Synthesis and SAR Studies of Praziquantel Derivatives with Activity against Schistosoma japonicum

Abstract
:1. Introduction
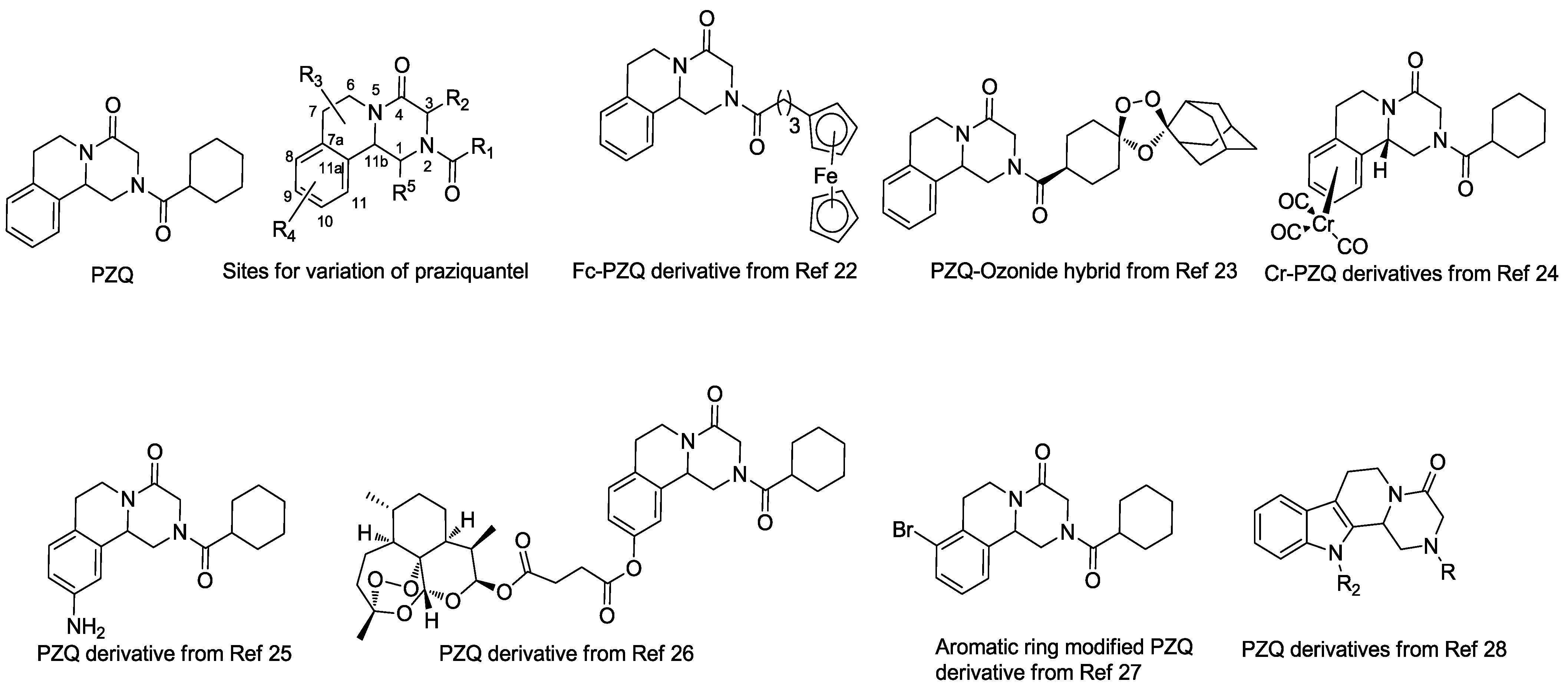
2. Results and Discussion
2.1. Chemistry
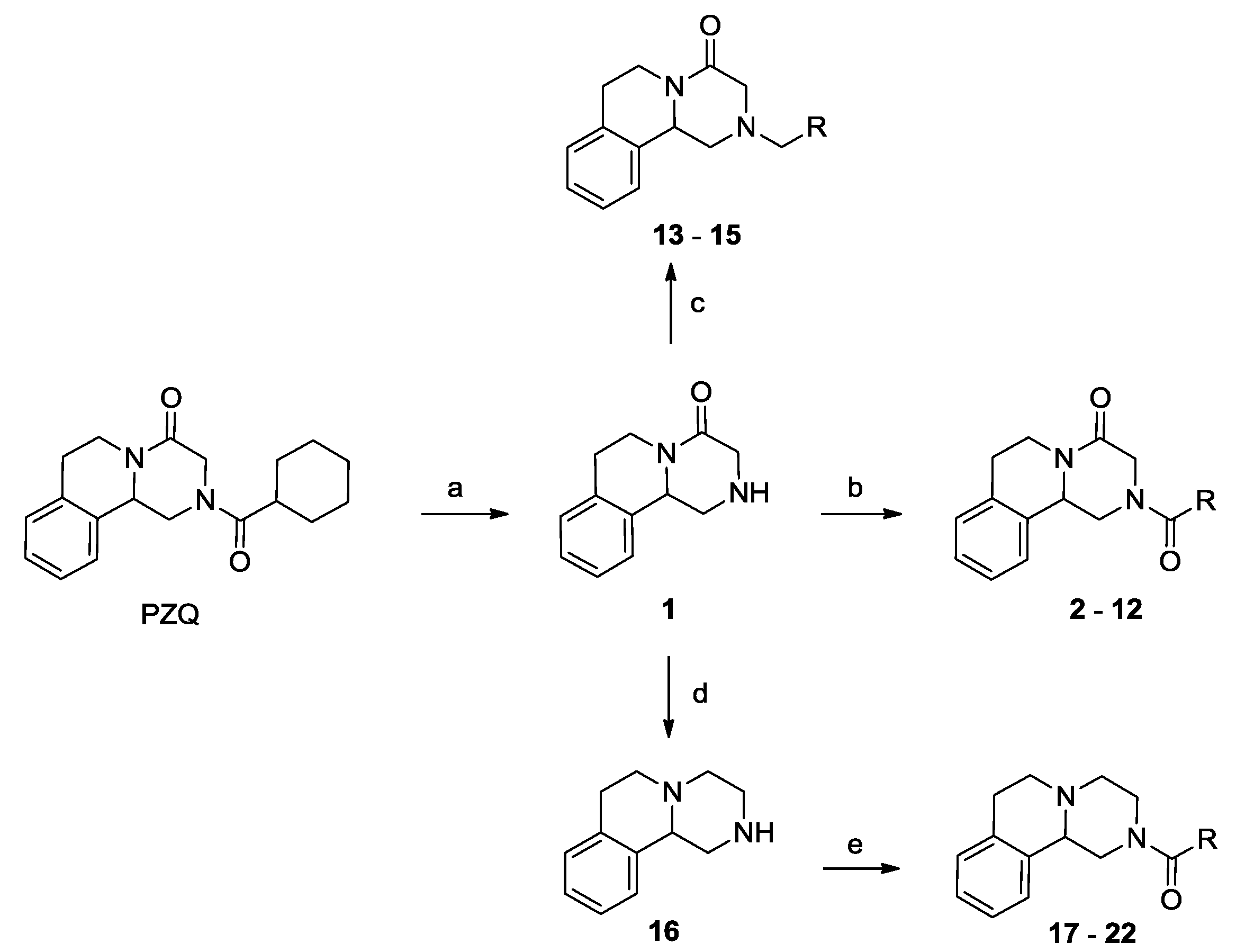
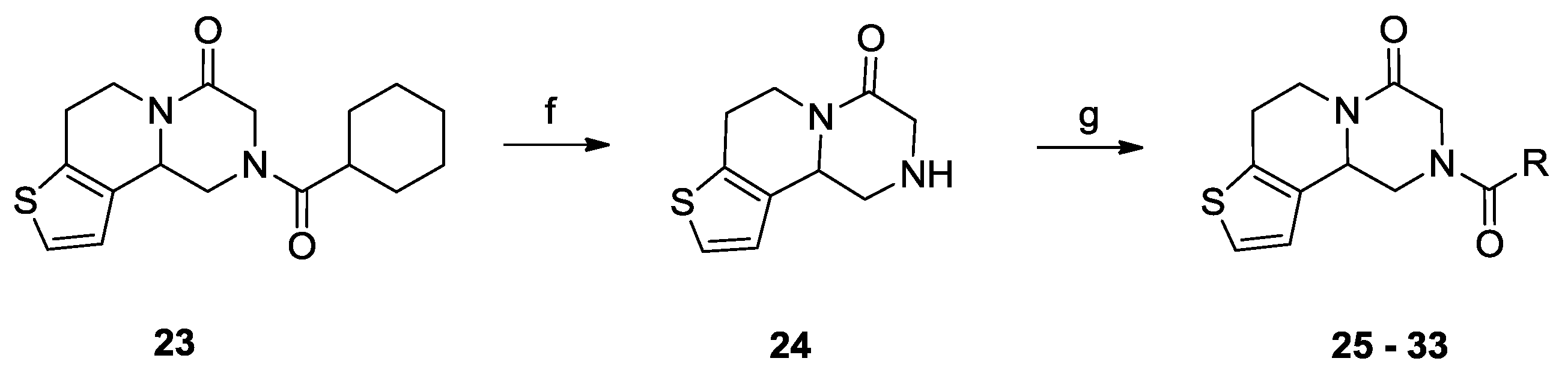
2.2. Biological Activities


{kind=link}
{kind=link}
{kind=link}
| Compound | R | X | Y | Z | Killing activity a | |||
|---|---|---|---|---|---|---|---|---|
| Conc (μM) b | 24 h | 48 h | 72 h | |||||
| Vehicle | n.e | n.e | n.e | |||||
| PZQ | 10 | 25.0%D | 25.0%D | 37.5%D | ||||
| 25 | 25.0%D | 25.0%D | 62.5%D | |||||
| 50 | 25.0%D | 37.5%D | 62.5%D | |||||
| 100 | 37.5%D | 50.0%D | 87.5%D | |||||
| 1 d, e | None | O | CH=CH | None | 100 | n.e. | n.e. | n.e. |
| 2 d, e |  | O | CH=CH | O | 100 | sluggish | 25.0% | 75.0% |
| 3 e |  | O | CH=CH | O | 10 | n.e. | n.e. | sluggish |
| 25 | n.e. | n.e. | 10.0%D | |||||
| 50 | n.e. | sluggish | 75.0%D | |||||
| 100 | sluggish | sluggish | 87.5%D | |||||
| 4 e |  | O | CH=CH | O | 10 | sluggish | sluggish | 37.5%D |
| 25 | sluggish | sluggish | 50.0%D | |||||
| 50 | sluggish | sluggish | 100%D | |||||
| 100 | 12.5%D | 12.5%D | 75.0%D | |||||
| 5 d, e | CH3 | O | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 6 d, e |  | O | CH=CH | O | 100 | n.e. | n.e. | 20.0%D |
| 7 c, e | ClCH2 | O | CH=CH | O | 1 | n.e. | n.e. | n.e. |
| 3 | n.e. | 25.0%D | 25.0%D | |||||
| 5 | 70.0%D | 90.0%D | 100%D | |||||
| 8 | 100%D | 100%D | 100%D | |||||
| 10 | 100%D | 100%D | 100%D | |||||
| 25 | 100%D | 100%D | 100%D | |||||
| 50 | 100%D | 100%D | 100%D | |||||
| 100 | 100%D | 100%D | 100%D | |||||
| 8 d, e |  | O | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 9 e |  | O | CH=CH | O | 10 | n.e. | n.e. | 55.6%D |
