Evaluation of Silica-H2SO4 as an Efficient Heterogeneous Catalyst for the Synthesis of Chalcones

 and
and
Abstract
:1. Introduction
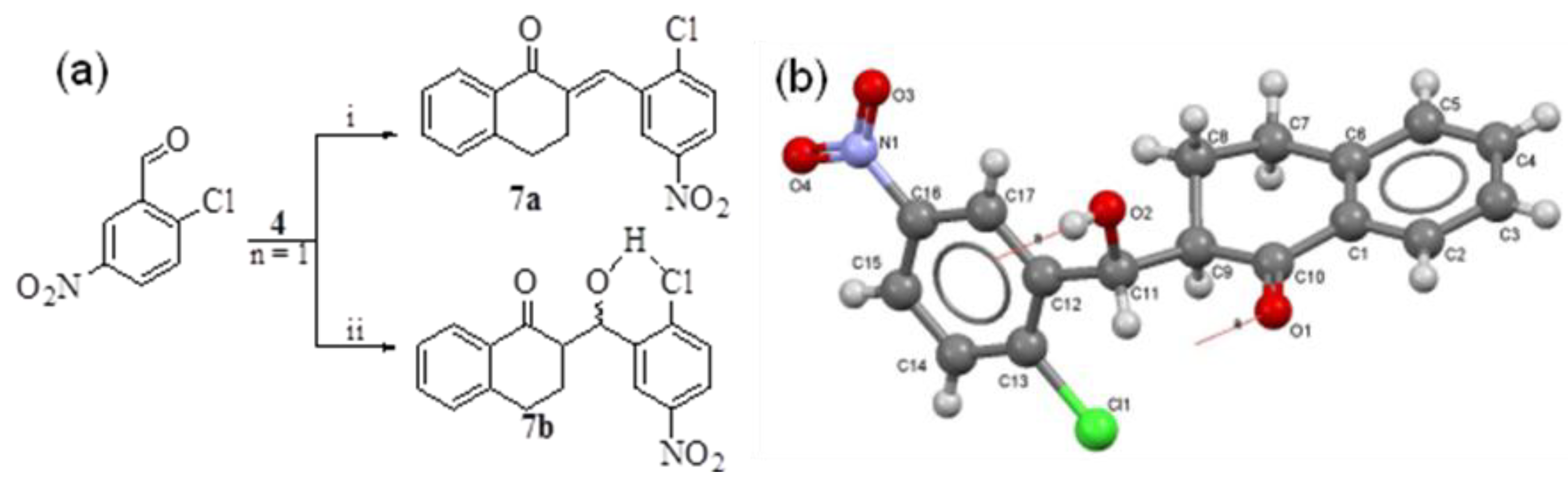
2. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
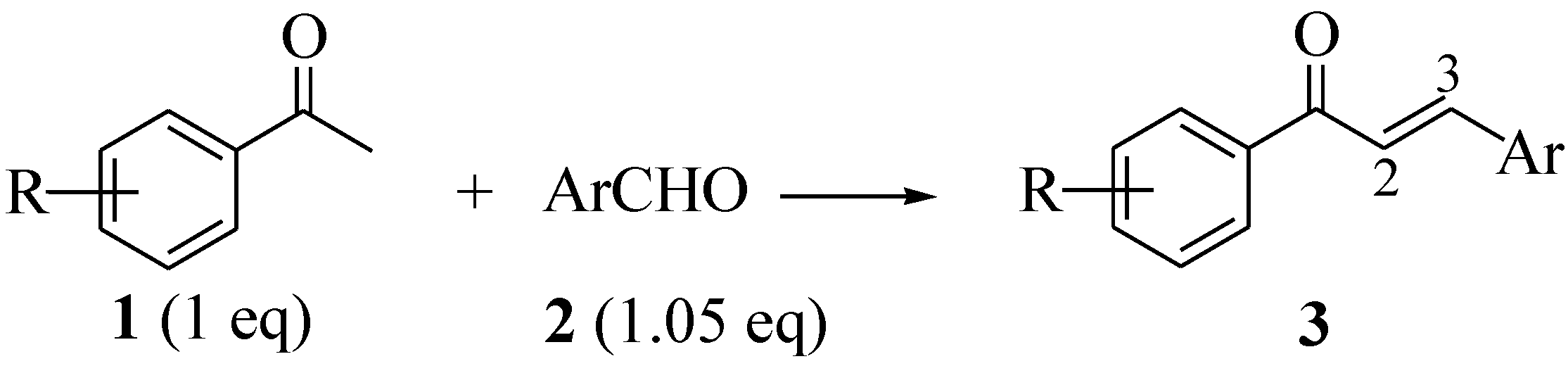{kind=link}
{kind=link}
| Entry | SSA (g) | Solvent | Time (Temperature, °C) | %Yield ¥ |
|---|---|---|---|---|
| 1 | 0.005 | MeOH | 4 h (reflux) | * |
| 2 | 0.01 | CH2Cl2 | 6 h (reflux) | * |
| 3 | 0.01 | - | 2 h (65) | 28 |
| 4 | 0.02 | - | 8 h (rt) | - |
| 5 | 0.02 | - | 1 h (65) | 91 |
| 6 | 0.02 | - | 0.5 h (100) | # |
| 7 | 0.05 | - | 0.5 h (65) | 94 |
| 8 | 0.1 | - | 0.5 h (65) | ^ |
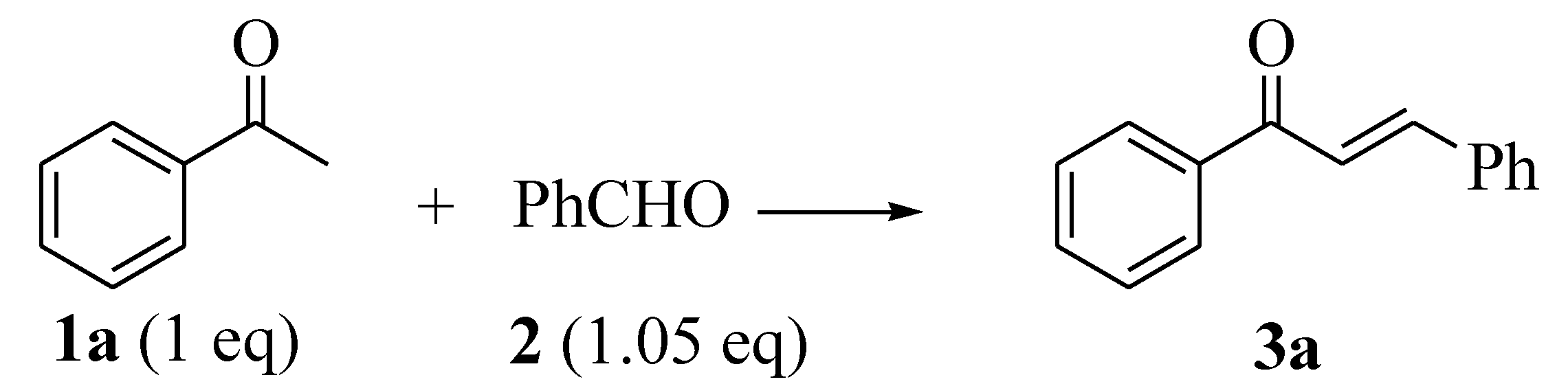
2.1. Synthesis of Open Chain Chalcones 3a–o
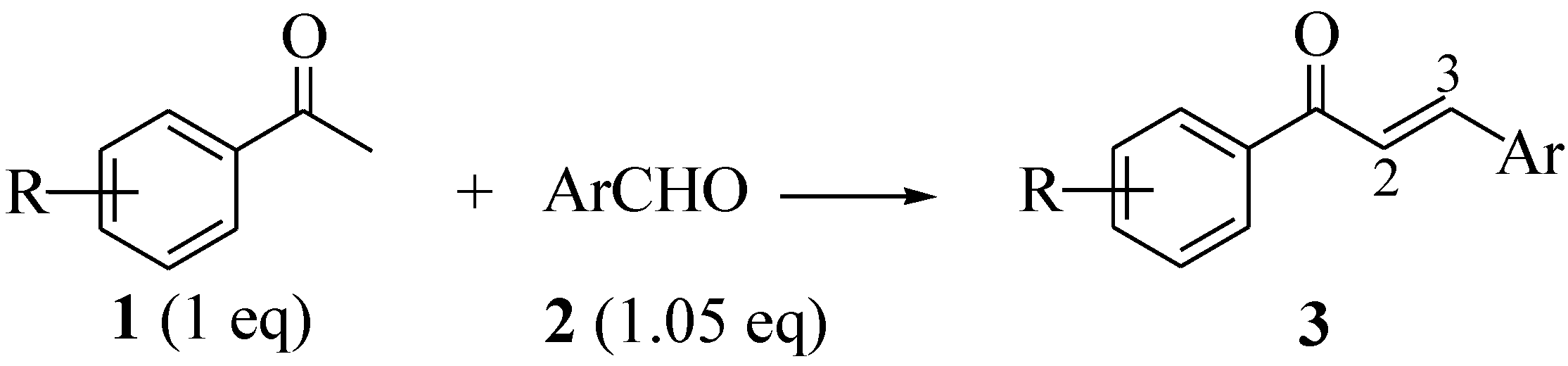
| Entry | R | Ar | 3 | %Yield | 1H-δ ! (J§) | ||||
|---|---|---|---|---|---|---|---|---|---|
| H2SO4* | NaOH # | SF^ | SSA ¥ | H2 | H3 | ||||
| a | H | Ph | 3a | 54 | 45 | 77 | 91 | 7.60 (16.4) | 8.12 (16.4) |
| b | 3′-OH | Ph | 3b | 25 | 51 | 82 | 95 | 7.58 (15.9) | 7.98 (15.9) |
| c | 3′-OH | 2-furyl | 3c | 38 | 43 | 71 | 83 | 7.35 (16.8) | 7.59 (16.8) |
| d | 4′-OH | Ph | 3d | 48 | 24 | 73 | 88 | 7.43 (15.6) | 7.81 (15.6) |
| e | 4′-OH | 2-furyl | 3e | 45 | 41 | 85 | 88 | 7.54 (17.1) | 7.83 (17.4) |
| f | 4′-OH | 4-MeOPh | 3f | 58 | 32 | 89 | 92 | 7.43 (15.4) | 7.79 (15.4) |
| g | 4′-Me | Ph | 3g | 62 | 73 | 92 | 89 | 7.56 (17.4) | 7.88 (17.4) |
| h | 4′-Me | 2-furyl | 3h | 54 | 79 | 86 | 94 | 7.55 (15.6) | 7.88 (15.6) |
| i | 4′-Me | 4-Me2NPh | 3i | <10 | 13 | 29 | 80 | 6.86 (14.9) | 7.58 (14.9) |
| j | 3′-NO2 | Ph | 3j | <10 | - | 35 | 83 | 7.62 (16.0) | 8.02 (16.0) |
| k | 3′-NO2 | 2-furyl | 3k | 15 | - | 40 | 87 | 7.50 (16.8) | 7.77 (16.8) |
