimental values when the aug-cc-pvTz basis set rather than the aug-cc-pvDz set was used (
Table 1). However, the O-C distance in furan is still overestimated by 0.008 Å even when the larger basis set is used [
32]. (From experimental data, the symmetry of the furan and thiophene molecules is C
2v, thus symmetry-restricted geometry optimizations were performed in these cases.) For thiophene, the calculated S-C distance improves by more than 0.02 Å with increasing basis sets. The bond angles are in good agreement with the experimental values at any calculation level [
33]. For the present purposes, however, the main goal is not achieving very high precision in individual geometries. As emphasized in the Introduction, this study aims to point out the trends of the changes in molecular conformations and relative free energies, affected by the solvent.
Table 1 indicates negligible solvent effect for ring structures.
Table 1.
C2v symmetry restricted optimized geometries at the B97D/aug-cc-pvXz level for furan and thiophene a.
Table 1.
C2v symmetry restricted optimized geometries at the B97D/aug-cc-pvXz level for furan and thiophene a.
| | Gas | CHCl3 | CH3CN | Exp b |
|---|
| | X = D | X = T | | X = T | X = T |
|---|
| Furan |
| OC | 1.375 | 1.370 | | 1.373 | 1.374 | 1.362 |
| C2C3 | 1.373 | 1.363 | | 1.363 | 1.364 | 1.361 |
| C3C4 | 1.442 | 1.435 | | 1.436 | 1.437 | 1.431 |
| CHα | 1.088 | 1.080 | | 1.082 | 1.082 | 1.075 |
| CHβ | 1.090 | 1.082 | | 1.083 | 1.084 | 1.077 |
| COC | 106.8 | 106.7 | | 106.7 | 106.7 | 106.6 |
| OCC | 110.4 | 110.4 | | 110.3 | 110.3 | 110.7 |
| CCC | 106.2 | 106.3 | | 106.3 | 106.4 | 106.1 |
| OCHα | 115.8 | 115.7 | | 115.8 | 115.8 | 115.9 |
| OCHβ | 126.3 | 126.3 | | 126.3 | 126.3 | 128.0 |
| | X = D | X = T | X = T + d | X = T + d | X = T + d | |
| Thiophene |
| SC | 1.745 | 1.730 | 1.723 | 1.724 | 1.725 | 1.714 |
| C2C3 | 1.381 | 1.373 | 1.374 | 1.374 | 1.375 | 1.370 |
| C3C4 | 1.434 | 1.426 | 1.426 | 1.427 | 1.427 | 1.423 |
| CHα | 1.091 | 1.082 | 1.082 | 1.084 | 1.085 | 1.078 |
| CHβ | 1.094 | 1.085 | 1.085 | 1.086 | 1.087 | 1.081 |
| CSC | 91.6 | 91.9 | 92.1 | 92.2 | 92.2 | 92.2 |
| SCC | 111.4 | 111.4 | 111.4 | 111.3 | 111.2 | 111.5 |
| CCC | 112.8 | 112.7 | 112.6 | 112.6 | 112.7 | 112.5 |
| SCHα | 119.8 | 120.1 | 120.2 | 120.2 | 120.2 | 119.9 |
| SCHβ | 123.2 | 123.2 | 123.3 | 123.3 | 123.2 | 124.3 |
Table 2,
Table S1 summarize the calculated characteristic geometric parameters for simple esters. All results were obtained from symmetry-unrestricted optimizations starting from some structure of C
1 symmetry. The symmetry of the converged
cis and
trans structures corresponds only
nearly to C
s, the torsional angles for the heavy atoms deviate from 0 or 180° by some hundredths of a degree (achievement of a perfect symmetry plane is almost impossible when the standard convergence thresholds are used in a symmetry-unrestricted energy minimization in Gaussian. The symmetry-unrestricted optimization assures, however, that the structure does not remain stuck in some transition state if the starting geometry is wrongly chosen; a possible source for defining a transiton state when the methyl group is set in an improper rotational position). It reveals from cases when comparison with experimental values is possible (
Table 2) that the gas-phase B97D single bonds are overestimated by up to 0.02 Å for methyl acetate [
34] and thioacetic acid S-methyl ester [
35]. The largest overestimation was noted for the C-C bond, which probably has, however, minor effect on the conformational equilibria. For the thioacetic acid S-methyl ester, the C-S and S-C distances are too long by up to 0.016 Å. The calculated bond angles for the
cis conformers agree within 1° with the corresponding experimental values.
Table 2.
Geometric parameters optimized at the B97D/aug-cc-pvTz and B97D/aug-cc-pv(T+d)z levels for methyl acetate and thioacetic acid S-methyl ester, respectively a.
Table 2.
Geometric parameters optimized at the B97D/aug-cc-pvTz and B97D/aug-cc-pv(T+d)z levels for methyl acetate and thioacetic acid S-methyl ester, respectively a.