| 25 | sluggish | 25.0%D | 87.5%D | |||||
| 50 | 18.2%D | 36.4%D | 45.5%D | |||||
| 100 | 45.5%D | 45.5%D | 81.8%D | |||||
| 10 d, e |  | O | CH=CH | O | 100 | n.e. | n.e. | sluggish |
| 11 e |  | O | CH=CH | O | 10 | n.e. | n.e. | 62.5%D |
| 25 | n.e. | n.e. | 62.5%D | |||||
| 50 | 25.0%D | 37.5%D | 87.5%D | |||||
| 100 | 14.3%D | 28.6%D | 87.5%D | |||||
| 12 d, e |  | O | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 13 d |  | O | CH=CH | H2 | 100 | n.e. | n.e. | n.e. |
| 14 d, e |  | O | CH=CH | H2 | 100 | n.e. | n.e. | n.e. |
| 15 d |  | O | CH=CH | H2 | 100 | n.e. | n.e. | n.e. |
| 16 d, e | None | H2 | CH=CH | None | 100 | n.e. | n.e. | n.e. |
| 17 d |  | H2 | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 18 |  | H2 | CH=CH | O | 10 | sluggish | sluggish | sluggish |
| 25 | sluggish | 18.2%D | 18.2%D | |||||
| 50 | 10.0%D | 20.0%D | 50.0%D | |||||
| 100 | 25.0%D | 25.0%D | 75.0%D | |||||
| 19 |  | H2 | CH=CH | O | 10 | n.e. | n.e. | n.e. |
| 25 | n.e. | 14.3%D | 57.1%D | |||||
| 50 | sluggih | 33.3%D | 66.7%D | |||||
| 100 | sluggish | sluggish | 85.7%D | |||||
| 20 d, e |  | H2 | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 21 d |  | H2 | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 22 | ClCH2 | H2 | CH=CH | O | 10 | n.e. | sluggish | 50.0%D |
| 25 | 87.5%D | 100%D | 100%D | |||||
| 50 | 100%D | 100%D | 100%D | |||||
| 100 | 100%D | 100%D | 100%D | |||||
| 23 e |  | O | S | O | 10 | sluggish | 75.0%D | 75.0%D |
| 25 | sluggish | 87.5%D | 87.5%D | |||||
| 50 | 25.0% | 75.0%D | 87.5%D | |||||
| 100 | 28.6%D | 71.4%D | 71.4%D | |||||
| 24 d | None | O | S | None | 100 | n.e. | n.e. | n.e. |
| 25 d |  | O | S | O | 100 | n.e. | 12.5%D | 37.5%D |
| 26 d |  | O | S | O | 100 | n.e. | sluggish | sluggish |