| l | 3′-NO2 | 4-Me2NPh | 3l | 23 | - | 25 | 76 | 7.54 (15.8) | 7.83 (15.8) |
| m | 3′-NO2 | 4-MeOPh | 3m | 19 | - | 33 | 74 | 7.38 (16.0) | 7.79 (16.0) |
| n | 4′-Cl | Ph | 3n | 78 | 68 | 83 | 96 | 7.61 (16.6) | 8.18 (16.5) |
| o | 4′-Cl | 2- MeOPh | 3o | 75 | 64 | 76 | 92 | 7.63 (16.1) | 8.03 (16.1) |
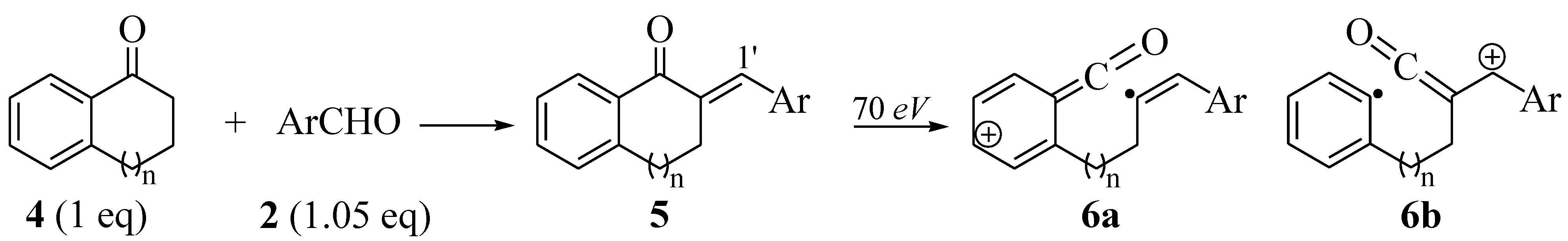
2.2. Synthesis of Tetralone- and Indanone-Based Chalcones 5a–m
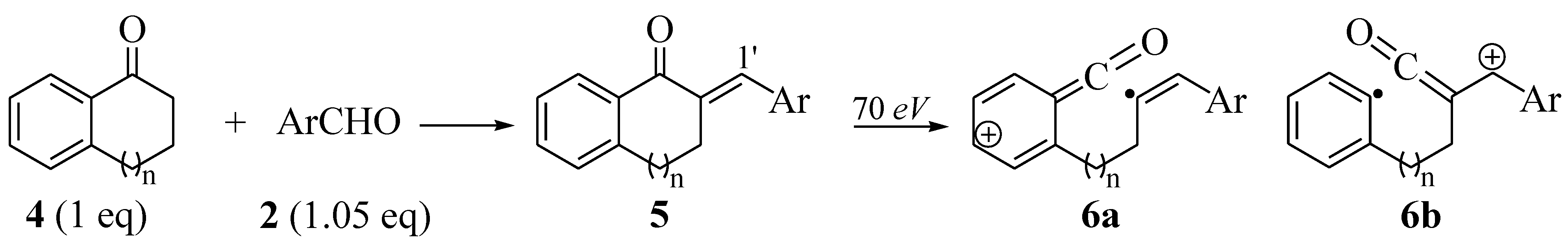
| Entry | n | Ar | 5 | %Yield | 1H-δ ! (H1′) | [6a]+ or [6b]+(% abundance) | |||
|---|---|---|---|---|---|---|---|---|---|
| H2SO4 * | NaOH# | SF^ | SSA ¥ | ||||||
| a | 0 | Ph | 5a | 35 | 41 | 83 | 87 | 7.93 | 220 (64) |
| b | 0 | 2-furyl | 5b | 48 | 52 | 78 | 91 | 7.44 | 210 (100) |
| c | 0 | 4-Me2NPh | 5c | 12 | - | 85 | 82 | 6.97 | 263 (100) |
| d | 0 | 4-MeOPh | 5d | 73 | 35 | 82 | 97 | 8.13 | 250 (76) |
| e | 0 | 3-MeOPh | 5e | 75 | 56 | 79 | 90 | 7.64 | 250 (100) |
| f | 0 | 3-NO2Ph | 5f | 13 | <10 | 53 | 72 | 8.53 | 265 (42) |
| g | 0 | 3,4-(OMe)2Ph | 5g | 75 | 63 | 80 | 86 | 7.18 | 280 (100) |
| h | 1 | Ph | 5h | 66 | 54 | 76 | 80 | 6.98 | 234 (56) |
| i | 1 | 2-furyl | 5i | 72 | 68 | 85 | 87 | 7.56 | 224 (100) |
| j | 1 | 4-Me2NPh | 5j | 21 | 17 | 62 | 94 | 7.82 | 277 (52) |
| k | 1 | 4-MeOPh | 5k | 76 | 71 | 85 | 89 | 6.69 | 264 (100) |
| l | 1 | 3-NO2Ph | 5l | 20 | 19 | 71 | 85 | 8.27 | 279 (29) |
| m | 1 | 3-ClPh | 5m | 78 | 42 | 79 | 87 | 6.81 | 268, 270 (38, 12) |
3. Experimental
3.1. Preparation of SSA
3.2. Representative Procedure for H2SO4 Catalyzed Synthesis of Chalcones under Reflux
3.3. Representative Procedure for NaOH Catalyzed Synthesis of Chalcones under Reflux
3.4. Representative Procedure for NaOH Catalyzed Synthesis of Chalcones under Solvent Free Conditions