| | Gas | CHCl3 | CH3CN | Exp. |
|---|
| CH3COOCH3, cis b |
| C-C | 1.514(1.500) | 1.511 | 1.510 | 1.496 |
| C=O | 1.212(1.212) | 1.217 | 1.219 | 1.206 |
| C-O | 1.363(1.351) | 1.357 | 1.354 | 1.357 |
| TS | 1.393 | 1.389 | 1.387 | |
| O-CH3 | 1.448(1.437) | 1.453 | 1.454 | 1.438 |
| C-C=O | 125.8(125.9) | 125.6 | 125.6 | |
| O=C-O | 123.8(123.3) | 123.6 | 123.6 | 123.0 |
| C-O-CH3 | 115.4(114.1) | 116.1 | 116.3 | 116.4 |
| TS | 114.6 | 114.5 | 114.5 | |
| HCC=O | 1.1(0.2) | 0.6 | 0.5 | |
| CH3COOCH3, trans |
| C-C | 1.519 | 1.514 | 1.511 | |
| C=O | 1.207 | 1.215 | 1.218 | |
| C-O | 1.370 | 1.361 | 1.358 | |
| O-CH3 | 1.440 | 1.449 | 1.453 | |
| C-C=O | 124.0 | 124.1 | 124.2 | |
| O=C-O | 118.5 | 118.0 | 117.8 | |
| C-O-CH3 | 120.4 | 120.3 | 120.3 | |
| HCC=O | 0.1 | 0.1 | 0.1 | |
| CH3COSCH3, cis c |
| C-C | 1.520(1.507) | 1.517 | 1.516 | 1.499 |
| C=O | 1.213(1.216) | 1.218 | 1.220 | 1.214 |
| C-S | 1.797(1.769) | 1.791 | 1.788 | 1.781 |
| rot 90° | 1.885 | 1.883 | 1.883 | |
| S-CH3 | 1.820(1.798) | 1.820 | 1.820 | 1.805 |
| C-C=O | 123.4(123.6) | 123.3 | 123.2 | 123.4 |
| O=C-S | 123.1(122.7) | 123.1 | 123.1 | 122.8 |
| C-S-CH3 | 99.9(98.5) | 100.7 | 101.1 | 99.2 |
| rot 90° | 95.9 | 96.3 | 96.4 | |
| HCC=O | 180.0(178.8) | 178.7 | 178.5 | 143.1 |
| CH3COSCH3, trans |
| C-C | 1.518 | 1.514 | 1.511 | |
| C=O | 1.212 | 1.218 | 1.221 | |
| C-S | 1.805 | 1.796 | 1.793 | |
| S-CH3 | 1.829 | 1.828 | 1.828 | |
| C-C=O | 123.9 | 124.0 | 124.0 | |
| O=C-S | 118.1 | 117.9 | 117.8 | |
| C-S-CH3 | 105.1 | 105.2 | 105.3 | |
| HCC=O | 0.0 | 0.6 | 0.6 | |
The most conspicuous changes of the geometric parameters upon the
cis to
trans transformation were noted for some bond angles. For the two esters in
Table 2, the O=C-X angles decrease and the C-X-CH
3 (X=O, S) bond angles increase by about 5° in the gas phase. For the C=S containing esters (
Table S1) the S=C-X angles decrease by 7–8°, and the S-CH
3 distance increases in the
trans conformer of the dithioacetic acid ester by more than 0.02 Å.
The H-C-C=O torsion angles are about 0 and 180° for the
cis CH
3COOCH
3 and CH
3COSCH
3 conformers, respectively, both at the B97D and MP2 levels. The experimental value for CH
3COOCH
3 was not provided by Pyckhout
et al. [
34]. The torsional angle is 143.1° (without refinement) for CH
3COSCH
3 from gas electron-diffraction measurements by Della Védova
et al. [
35]. This torsion angle was calculated at 180° and 178.8° using the B97D and MP2 method, respectively, and applying the aug-cc-pv(t+d)z basis set. It is remarkable that the H-C-C=O torsion angle changes to 0° for the
trans conformer.
The solvent effects are small and consistent for any geometric parameters. By consistency the finding is meant that the geometric parameters monotonically decrease or increase with increasing polarity of the environment in the gas-phase, chloroform and acetonitrile series. Remarkable changes were found for some atom distances, which were larger by up to 0.017 Å in acetonitrile compared to those in the gas phase.
3.2. Energy Results for Esters
Relative energy/free energy components of ΔG
tot for the studied esters are compared in
Table 3. Due to the practically achieved symmetry plane for the molecules in their
cis and
trans conformations, and the energy equivalence of the clockwise and anticlockwise rotation about the C-Y bond, these structures must correspond to either a local energy minimum or a transition state. Non-planar structures exist in two mirror image forms, and for their ΔG
tot value the entropy of mixing term, -RT ln 2 = −0.41 kcal/mol is to be considered.
Table 3,
Table 4 include the ΔG
th values for the local-energy- minimum and TS structures in both solvents, and accordingly the correct ΔG
tot = ΔE
int + ΔG
th + ΔG(solv) was calculated. For torsional rotamers corresponding neither to energy minima nor TS structures, only the ΔE
int + ΔG(solv) term could be reasonably compared.
The theoretical results could be summarized as follows. The X=C-Y-CH
3 cis conformers are more stable than the
trans forms for all four studied esters in the gas phase. ΔE
int is considerably larger for OCH
3 esters compared with SCH
3 esters. The B97D/aug-cc-pvtz level, however, probably underestimates the
cis-trans energy difference for methyl acetate as of ΔH = ΔE
int + ΔH
th = 5.71 − 0.07 = 5.64 kcal/mol. Here ΔH
th is the thermal enthalpy correction (including relative zero-point energies) at T = 298 K° and p = 1 atm. The total relative free energy is remarkably larger, 5.71 + 0.89 = 6.60 kcal/mol. Blom and Günthard [
36] estimated the enthalpy difference as 8.5 ± 1 kcal/mol and commented that there was a very small band in the matrix IR spectra, and “it is possible that this band is due to the methyl acetate”. The equilibrium composition depends on the relative total free energy. If, as calculated here, ΔG
tot is as large as 6.6 kcal/mol, the trans conformer could be hardly observed. Pyckhout
et al. [
34] calculated ΔE = 10.3 kcal/mol at the
ab initio 4-21G level. Nagy
et al. [
7] obtained ΔH = 7.8–8.2 kcal/mol at the MP2 and B3LYP levels, using the 6-311++G** basis sets, whereas the G3B3 [
37] ΔH value by Terhorst and Jorgensen is 7.46 kcal/mol [
8]. Although the aug-cc-pvtz basis set was applied presently in geometry optimizations, which is considerably larger than in any former study, the author has no good explanation for the underestimation of ΔH for methyl acetate.