| 27 d |  | O | S | O | 100 | n.e. | n.e. | sluggish |
| 28 d |  | O | S | O | 100 | n.e. | n.e. | n.e. |
| 29 d |  | O | S | O | 100 | sluggish | sluggish | sluggish |
| 30 e |  | O | S | O | 10 | n.e. | n.e. | n.e. |
| 25 | n.e. | sluggish | sluggish | |||||
| 50 | n.e. | 14.3%D | 14.3%D | |||||
| 100 | sluggish | 37.5%D | 75.0%D | |||||
| 31 d, e |  | O | S | O | 100 | n.e. | n.e. | n.e. |
| 32 d, e |  | O | S | O | 100 | n.e. | n.e. | n.e. |
| 33 c | ClCH2 | O | S | O | 1 | n.e. | n.e. | n.e. |
| 3 | n.e. | n.e. | n.e. | |||||
| 5 | sluggish | 25%D | 37.5%D | |||||
| 8 | 75%D | 87.5%D | 87.5%D | |||||
| 10 | 100%D | 100%D | 100%D | |||||
| 25 | 100%D | 100%D | 100%D | |||||
| 50 | 100%D | 100%D | 100%D | |||||
| 100 | 100%D | 100%D | 100%D | |||||
3. Experimental
3.1. Chemistry
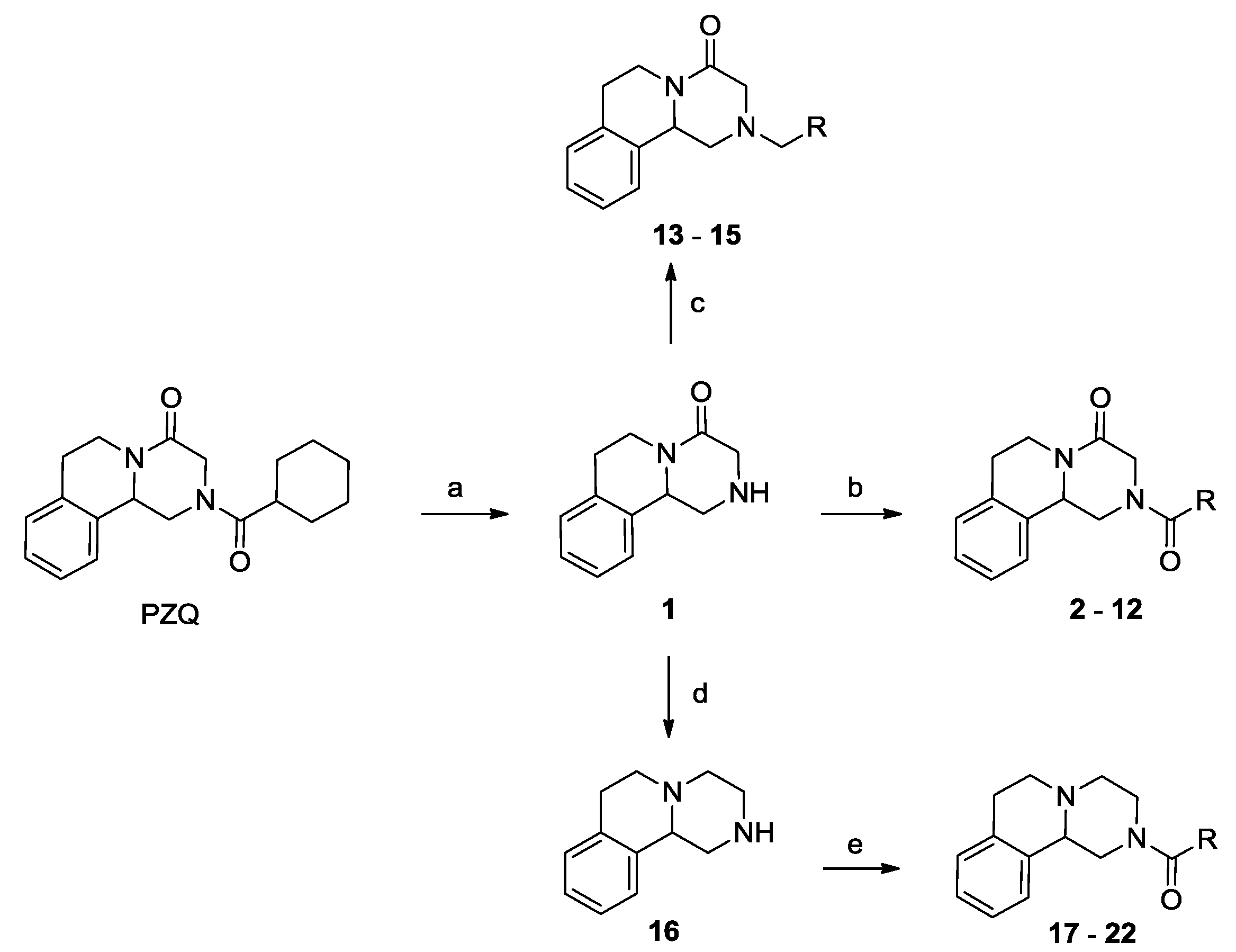
3.1.1. Procedure for the Preparation of Compound 1
3.1.2. Procedure for the Preparation of Compound 8
3.1.3. General Procedure for the Preparation of Derivatives 2–7 and 9–12
3.1.4. General Procedure for the Preparation of Derivatives 13–15
3.1.5. Procedure for the Preparation of Compound 16
3.1.6. General Procedure for the Preparation of Derivatives 17–22
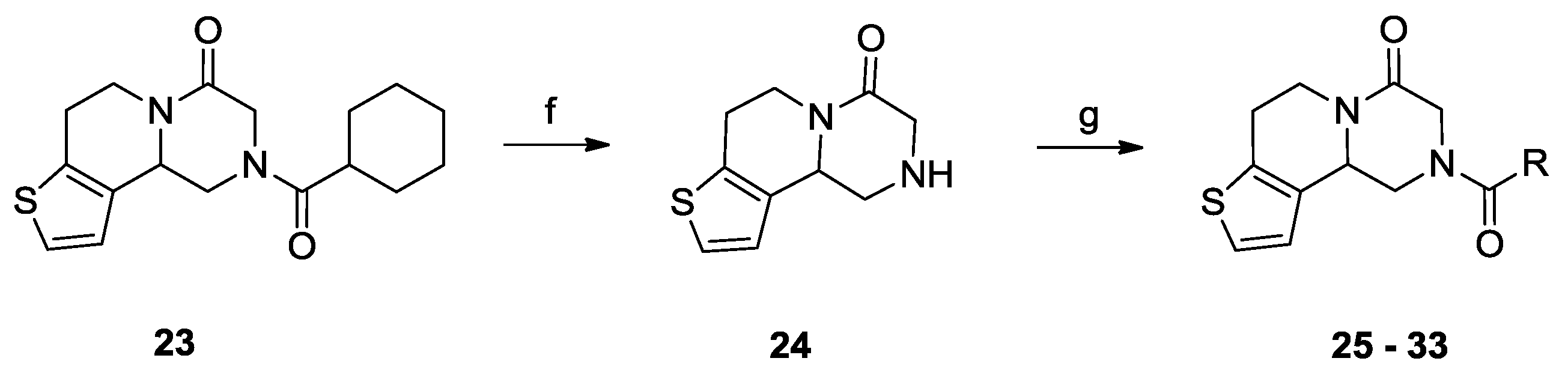
3.1.7. The Procedure for the Preparation of Compound 24
3.1.8. General Procedure for the Preparation of Compound 25–33
3.2. Killing Activity of Compounds 1–33 on S. Japonicum Adult Worms in Vitro [36]
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Danso-Appiah, A.; Olliaro, P.L.; Donegan, S.; Sinclair, D.; Utzinger, J. Drugs for treating Schistosoma mansoni infection. Cochrane. Database. Syst. Rev. 2013, 2, CD000528. [Google Scholar]