3.5. Representative Procedure for the SSA Catalysed Synthesis of Chalcones
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Hsieh, H.K.; Lee, T.H.; Wang, J.P.; Wang, J.J.; Kin, C.N. Synthesis and anti-inflammatory effects of chalcones and related compounds. Pharm. Res. 1998, 15, 39–46. [Google Scholar] [CrossRef]
- Torigoo, T.; Arisawa, M.; Iloch, S.; Fujiu, M.; Mayuyama, H.B. Antimutagenic chalcones: Antagonizing the mutagenicity of benzo(a)pyrene in Salmonella typhymurium. Biochem. Biophys. Res. Commun. 1983, 112, 833–842. [Google Scholar] [CrossRef]
- Haraguchi, H.; Ishikawa, H.; Mizutani, K.; Tamura, Y.; Kinoshira, T. Antioxidant and superoxide scavenging activities of retrochalcones in Glycyrrhiza inflate. Bioorg. Med. Chem. 1998, 6, 339–347. [Google Scholar] [CrossRef]
- De Vincenzo, R.; Ferlini, C.; Distefeno, M.; Gaggini, C.; Riva, A.; Bombardelli, E.; Morazzini, P.; Belluti, F.; Ranelletti, F.O.; Mancuso, S.; et al. In vitro evaluation of newly developed chalcone analogues in human cancer cells. Cancer Chem. Pharmacol. 2000, 46, 305–312. [Google Scholar] [CrossRef]
- Geissman, T.A.; Clinton, R.O. Flavanones and related compounds. I. The preparation of polyhydroxychalcones and flavanones. J. Am. Chem. Soc. 1946, 68, 697–700. [Google Scholar] [CrossRef]
- Russel, A.; Todd, S. The constitution of natural tannins. VI.1 Coloring matters derived from 2,5-dihydroxyacetophenone. J. Am. Chem. Soc. 1939, 61, 2651–2658. [Google Scholar] [CrossRef]
- Zwaagstra, M.E.; Timmerman, H.; Tamura, M.; Tohma, T.; Wada, Y.; Onogi, K.; Zhang, M. Synthesis and structure activity relationships of carboxylated chalcones: A novel series of CysLT1 (LTD4) receptor antagonists. J. Med. Chem. 1997, 40, 1075–1089. [Google Scholar] [CrossRef]