Table 3.
Relative energies for esters as the function of the rotation about the C-O and C-S bonds a.
Table 3.
Relative energies for esters as the function of the rotation about the C-O and C-S bonds a.
| | Gas | Chloroform | Acetonitrile |
|---|
| | ΔEint | ΔEint + ΔG(solv) | ΔEint + ΔG(solv) |
|---|
| O=C-O-C |
| 0 | 0.0 | 0.0 + 0.0 = 0.0 | 0.0 + 0.0 = 0.0 |
| 30 | 2.88 | 2.91 − 0.04 = 2.87 | 2.95 − 0.12 = 2.83 |
| 60 | 8.76 | 8.85 − 0.13 = 8.72 | 8.96 − 0.35 = 8.61 |
| TS(95.4, 92.2, 90.7) | 12.40 | 12.59 − 0.75 = 11.84 | 12.80 − 1.29 = 11.51 |
| 120 | 11.15 | 11.54 − 1.57 = 9.97 | 11.98 − 2.61 = 9.37 |
| 150 | 7.00 | 7.96 − 2.48 = 5.48 | 8.59 − 4.02 = 4.57 |
| 180 | 5.71 | 6.37 − 2.86 = 3.51 | 7.09 − 4.59 = 2.50 |
| ΔGth | 0.89 | 1.09 | 1.10 |
| ΔGth(TS) | 0.44 | 0.48 | 0.40 |
| O=C-S-C |
| 0 | 0.0 | 0.0 + 0.0 = 0.0 | 0.0 + 0.0 = 0.0 |
| 30 | 2.72 | 2.76 + 0.02 = 2.78 | 2.80 − 0.03 = 2.77 |
| 60 | 8.86 | 8.93 − 0.12 = 8.81 | 9.00 − 0.32 = 8.68 |
| 90 | 12.16 | 12.34 − 0.61 = 11.73 | 12.55 − 1.17 = 11.38 |
| 120 | 10.27 | 10.60 − 1.10 = 9.50 | 10.89 − 1.87 = 9.02 |
| 150 | 5.63 | 6.08 − 1.62 = 4.46 | 6.57 − 2.72 = 3.85 |
| 180 | 3.35 | 3.86 − 1.89 = 1.97 | 4.42 − 3.12 = 1.30 |
| ΔGth | 1.18 | 1.29 | 1.31 |
| S=C-O-C |
| 0 | 0.0 | 0.0 + 0.0 = 0.0 | 0.0 + 0.0 = 0.0 |
| 30 | 2.83 | 2.84 + 0.00 = 2.84 | 2.86 − 0.04 = 2.82 |
| 60 | 8.95 | 8.98 + 0.00 = 8.98 | 9.02 − 0.11 = 8.91 |
| 90 | 12.94 | 13.04 − 0.35 = 12.69 | 13.17 − 0.72 = 12.45 |
| 120 | 11.61 | 11.94 − 1.37 = 10.57 | 12.33 − 2.32 = 10.01 |
| 150 | 7.81 | 8.44 − 2.64 = 5.80 | 9.14 − 4.30 = 4.84 |
| 180 | 6.25 | 7.00 − 3.19 = 3.81 | 7.83 − 5.13 = 2.70 |
| ΔGth | 0.10 | 0.42 | 0.51 |
| S=C-S-C |
| 0 | 0.0 | 0.0 + 0.0 = 0.0 | 0.0 + 0.0 = 0.0 |
| 30 | 3.02 | 3.00 − 0.15 = 2.85 | 3.03 − 0.28 = 2.75 |
| 60 | 9.96 | 9.86 + 0.12 = 9.98 | 9.76 + 0.16 = 9.92 |
| TS(90.6, 89.7, 89.0) | 14.30 | 14.27 + 0.07 = 14.34 | 14.24 − 0.10 = 14.14 |
| 120 | 11.40 | 11.52 − 0.93 = 10.59 | 11.71 − 1.55 = 10.16 |
| 150 | 5.60 | 6.03 − 1.92 = 4.11 | 6.62 − 3.20 = 3.42 |
| 180 | 3.10 | 3.65 − 2.05 = 1.60 | 4.30 − 3.42 = 0.88 |
| ΔGth | 0.16 | 0.80 | 1.10 |
| ΔGth(TS) | 0.47 | 0.85 | 1.16 |
Table 4.
Torsion barrier energies relative to the cis conformation in the gas phase a.
Table 4.
Torsion barrier energies relative to the cis conformation in the gas phase a.
| | CH3COOCH3 | CH3COSCH3 |
|---|
| B3LYP/6-311++G** b | 13.35 | 11.80 |
| B97D/aug-cc-pvtz c | 12.40 | 12.16 |
| MP2/6-311++G** b | 13.45 | 11.18 |
| MP2/aug-cc-pv(t+d)z | | 11.98 |
| INDO d | 29.10 | 14.06 |
| PM3 | | 6.05 |
| PM6 | | 6.53 |
| PDDG/PM3 | | 4.87 |
Della Vedova found only one conformation in the sample of CH
3COSCH
3, assigning it to the
cis conformation [
35].