- Gryseels, B.; Polman, K.; Clerinx, J.; Kestens, L. Human schistosomiasis. Lancet 2006, 368, 1106–1118. [Google Scholar] [CrossRef]
- Zheng, H.; Zhang, L.J.; Zhu, R.; Xu, J.; Li, S.Z.; Guo, J.G.; Xiao, N.; Zhou, X.N. Schistosomiasis situation in People's Republic of China in 2011. Chin. J. Schisto. Control. 2012, 24, 621–626. [Google Scholar]
- Caffrey, C.R.; Secor, W.E. Schistosomiasis: From drug deployment to drug development. Curr. Opin. Infect. Dis. 2011, 24, 410–417. [Google Scholar] [CrossRef]
- Stelma, F.F.; Talla, I.; Sow, S.; Kongs, A.; Niang, M.; Polman, K.; Deelder, A.M.; Gryseels, B. Efficacy and side effects of praziquantel in an epidemic focus of Schistosoma mansoni. Am. J. Trop. Med. Hyg. 1995, 53, 167–170. [Google Scholar]
- Wang, W.; Wang, L.; Liang, Y.S. Susceptibility or resistance of praziquantel in human schistosomiasis: a Review. Parasitol. Res. 2012, 111, 1871–1877. [Google Scholar] [CrossRef]
- Caixeta, S.C.; Magalhaes, L.G.; de Melo, N.I.; Wakabayashi, K.A.; Aguiar Gde, P.; Aguiar Dde, P.; Mantovani, A.L.; Alves, J.M.; Oliveira, P.F.; Tavares, D.C.; et al. Chemical composition and in vitro schistosomicidal activity of the essential oil of Plectranthus neochilus grown in Southeast Brazil. Chem. Biodivers. 2011, 8, 2149–2157. [Google Scholar] [CrossRef]
- Moraes, J.; Almeida, A.A.; Brito, M.R.; Marques, T.H.; Lima, T.C.; Sousa, D.P.; Nakano, E.; Mendonca, R.Z.; Freitas, R.M. Anthelmintic activity of the natural compound (+)-limonene epoxide against Schistosoma mansoni. Planta Med. 2013, 79, 253–258. [Google Scholar] [CrossRef]
- Porto, T.S.; da Silva Filho, A.A.; Magalhaes, L.G.; dos Santos, R.A.; Furtado, N.A.; Arakawa, N.S.; Said, S.; de Oliveira, D.C.; Gregorio, L.E.; Rodrigues, V.; et al. Fungal transformation and schistosomicidal effects of pimaradienoic acid. Chem. Biodivers. 2012, 9, 1465–1474. [Google Scholar] [CrossRef]