- Davey, W.; Tivey, D.J. Chalcones and related compounds. Part IV. Addition of hydrogen cyanide to chalcones. J. Chem. Soc. 1958, 1958, 1230–1236. [Google Scholar] [CrossRef]
- Jadhav, G.V.; Kulkarni, V.G. Borax as a new condensing agent for the synthesis of chalkones. Curr. Sci. 1951, 20, 42–43. [Google Scholar]
- Matsushima, R.; Murakami, T. Photoreactions of 3-(2-Hydroxyphenyl)-1-substituted phenyl-2-propen-1-ones (Substituted 2-Hydroxychalcones) in organic solvents in the presence and absence of acid. Bull. Chem. Soc. 2000, 73, 2215–2219. [Google Scholar] [CrossRef]
- Breslow, D.S.; Hauser, C.R. Condensations. 1 XI. Condensations of certain active hydrogen compounds effected by BF3 and AlCl3. J. Am. Chem. Soc. 1940, 62, 2385–2388. [Google Scholar] [CrossRef]
- Guthrie, J.L.; Rabjohm, N. Some reactions effected by means of bromomagnesium t-alkoxides. J. Org. Chem. 1957, 22, 176–179. [Google Scholar] [CrossRef]
- Jia, H.P.; Dreyer, D.R.; Bielawski, C.W. Graphite oxide as an auto-tandem oxidation-hydration-aldol coupling catalyst. Adv. Synth. Catal. 2011, 353, 528–532. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Bielawski, C.W. Carbocatalysis: Heterogeneous carbons finding utility in synthetic chemistry. Chem. Sci. 2011, 2, 1233–1240. [Google Scholar] [CrossRef]
- Solhy, A.; Tahir, R.; Sebti, S.; Skouta, R.; Bousmina, M.; Zahouily, M.; Larzek, M. Efficient synthesis of chalcone derivatives catalyzed by re-usable hydroxyapatite. Appl. Catal. A 2010, 374, 189–193. [Google Scholar] [CrossRef]
- Sebti, S.; Solhy, A.; Tahir, R.; Smahi, A. Modified hydroxyapatite with sodium nitrate: an efficient new solid catalyst for the Claisen-Schmidt condensation. Appl. Catal. A 2002, 335, 273–281. [Google Scholar]