Table 3 predicts ΔG
tot = 4.53 kcal/mol for the
trans conformer (4.60 kcal/mol in ref. [
8]), a value large enough for preventing the presence of this conformer at an experimentally observable concentration in the gas-phase mixture. Overall, gas-phase calculation results suggest that ΔG
tot is prohibitively large for the observation of the
trans CH
3 position in any studied X=C-Y- CH
3 ester moiety. The question is, whether the solvent effects could modify this equilibrium ratio.
The conclusion about the solvent effect for esters (always talking about the four studied prototypes) is that the
cis conformer remains the prevailing conformation, although the
cis-trans free energy difference could be largely decreased in comparison with the gas phase. The 0° and 180° torsion angles are underscored in
Table 3, indicating local energy minima both in chloroform and the acetonitrile solvents. Adding ΔG
th to the 180° ΔE
int + ΔG(solv) value, the obtained ΔG
tot (in the order of the appearance of the structures in
Table 3) are 4.60, 3.26, 4.23, and 2.40 kcal/mol in chloroform, and 3.60, 2.61, 3.21, and 1.98 kcal/mol in acetonitrile. The in-acetonitrile values are consistently smaller than those with chloroform solvent, but even the smallest calculated ΔG
tot value for CH
3CSSCH
3 in acetonitrile allows for a
trans-cis ratio of only about 4:96 at T = 298 K. This ratio was calculated by using the relationship −RT ln K = ΔG°, where ΔG° was accepted to be equal to the calculated ΔG
tot, and the concentration related activity coefficients were accepted to be equal for the two conformers in very dilute solution as modeled by IEF-PCM. [
39,
40]. Thus one may conclude that the effect of a non-protic solvent on the
cis-trans equilibrium of esters is small, but may not be negligible for esters mainly with the –SCH
3 group.
Another important question is: how can the esters reach the equilibrium composition? For compounds with a polar hydrogen atom, even conformational equilibration could be reached by the catalytic involvement of a protic solvent [
41,
42]. For esters in a non-protic solvent, this reaction path does not seem to be travelable. The straightforward mechanism is, of course, the rotation about the C-Y (Y = O, S) bond. This is a simple mechanism if the barrier to the rotation is energetically affordable. The transition states were identified for CH
3COOCH
3 and CH
3CSSCH
3 in all three environments, and the energy values for the barrier scatter in the 11.5–14.3 kcal/mol range. The barrier tops are close to 90°. The activation free energy for passing the barrier can be obtained by adding ΔG
th(TS) to the TS energy, and subtracting 0.41 kcal/mol for the entropy of mixing for the TS optical antipodes. Still one is left in the range of ΔG
tot = 11–15 kcal/mol. For the other two molecules, the 90° relative energies are also in this latter range. This activation free energy may be considered fairly large, although, e.g., Lunazzi
et al. [
43] found experimentally the successful annular tautomerizations of azoles in DMSO, THF and CD
2Cl
2, with activation free energies of 11–14 kcal/mol. Even though the chemical problem studied by Lunazzi
et al. is quite different, an activation free energy in the indicated range seems to be affordable for structural transformations in solution. Then this author considers the
cis-trans equilibration for simple esters in solution via the rotation of the methyl group about the C-Y bond.
Components of the ΔE
int + ΔG (solv) term as the function of the X=C-Y-CH
3 torsion angle are provided in the 0–180° range in
Table 3. ΔE
int, compared with the corresponding gas-phase value, changes only slightly up to about 90° torsion, but conspicuously and unanimously increases after then in solution in comparison with the gas phase. What could be the explanation?
The PCM optimization procedure (keeping only the X=C-Y-CH3 torsion angle at the set value with the exception for the local-minimum-energy conformers) seeks the structure corresponding to the minimum of the Eint+Gelst term. The solvent polarizes the solute’s electron distribution and slightly modifies the geometry throughout the optimization. The internal energy itself, Eint necessarily increases, whereas a more and more negative Gelst is being developed in parallel, and the Eint+Gelst minimum for the considered conformer is ultimately reached. As long as ΔG(solv) is only of a few tenths of a kcal/mol for different conformers as calculated in the 0–60° torsion range, ΔEint changes only moderately compared with it in the gas phase. However, the stiffness of the PES depends on the course of ΔEint in the given environment and could be considered moderate only up to about 30°. ΔEint is here about 3 kcal/mol for all studied esters both in the gas phase and solution, but increases to 8.8–10.0 kcal/mol at torsion angle of 60°.
ΔE
int(solution), as the relative internal energy in solution, could both increase and decrease compared with ΔE
int(gas) depending on the sensitivity of the internal energy itself on the solvent effect for the
cis conformer compared with a torsioned structure. When, however, ΔG
elst can become remarkable, due to, e.g., the increase of dipole moment, the molecular structure gets distorted accordingly.
Table 3 shows that ΔG(solv) is at least about –1 kcal/mol starting at X=C-Y-CH
3 = 120°.
Overall, the question can be raised: how reliable is the calculated torsion potential curve for ΔE
int? This is an important issue, since molecular modelers may want to develop torsional potential parameters for esters/ethers to be utilized in Monte Carlo and/or molecular mechanics/dynamic theoretical binding studies. Unfortunately, no experimental torsion barrier data are available for the molecules under scrutiny. To get some information about the stability of the calculated data, MP2/aug-cc-pv(t+d)z calculations in addition to the B97D calculations have been carried out for the
cis and 90° rotated CH
3COSCH
3 structures in the gas phase and are compared with PM3, PM6 and PDDG/PM3 results based on symmetry-unrestricted geometry optimizations. Former INDO [
38] and B3LYP/MP2 studies [
7] have been also considered.