- Ndjonka, D.; Rapado, L.N.; Silber, A.M.; Liebau, E.; Wrenger, C. Natural Products as a Source for Treating Neglected Parasitic Diseases. Int. J. Mol. Sci. 2013, 14, 3395–3439. [Google Scholar] [CrossRef]
- Jao, S.C.; Chen, J.; Yang, K.; Li, W.S. Design of potent inhibitors for Schistosoma japonica glutathione S-transferase. Bioorg. Med. Chem. 2006, 14, 304–318. [Google Scholar] [CrossRef]
- Rai, G.; Sayed, A.A.; Lea, W.A.; Luecke, H.F.; Chakrapani, H.; Prast-Nielsen, S.; Jadhav, A.; Leister, W.; Shen, M.; Inglese, J.; et al. Structure Mechanism Insights and the Role of Nitric Oxide Donation Guide the Development of Oxadiazole-2-Oxides as Therapeutic Agents against Schistosomiasis. J. Med. Chem. 2009, 52, 6474–6483. [Google Scholar] [CrossRef]
- Rai, G.; Thomas, C.J.; Leister, W.; Maloney, D.J. Synthesis of oxadiazole-2-oxide analogues as potential antischistosomal agents. Tetrahedron Lett. 2009, 50, 1710–1713. [Google Scholar] [CrossRef]
- Sayed, A.A.; Simeonov, A.; Thomas, C.J.; Inglese, J.; Austin, C.P.; Williams, D.L. Identification of oxadiazoles as new drug leads for the control of schistosomiasis. J. Nat. Med. 2008, 14, 407–412. [Google Scholar] [CrossRef]
- Liu, J.; Dyer, D.; Wang, J.; Wang, S.; Du, X.; Xu, B.; Zhang, H.; Wang, X.; Hu, W. 3-Oxoacyl-ACP Reductase from Schistosoma japonicum: Integrated In Silico-In Vitro Strategy for Discovering Antischistosomal Lead Compounds. PLoS One 2013, 8, e64984. [Google Scholar]
- Domling, A.; Khoury, K. Praziquantel and schistosomiasis. Chemmedchem 2010, 5, 1420–1434. [Google Scholar] [CrossRef]
- Woelfle, M.; Seerden, J.P.; de Gooijer, J.; Pouwer, K.; Olliaro, P.; Todd, M.H. Resolution of praziquantel. PLoS. Negl. Trop. Dis. 2011, 5, e1260. [Google Scholar] [CrossRef]