- Sebti, S.; Solhy, A.; Tahir, R.; Abdelatif, S.; Boulaajaj, S.; Mayoral, J.A.; Garcı́a, J.I.; Fraile, J.M.; Kossir, A.; Oumimoun, H. Application of natural phosphate modified with sodium nitrate in the synthesis of chalcones: a soft and clean method. J. Catal. 2003, 213, 1–6. [Google Scholar] [CrossRef]
- Riadi, Y.; Abrouki, Y.; Mamouni, R.; El Haddad, M.; Routier, S.; Guillaumet, G.; Lazar, S. New eco-friendly animal bone meal catalysts for preparation of chalcones and aza-Michael adducts. Chem. Cent. J. 2012, 6, 60–71. [Google Scholar] [CrossRef]
- Gupta, R.; Paul, S.; Gupta, A. Improved microwave-induced synthesis of chalcones and related enones. Ind. J. Chem. 1995, 34, 61–62. [Google Scholar]
- Boss, A.K.; Manhas, M.S.; Gosh, M.S. Microwave-induced organic reaction enhancement chemistry. 2. Simplified techniques. J. Org. Chem. 1991, 56, 6968–6970. [Google Scholar] [CrossRef]
- Seedhar, N.Y.; Jayapal, M.R.; Prasad, K.S.; Prasad, P.R. Synthesis and characterization of 4-hydroxy chalcones using PEG-400 as a recyclable solvent. Res. J. Pharm. Biol. Chem. Sci. 2010, 1, 480–485. [Google Scholar]
- Boukhvalov, D.W.; Dreyer, D.R.; Bielawski, C.W.; Son, Y.W. A computational investigation of the catalytic properties of graphene oxide: Exploring mechanisms by using DFT methods. Chem. Cat. Chem. 2012, 4, 1844–1849. [Google Scholar]
- Landarani-Isfahani, A.; Safari, J.; Ghotbinejad, M.; Gandomi-Ravandi, S.; Moshtael. Silica sulfuric acid (SSA), a novel catalyst for synthesis of some-α-phenylhydrazone-2-ketomethylquinolines. Org. Chem. An. Indian J. 2009, 5, 39–42. [Google Scholar]