INDO geometries are fairly good for the
cis CH
3COOCH
3, but cannot be reasonably judged for CH
3COSCH
3, where the authors did not optimize the S-C bond length and the C-S-C bond angle. Using the sp basis set, the C(O)-S bond length was underestimated by 0.05 Å, with the spd set the C=O became even further away from the experimental value. In the torsion energy barrier calculation they considered a rigid rotation, whereas
Table 2 of this paper shows that the C-S bond length increases by about 0.09 Å. PM3 finds the C-S bond length longer than that for S-C and the CSC angle is too large by about 6° for the
cis ester conformer. With PM6, the two C-S bonds are still only equally long, the CSC angle is too large by 4°, and the CCO angle was overestimated by about 5°. All these parameters are close to the experimental values from PDDG/PM3 optimizations, although the
trans hydrogens of the ester methyl group was found 23° out of the heavy-atom plane. Furthermore, all three optimizations predicted eclipsed HCCO arrangements in contrast to the experimental.
Still the main problem is related to the predicted torsion barrier. The results indicate (
Table 4) that the DFT and MP2 energy barriers are similar; they scatter in a range of about 1 kcal/mol. In contrast, the INDO barrier is about the double of the DFT/MP2 values for CH
3COOCH
3, whereas PM3, PM6, PDDG/PM3 give barrier heights about the half of the DFT/MP2 values. All these findings together suggest that consistent, numerically stable potential curves could be probably only obtained based on large-basis-set calculations.
3.3. Energy Results for Ethers
Relative energies are provided in
Table 5 with respect to the actually lowest-energy conformer. In contrast to esters, the relative energy/free energy of two local energy minimum structures are relatively small, up to about 2 kcal/mol. The solvent can both increase (OCH
3) and decrease (SCH
3) the energy separations for the local-minimum-energy ether structures in comparison with the gas phase. The X-C-Y-C torsional angle in the lowest-energy conformation is 180° for the 2-OCH
3 ethers. The second local energy minimum is, however, largely different: XCOC is about 50° and 6° for the furan and thiophene derivative, respectively. The most stable conformation for the furan-2SCH
3 thioether is a
gauche form with OCSC = 69–76°, whereas for the second stable structure OCSC = 180°. The most complicated equilibrium was found for the thiophene-2SCH
3 thioether. None of the stable conformers adopt a coplanar heavy-atom arrangement. As mentioned above, if conformers with a molecular symmetry plane, as in this case with XCYC torsion angles of 0° and 180°, do not correspond to local energy minima, then they must exhibit a transition state structure due to the equivalence of the clockwise and anti-clockwise rotation. For the 2-SCH
3 thiophene, in-solution minimum-energy structures adopt XCYC torsion angles of 83–84° and ~172°. There must be a local energy maximum around 120°–150° (see
Table 5), thus this potential curve has two minima and three TS structures in the 0–180° range of the XCYC rotation. The subtle effect of the solvents indicates that the marginal ΔE
int+ΔG(solv) preference for the two minimum-energy structures is reversed in the two solvents.
In general, ΔG
tot is small for any studied ether in solution. Considering ΔG
th and –0.41 kcal/mol for the entropy of mixing for non-planar structures, ΔG
tot for the less stable conformer is (in the order of the structure appearance in
Table 5) 0.34, 1.11, 0.39, and 0.64 kcal/mol in chloroform, and 1.10, 1.40, 0.24, and 0.58 kcal/mol in acetonitrile. Considering the moderate rotational barriers, equilibration of the stable conformers via C-Y rotation at T = 298 K° would not be hindered. Provided the thermodynamic control for the conformer equilibration, structures with different orientations for the lone pairs of the O and S atoms in the methyl ether and thiomethyl ether groups are expected in the solution. This is a remarkable difference in comparison with esters, where the prevailing lone pair arrangements are determined by the XCYC cis conformation. For esters, the lone pairs of the X atom point approximately toward northwest and east, and those for Y about toward southwest if the CH
3-C axis points from west to east (
Figure 1). Thus the lone pairs point in fairly opposite directions in this case. For furan and thiophene ethers, the lone pair of the X heteroatom (nearly) in the ring plane points south. Due to fairly unhindered rotation of the CH
3 group about the C-Y bond, the lone pairs of Y would point in many different directions, including orientations basically parallel or antiparallel with those on X. This variety of the lone pair directionality could be expediently utilized through drug design, as hinted for in the Introduction section.
Table 5.
Relative energies for ethers as the function of the rotation about the C-O and C-S bonds a.
Table 5.