- Meyer, T.; Sekljic, H.; Fuchs, S.; Bothe, H.; Schollmeyer, D.; Miculka, C. Taste, a new incentive to switch to (R)-praziquantel in schistosomiasis treatment. PLoS Negl. Trop. Dis. 2009, 3, e357. [Google Scholar] [CrossRef]
- Cao, H.; Liu, H.; Domling, A. Efficient multicomponent reaction synthesis of the schistosomiasis drug praziquantel. Chemistry 2010, 16, 12296–12298. [Google Scholar] [CrossRef]
- Liu, H.; William, S.; Herdtweck, E.; Botros, S.; Domling, A. MCR synthesis of praziquantel derivatives. Chem. Biol. Drug. Des. 2012, 79, 470–477. [Google Scholar] [CrossRef]
- Andrews, P.; Thomas, H.; Pohlke, R.; Seubert, J. Praziquantel. Med. Res. Rev. 1983, 3, 147–200. [Google Scholar] [CrossRef]
- Patra, M.; Ingram, K.; Pierroz, V.; Ferrari, S.; Spingler, B.; Keiser, J.; Gasser, G. Ferrocenyl derivatives of the anthelmintic praziquantel: Design, synthesis, and biological evaluation. J. Med. Chem. 2012, 55, 8790–8798. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.X.; Chollet, J.; Vargas, M.; Mansour, N.R.; Bickle, Q.; Alnouti, Y.; Huang, J.G.; Keiser, J.; Vennerstrom, J.L. Praziquantel analogs with activity against juvenile Schistosoma mansoni. Bioorg. Med. Chem. Lett. 2010, 20, 2481–2484. [Google Scholar] [CrossRef]
- Patra, M.; Ingram, K.; Pierroz, V.; Ferrari, S.; Spingler, B.; Gasser, R.B.; Keiser, J.; Gasser, G. [(eta(6)-Praziquantel)Cr(CO)3] derivatives with remarkable in vitro anti-schistosomal activity. Chemistry 2013, 19, 2232–2235. [Google Scholar] [CrossRef]
- Ronketti, F.; Ramana, A.V.; Chao-Ming, X.; Pica-Mattoccia, L.; Cioli, D.; Todd, M.H. Praziquantel derivatives I: Modification of the aromatic ring. Bioorg. Med. Chem. Lett. 2007, 17, 4154–4157. [Google Scholar] [CrossRef]