- Mobinikhaledi, A.; Foroughifar, N.; Khodaei, H. Synthesis of octahydroquinazolinone derivatives using silica sulfuric acid as an efficient catalyst. Eur. J. Chem. 2010, 1, 291–293. [Google Scholar] [CrossRef]
- Azizian, J.; Mohammadi, A.A.; Soleimani, E.; Karimi, A.R.; Mohammadizadeh, M.R. A stereoselective three-component reaction: One-pot synthesis of cis-isoquinolonic acids catalyzed by silica sulfuric acid under mild and heterogeneous conditions. J. Heterocycl. Chem. 2006, 43, 187–190. [Google Scholar] [CrossRef]
- Wu, H.; Lin, W.; Wan, Y.; Xin, H.Q.; Shi, D.Q.; Shi, Y.H.; Yuan, R.; Bo, R.C.; Yin, W. Silica gel-catalyzed one-pot synthesis in water and fluoroscene properties studies of 5-amino-2-aryl-3H-chromeno[4,3,2-de][1,8]naphthyridine-4-carbonitriles and 5-amino-2-aryl-3H-quinolino [4,3,2-de][1,6]naphthyridine-4-carbonitriles. J. Comb. Chem. 2010, 12, 31–34. [Google Scholar] [CrossRef]
- Cao, C.; Xu, C.; Lin, W.; Li, X.; Hu, M.; Wang, J.; Huang, Z.; Shi, D.; Wang, Y. Microwave-assisted improved synthesis of pyrrolo[2,3,4-kl]acridine and dihydropyrrolo[2,3,4-kl]acridine derivatives catalyzed by silica sulfuric acid. Molecules 2013, 18, 1613–1625. [Google Scholar] [CrossRef]
- Ziarani, G.M.; Badiei, A.; Abbasi, A.; Farahani, Z. Cross-aldol condensation of cycloalkanones and aromatic aldehydes in the presence of nanoporous silica-based sulfonic acid (SiO2-Pr-SO3H) under solvent free conditions. Chin. J. Chem. 2009, 27, 1537–1542. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, Y.Q.; Guo, S.R. Silica sulfuricacid promotes Aza-Michael addition reactions under solvent-free condition as a heterogeneous and reusable catalyst. Molecules 2009, 14, 4779–4789. [Google Scholar] [CrossRef]