Relative energies for ethers as the function of the rotation about the C-O and C-S bonds a.
| | Gas | Chloroform | Acetonitrile |
|---|
| | ΔEint | ΔEint + ΔG(solv) | ΔEint + ΔG(solv) |
|---|
| Furan, O-C-O-C |
| 0 | 1.53 | 1.38 + 0.72 = 2.10 | 1.18 + 1.16 = 2.32 |
| 45.1 | 1.43 | | |
| 49.9 | | 1.31 + 0.74 = 2.05 | |
| 53.9 | | | 1.18 + 1.11 = 2.29 |
| 90 | 2.12 | 2.05 + 0.58 = 2.63 | 1.97 + 0.79 = 2.76 |
| 120 | 2.41 | 2.39 + 0.23 = 2.62 | 2.37 + 0.27 = 2.64 |
| 150 | 1.11 | 1.11 + 0.05 = 1.16 | 1.11 + 0.04 = 1.15 |
| 180 | 0.0 | 0.0 + 0.0 = 0.0 | 0.0 + 0.0 = 0.0 |
| ΔGth | | −1.30 | −0.78 |
| Furan, O-C-S-C |
| 0 | 1.92 | 1.83 − 0.06 = 1.77 | 1.70 + 0.10 = 1.80 |
| 30 | 1.14 | 1.06 +0.03 = 1.03 | 0.95 + 0.19 = 1.14 |
| 69.2 | 0.0 | | |
| 73.7 | | 0.0 + 0.0 = 0.0 | |
| 75.9 | | | 0.0 + 0.0 = 0.0 |
| 90 | 0.31 | 0.32 − 0.09 = 0.23 | 0.33 − 0.15 = 0.18 |
| 120 | 1.35 | 1.39 − 0.35 = 1.04 | 1.45 − 0.54 = 0.91 |
| 150 | 1.45 | 1.49 − 0.60 = 0.89 | 1.56 − 0.85 = 0.71 |
| 180 | 0.92 | 0.96 − 0.60 = 0.36 | 1.01 − 0.80 = 0.21 |
| ΔGth | | 0.34 | 0.78 |
| Thiophene, S-C-O-C |
| 0 | 0.87 | 0.81 + 0.17 = 0.98 | 0.70 + 0.36 = 1.06 |
| 6.3 | | 0.81 + 0.15 = 0.96 | |
| 5.1 | | | 0.70 + 0.35 = 1.05 |
| 30 | 1.10 | 1.04 + 0.22 = 1.26 | 0.94 + 0.40 = 1.34 |
| 60 | 1.58 | 1.52 + 0.03 = 1.55 | 1.44 + 0.60 = 2.04 |
| 90 | 1.89 | 1.86 + 0.05 = 1.91 | 1.80 + 0.49 = 2.29 |
| 120 | 1.98 | 1.97 − 0.03 = 1.94 | 1.95 + 0.29 = 2.24 |
| 150 | 0.90 | 0.90 − 0.01 = 0.89 | 0.89 + 0.0 = 0.89 |
| 180 | 0.0 | 0.0 + 0.0 = 0.0 | 0.0 + 0.0 = 0.0 |
| ΔGth | | −0.16 | −0.40 |
| Thiophene, S-C-S-C |
| 0 | 1.10 | 1.06 − 0.52 = 0.54 | 1.01 − 0.55 = 0.46 |
| 30 | 0.78 | 0.75 − 0.32 = 0.43 | 0.70 − 0.34 = 0.36 |
| 60 | 0.21 | 0.19 − 0.10 = 0.09 | 0.16 − 0.08 = 0.08 |
| 82.7 | 0.0 | | |
| 83.3 | | 0.0 + 0.0 = 0.0 | |
| 83.8 | | | 0.0 + 0.0 = 0.0 |
| 120 | 0.66 | 0.69 − 0.29 = 0.40 | 0.72 − 0.42 = 0.30 |
| 150 | 0.99 | 1.02 − 0.59 = 0.43 | 1.08 − 0.82 = 0.26 |
| 171.9 | | 0.85 − 0.73 = 0.12 | |
| 172.1 | | | 0.88 − 0.97 = -0.09 |
| 180 | 0.81 | 0.84 − 0.67 = 0.17 | 0.88 − 0.92 = -0.04 |
| ΔGth | | 0.52 | 0.67 |
3.4. Atomic Charges
In molecular mechanics, Monte Carlo or molecular dynamics studies utilizing some effective pair potential for calculating the interaction energy for a ligand-macromolecule complex, one of the most important issue is the proper estimation of the electrostatic component. This energy is generally represented, at least as the leading term of an expanded series, by the sum of Coulomb energies of effective net atomic charges. There are a number of different methods known in the literature for deriving these charges, and the applied force field parameters must be harmonized with the actually derived charge sets. The charges presented in
Table 6 were derived by means of the CHELPG procedure [
44], fitting the in-solution atomic charges to the corresponding molecular electrostatic potential.
Table 6.
Net atomic charges fitted to the B97D/aug-cc-pv(t+d)z molecular electrostatic potential by means of the CHELPG procedure a.
Table 6.
Net atomic charges fitted to the B97D/aug-cc-pv(t+d)z molecular electrostatic potential by means of the CHELPG procedure a.