- Duan, W.W.; Qiu, S.J.; Zhao, Y.; Sun, H.; Qiao, C.; Xia, C.M. Praziquantel derivatives exhibit activity against both juvenile and adult Schistosoma japonicum. Bioorg. Med. Chem. Lett. 2012, 22, 1587–1590. [Google Scholar] [CrossRef]
- Wang, Z.X.; Chen, J.L.; Qiao, C. Praziquantel Derivatives with Anti-Schistosomal Activity: Aromatic ring Modification. Chem. Biol. Drug. Des. 2013, 82, 216–225. [Google Scholar] [CrossRef]
- Sadhu, P.S.; Kumar, S.N.; Chandrasekharam, M.; Pica-Mattoccia, L.; Cioli, D.; Rao, V.J. Synthesis of new praziquantel analogues: Potential candidates for the treatment of schistosomiasis. Bioorg. Med. Chem. Lett. 2012, 22, 1103–1106. [Google Scholar] [CrossRef]
- Frehel, D.; Maffrand, J.P. Synthesis of the new tricyclic system thieno[3',2':3,4]pyrido[1,2-a]pyrazin-4-one. Heterocycles 1984, 22, 143–149. [Google Scholar] [CrossRef]
- Todd, M.H.; Ndubaku, C.; Bartlett, P.A. Amino acid derived heterocycles: Lewis acid catalyzed and radical cyclizations from peptide acetals. J. Org. Chem. 2002, 67, 3985–3988. [Google Scholar] [CrossRef]
- Bissinger, E.M.; Heinke, R.; Spannhoff, A.; Eberlin, A.; Metzger, E.; Cura, V.; Hassenboehler, P.; Cavarelli, J.; Schule, R.; Bedford, M.T.; et al. Acyl derivatives of p-aminosulfonamides and dapsone as new inhibitors of the arginine methyltransferase hPRMT1. Bioorg. Med. Chem. 2011, 19, 3717–3731. [Google Scholar] [CrossRef]
- Kim, D.J.; Reddy, K.; Kim, M.O.; Li, Y.; Nadas, J.; Cho, Y.Y.; Kim, J.E.; Shim, J.H.; Song, N.R.; Carper, A.; et al. (3-Chloroacetyl)-indole, a Novel allosteric AKT inhibitor, Suppresses colon cancer growth in vitro and in vivo. Cancer Prev. Res. 2011, 4, 1842–1851. [Google Scholar] [CrossRef]