- Wu, H.; Shen, Y.; Fan, L.Y.; Wan, Y.; Wang, W.X.; Shi, D.Q. Solid silica sulfuric acid (SSA) as a novel and efficient catalyst for acetylation of aldehydes and sugars. Tetrahedron 2006, 62, 7995–7998. [Google Scholar] [CrossRef]
- Kiasat, A.R.; Kazemi, F.; Mehrjardi, M.F. Protection of carbonyl groups as 2,4-dinitro-phenyldrazone catalyzed by silica sulfuric acid. Asian J. Chem. 2006, 18, 969–972. [Google Scholar]
- Aoyama, T.; Kubota, S.; Takido, T.; Kodomari, M. Silica sulfuric acid-promoted deacylation of α-bromo-β-diketones. Chem. Lett. 2011, 40, 484–485. [Google Scholar] [CrossRef]
- Ghorbani-Choghamarani, A.; Zamani, P. Ammonium bromide as an effective and viable catalyst in the oxidation of sulfides using nitro urea and silica sulfuric acid. J. Iran. Chem. Soc. 2011, 8, 142–148. [Google Scholar] [CrossRef]
- Maleki, B.; Shirvan, H.K.; Taimazi, F.; Akbaradeh, E. Sulfuric acid immobilized on silica gel as highly efficient and heterogeneous catalyst for the one-pot synthesis of 2,4,5-triaryl-1H-imidazoles. Int. J. Org. Chem. 2012, 2, 93–99. [Google Scholar] [CrossRef]
- Zolfigol, M.A. Silica sulfuric acid/NaNO2 as a novel heterogeneous system for production of thionitrites and disulfides under mild conditions. Tetrahedron 2001, 57, 9509–9511. [Google Scholar] [CrossRef]
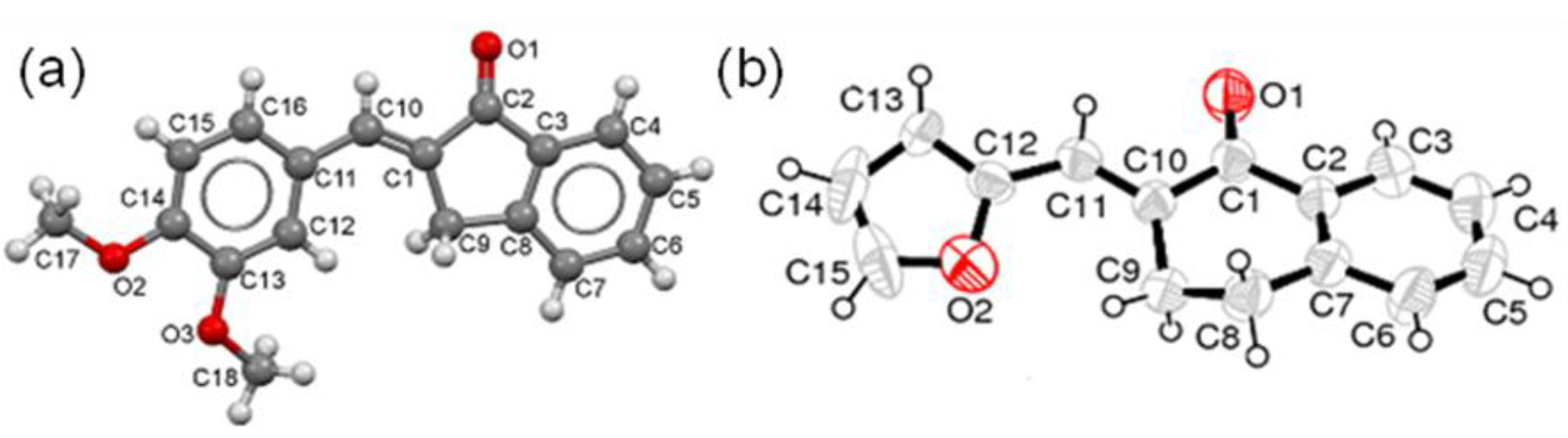
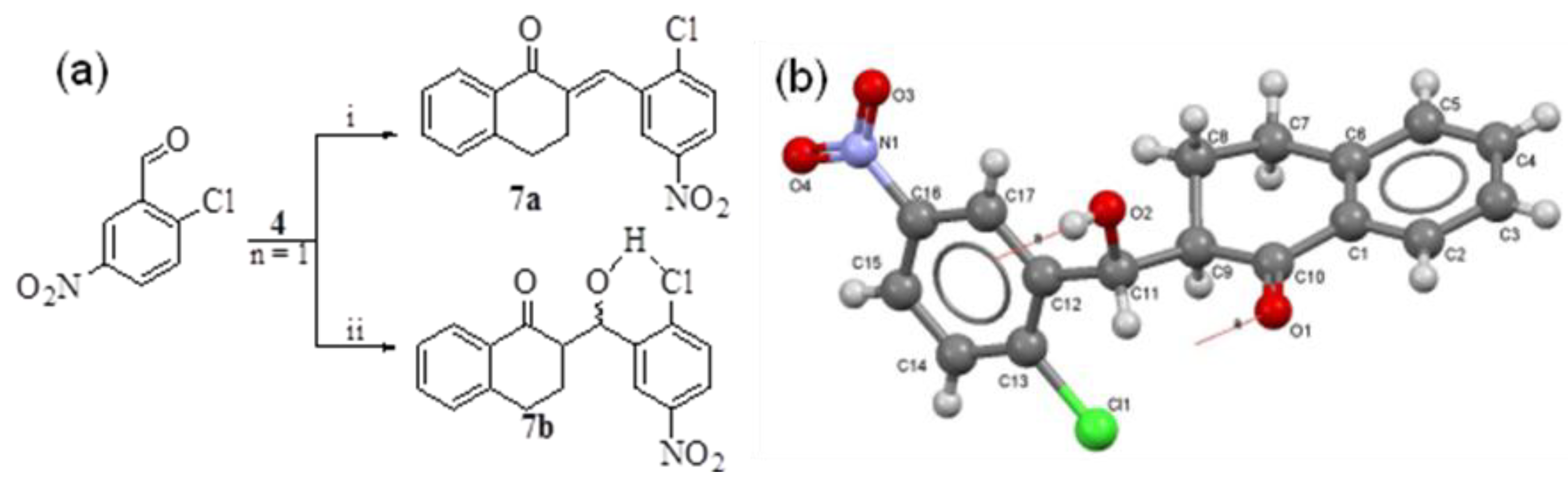
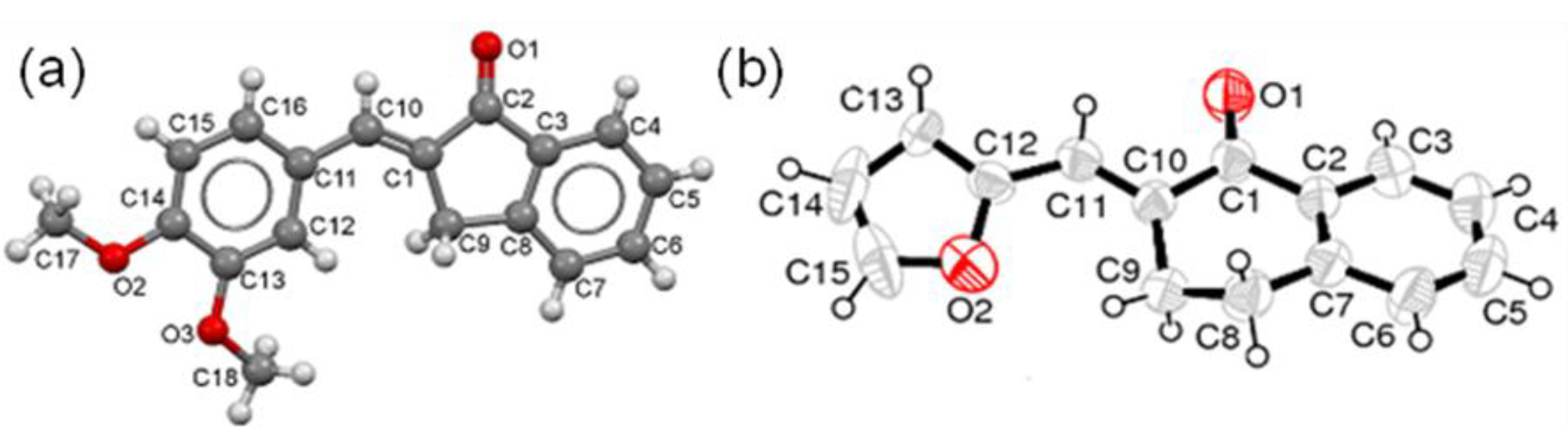
- Crystallographic data of 5g have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication No. 950124. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- Crystallographic data of 7b have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. 950123. Both X-ray structures were obtained by Professor Muhammad Nawaz Tahir, Department of Physics, University of Sargodha, Pakistan.