| | Gas | CHCl3 | CH3CN |
|---|
| X | Y | DM | X | Y | DM | X | Y | DM |
|---|
| CH3C(O)OCH3 |
| cis | −0.55 | −0.40 | 1.87 | −0.61 | −0.41 | 2.33 | −0.63 | −0.42 | 2.53 |
| trans | −0.55 | −0.39 | 4.43 | −0.63 | −0.43 | 5.52 | −0.66 | −0.44 | 5.99 |
| CH3C(O)SCH3 |
| cis | −0.45 | −0.25 | 1.30 | −0.51 | −0.26 | 1.71 | −0.53 | −0.26 | 1.93 |
| trans | −0.45 | −0.19 | 4.16 | −0.52 | −0.23 | 5.25 | −0.55 | −0.25 | 5.74 |
| CH3C(S)OCH3 |
| cis | −0.32 | −0.29 | 2.32 | −0.39 | −0.30 | 3.07 | −0.42 | −0.30 | 3.41 |
| trans | −0.33 | −0.35 | 4.67 | −0.43 | −0.38 | 6.15 | −0.47 | −0.39 | 6.80 |
| CH3C(S)SCH3 |
| cis | −0.28 | −0.16 | 2.01 | −0.35 | −0.15 | 2.84 | −0.39 | −0.13 | 3.27 |
| trans | −0.26 | −0.15 | 4.35 | −0.35 | −0.17 | 5.85 | −0.40 | −0.17 | 6.55 |
| Furan-2OCH3 |
| OCOC = 45–54° | −0.14 | −0.37 | 1.13 | −0.15 | −0.41 | 1.35 | −0.16 | −0.43 | 1.49 |
| OCOC = 180° | −0.19 | −0.36 | 1.94 | −0.21 | −0.39 | 2.40 | −0.23 | −0.40 | 2.62 |
| Furan-2SCH3 |
| OCSC = 69–76° | −0.17 | −0.25 | 1.56 | −0.20 | −0.30 | 2.04 | −0.21 | −0.33 | 2.29 |
| OCSC = 180° | −0.13 | −0.17 | 1.95 | −0.15 | −0.21 | 2.50 | −0.17 | −0.23 | 2.77 |
| Thiophene-2OCH3 |
| SCOC = 0–6° | −0.04 | −0.31 | 1.11 | −0.05 | −0.34 | 1.30 | −0.04 | −0.35 | 1.37 |
| SCOC = 180° | −0.03 | −0.26 | 1.85 | −0.05 | −0.28 | 2.29 | −0.06 | −0.29 | 2.51 |
| Thiophene-2SCH3 |
| SCSC = 83–84° | −0.03 | −0.24 | 1.63 | −0.03 | −0.30 | 2.10 | −0.03 | −0.32 | 2.33 |
| SCSC = 171–172° | 0.01 | −0.14 | 1.86 | −0.02 | −0.19 | 2.36 | −0.02 | −0.20 | 2.61 |
The derived charges are, however, fitting method and theoretical level dependent. In a recent publication by Nagy
et al. [
27], the CHELPG charges were compared for 3,5-dimethoxy-1,2,4-thiadiazole calculated in tetrahydrofuran solvent. Non-negligible differences were noticed in the charges derived at different theoretical levels, although the trends were generally preserved. Monti and Nagy [
21,
45] compared CHELPG and RESP [
46] atomic charges for the trimethyl- and dimethylammonium cations at different theoretical levels. Non-negligible differences were noticed in some cases also in these studies. These findings confirm that the charge derivation by different methods and/or at different theoretical levels generally lead more or less different charge sets. Charges in
Table 6 were derived on the basis of large-basis-set calculations, where the molecular electrostatic potential utilized in the atom-charge fitting process was generated by means of a wave function reflecting the electron correlation for the target molecule. Although the present study represents high-level calculations and probably provides fairly relevant charges, the primary goal of the comparison in
Table 6 is to point out the
sensitivity of the obtained charges to the conformation and solvent effects.
A physically measurable structure characteristic is the dipole moment (DM). No proper experimental data have been found for the present molecules, the only available experimental value is 1.72 ± 0.09 D for the liquid methyl acetate, probably assignable to the
cis form [
47].
Table 6 shows that the quantum-mechanical DM is both conformation and environment dependent. The dipole moment gradually increases when the dielectric constant of the solvent increases compared to the gas phase (with unit dielectric constant). This finding emphasizes the polarization effect of the environment because geometries have only slightly changed upon solvation. The conformation is inherently a major determinant of the molecular dipole, as reveals from their large changes mainly for esters.
The dipole moment is capable only for an overall characterization of the molecule in the given environment, whereas effective pair-potentials need individual atom charges. It is to be emphasized, however, that atom charges have no physical meaning; they facilitate a simple way to calculate molecular electrostatic interactions in the system. It is important to know, how well they would reproduce the overall, physically meaningful dipole moment of the molecule calculated quantum mechanically. Not indicated in
Table 6, but the dipoles calculated by means of net atomic charges deviate by less than 0.13 D from the corresponding exact B97D/aug-cc-pvtz values (as an exception, the difference amounted to 0.17 D for 2SCH
3 thiophene in solution). Thus the dipoles calculated by means of net atomic charges approach the exact values satisfactorily in general, crediting the underlying atom-charge distribution.
Changes in the corresponding atomic charge values (if the change is larger than 0.01 units thus larger than the rounding error) are gradual: from left to right the negative charges monotonically vary; generally increase in accord with the increasing dipole moments. The trend in the change for a specific atom in different conformations (upper line compared to the lower line) is generally the same in all three environments. All these findings together indicate that one has to consider conformation dependent charges, and they have to be determined in the specific solvent for providing Monte Carlo or molecular dynamics atom-charge parameters for explicit solvent simulations.
3.5. Vibrational Frequencies
Hernandez
et al. [
48] presented some parts of the IR spectra for methyl acetate and its deuterated forms recorded in dilute CCl
4. These authors also performed 6-31** theoretical calculations, and assigned the six C-H stretching frequencies and IR intensities to the acetyl methyl and the ester methyl groups. The dielectric constant of the used solvent is 2.23 [
47], thus the results could be relevantly compared with our calculated C-H stretching frequencies for the gas phase and in chloroform solution with ε = 4.71.
The presently calculated B97D frequencies for CH
3COOCH
3 and CH
3CSSCH
3, both for their
cis and
trans conformations are summarized in
Table 7. The X=C-Y-C torsion frequencies for the
cis forms are also provided. The corresponding vibrations in the
trans forms are strongly coupled with both methyl torsions.