- Syam, S.; Abdelwahab, S.I.; Al-Mamary, M.A.; Mohan, S. Synthesis of chalcones with anticancer activity. Molecules 2012, 17, 6179–6195. [Google Scholar] [CrossRef]
- Zheng, C.J.; Jiang, S.M.; Chen, Z.H.; Ye, B.J.; Piao, H.R. Synthesis and anti-bacterial activity of some heterocyclic chalcone derivatives bearing thiofuran, furan, and quinoline moieties. Arch. Pharm. 2011, 344, 689–695. [Google Scholar] [CrossRef]
- Sharma, B. Comparative study of conventional and microwave assisted synthesis of chalcones. Asian J. Chem. 2011, 23, 2468–2470. [Google Scholar]
- Li, J.P.; Zhang, Y.X.; Ji, Y. Selective 1,4-reduction of chalcones with Zn/NH4Cl/C2H5OH/ H2O. J. Chin. Chem. Soc. 2008, 55, 390–393. [Google Scholar]
- Camps, P.; Domingo, L.R.; Formosa, X.; Galdeano, C.; González, D.; Muñoz-Torrero, D.; Segalés, S.; Font-Bardia, M.; Solans, X. Highly diastereoselective one-pot synthesis of spiro{cyclopenta[a]indene-2,2′-indene}diones from 1-indanones and aromatic aldehydes. J. Org. Chem. 2006, 71, 3464–347. [Google Scholar] [CrossRef]
- Gazzetta Chimica Italiana; Italian Chemical Society: Roma, Italy, 1975; Volume 105, pp. 971, 975–976, 980–981.
- El-Rayyes, N.; Al-Qatami, S.; Edun, M. Heterocycles. 14. Synthesis of 5H-indenopyrimidines. J. Chem. Eng. Data 1987, 32, 481–483. [Google Scholar] [CrossRef]
- Rothenberg, G.; Downie, A.P.; Raston, C.L.; Scott, J.L. Understanding solid/solid organic reactions. J. Am. Chem. Soc. 2001, 123, 8701–8708. [Google Scholar]
- Kamakshi, R.; Latha, S.S.; Reddy, B.S.R. An efficient synthesis of bio-active flourescent benzylidene tetralones. Indian J. Chem. 2010, 49B, 944–947. [Google Scholar]
- Sultan, A.; Raza, A.R.; Tahir, M.N. Free radical mediated chemoselective reduction of enones. Synth. Commun. 2013. submitted. [Google Scholar]
- Sample Availability:Samples of the compounds 3a–o, 5a–m and 7a–b are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sultan, A.; Raza, A.R.; Abbas, M.; Khan, K.M.; Tahir, M.N.; Saari, N. Evaluation of Silica-H2SO4 as an Efficient Heterogeneous Catalyst for the Synthesis of Chalcones. Molecules 2013, 18, 10081-10094. https://doi.org/10.3390/molecules180810081
Sultan A, Raza AR, Abbas M, Khan KM, Tahir MN, Saari N. Evaluation of Silica-H2SO4 as an Efficient Heterogeneous Catalyst for the Synthesis of Chalcones. Molecules. 2013; 18(8):10081-10094. https://doi.org/10.3390/molecules180810081
Chicago/Turabian StyleSultan, Aeysha, Abdul Rauf Raza, Muhammad Abbas, Khalid Mohammed Khan, Muhammad Nawaz Tahir, and Nazamid Saari. 2013. "Evaluation of Silica-H2SO4 as an Efficient Heterogeneous Catalyst for the Synthesis of Chalcones" Molecules 18, no. 8: 10081-10094. https://doi.org/10.3390/molecules180810081