Table 7.
B97D frequencies and IR intensities for CH3COOCH3 and CH3CSSCH3 a.
Table 7.
B97D frequencies and IR intensities for CH3COOCH3 and CH3CSSCH3 a.
| | Gas | | CHCl3 | | CH3CN | | Exp b |
|---|
| | ω | Int | ω | Int | ω | Int | |
|---|
| CH3COOCH3,
cis |
| C-H stretching | | | | | | | |
| acetyl methyl | 3088 | 8.3 | 3080 | 9.2 | 3077 | 5.2 | 2844–3026 |
| | 3041 | 6.5 | 3034 | 3.2 | 3031 | 1.9 | |
| | 2967 | 5.8 | 2960(c) | 5.0 | 2956(c) | 13.3 | |
| ester methyl | 3080 | 15.3 | 3079 | 10.2 | 3079 | 12.3 | |
| | 3045 | 23.1 | 3046 | 20.8 | 3046 | 20.1 | |
| | 2961 | 33.0 | 2957 | 26.5 | 2955(c) | 15.5 | |
| torsional | | | | | | | |
| O=C-O-CH3 | 177 | 6.0 | 196 | 8.4 | 197 | 9.5 | |
| CH3COOCH3,
trans |
| C-H stretching | | | | | | | |
| acetyl methyl | 3092 | 6.0 | 3085 | 4.3 | 3082 | 3.4 | |
| | 3031(c) | 14.7 | 3026 | 0.9 | 3022 | 0.3 | |
| | 2961 | 7.7 | 2955 | 3.0 | 2952(c) | 4.4 | |
| ester methyl | 3067 | 18.5 | 3073 | 13.5 | 3074 | 11.6 | |
| | 3020(c) | 24.8 | 3036 | 22.5 | 3041 | 15.9 | |
| | 2941 | 35.6 | 2948 | 27.9 | 2949 | 21.0 | |
| CH3CSSCH3,
cis |
| C-H stretching | | | | | | | |
| acetyl methyl | 3042 | 5.1 | 3032 | 7.4 | 3027 | 10.7 | |
| | 3025 | 9.4 | 3022 | 8.5 | 3021 | 5.3 | |
| | 2945 | 9.4 | 2939 | 3.4 | 2936 | 1.9 | |
| ester methyl | 3070 | 4.0 | 3064 | 1.9 | 3061 | 1.0 | |
| | 3063 | 3.0 | 3058 | 1.4 | 3056 | 0.8 | |
| | 2969 | 11.7 | 2953 | 5.0 | 2950 | 2.8 | |
| torsional | | | | | | | |
| S=C-S-CH3 | 220 | 4.3 | 191 | 7.3 | 188 | 8.3 | |
| CH3CSSCH3,
trans |
| C-H stretching | | | | | | | |
| acetyl methyl | 3069 | 3.7 | 3063(c) | 2.8 | 3061(c) | 2.8 | |
| | 3003 | 7.8 | 2999 | 2.7 | 2995 | 0.8 | |
| | 2938 | 10.5 | 2933 | 2.6 | 2929 | 0.3 | |
| ester methyl | 3064 | 5.6 | 3062(c) | 2.7 | 3061 | 0.6 | |
| | 3061 | 6.9 | 3062(c) | 2.5 | 3060(c) | 1.8 | |
| | 2960 | 2960 | 2955 | 5.2 | 2952 | 1.9 | |
Our frequencies have been calculated in the harmonic oscillator approximation [
22]. As it is known, the calculated high-frequency stretching values are generally larger than the anharmonic frequencies [
28], which are probably observed in the experimental spectra. Then it is not surprising that the present C-H stretching frequencies are higher by 60–120 cm
−1 than the experimental values. Although Cappelli
et al. [
49] developed recently a perturbative methodology considering nonequibrium solvation conditions for evaluating anharmonic vibrational frequencies in solution, the method is not available in Gaussian 09. Nonetheless, assignment of the C-H stretching frequencies to vibrations of the two methyl groups is still possible.
Solvation generally results in a gradual decrease of the C-H frequencies. Shifts in ω are up to about 20 cm−1. However, these frequencies for the ester methyl group of CH3COOCH3 are hardly sensitive to the solvent effects in the cis conformation, and remarkably increase in the trans form with increasing solvent polarity. No such “out of trend” behavior has been calculated for CH3CSSCH3, where, however, the ester group shows insensitivity for the solvent effects in the trans conformation.
In accord with the conclusions of Hernandez
et al. above, the IR intensities of the C-H stretching frequencies are consistently higher for the ester rather than for the acetyl methyl group for CH
3COOCH
3. This conclusion is valid for the series calculated both for the
cis and the
trans conformers, both in the gas-phase and in solution. For CH
3CSSCH
3, no such clear-cut trend was calculated, the IR intensities are of the same order of magnitude for the C-H vibrations of the two methyl groups. In comparison with the experiment for CH
3COOCH
3 it has to be mentioned, however, that the largest intensities were observed in the middle of the spectrum, whereas the largest intensities were theoretically calculated at the low-frequency site both here and in ref. [
48].
The X=C-Y-CH3 torsional frequencies were assigned to 177–197 cm−1 vibrations for CH3COOCH3 and to 188–220 cm−1 for CH3CSSCH3. The IR intensities increase for both molecules in their cis form with increasing polarity of the environment. However, the theoretical frequencies increase in this series for CH3COOCH3 but decreases for CH3CSSCH3, further emphasizing the structural differences for XY=OO and XY=SS esters.



{kind=link}