- Nam, K.N.; Koketsu, M.; Lee, E.H. 5-Chloroacetyl-2-amino-1,3-selenazoles attenuate microglial inflammatory responses through NF-kappa B inhibition. Eur. J. Pharmacol. 2008, 589, 53–57. [Google Scholar] [CrossRef]
- Dezube, B.J.; Von Roenn, J.H.; Holden-Wiltse, J.; Cheung, T.W.; Remick, S.C.; Cooley, T.P.; Moore, J.; Sommadossi, J.P.; Shriver, S.L.; Suckow, C.W.; et al. Fumagillin analog in the treatment of Kaposi’s sarcoma: A phase I AIDS Clinical Trial Group Study. J. Clin. Oncol. 1998, 16, 1444–1449. [Google Scholar]
- Kruger, E.A.; Figg, W.D. TNP-470: An Angiogenesis inhibitor in clinical development for cancer. Expert Opin. Invest. Drug 2000, 9, 1383–1396. [Google Scholar] [CrossRef]
- Song, L.J.; Li, J.H.; Xie, S.Y.; Qian, C.Y.; Wang, J.; Zhang, W.; Yin, X.R.; Hua, Z.C.; Yu, C.X. Thioredoxin Glutathione Reductase as a Novel Drug Target: Evidence from Schistosoma japonicum. PLoS One 2012, 7, e31456. [Google Scholar]
- Sample Availability: Samples of the compounds 1–33 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, W.-L.; Song, L.-J.; Chen, X.; Yin, X.-R.; Fan, W.-H.; Wang, G.-P.; Yu, C.-X.; Feng, B. Synthesis and SAR Studies of Praziquantel Derivatives with Activity against Schistosoma japonicum. Molecules 2013, 18, 9163-9178. https://doi.org/10.3390/molecules18089163
Wang W-L, Song L-J, Chen X, Yin X-R, Fan W-H, Wang G-P, Yu C-X, Feng B. Synthesis and SAR Studies of Praziquantel Derivatives with Activity against Schistosoma japonicum. Molecules. 2013; 18(8):9163-9178. https://doi.org/10.3390/molecules18089163
Chicago/Turabian StyleWang, Wen-Long, Li-Jun Song, Xia Chen, Xu-Ren Yin, Wen-Hua Fan, Gu-Ping Wang, Chuan-Xin Yu, and Bainian Feng. 2013. "Synthesis and SAR Studies of Praziquantel Derivatives with Activity against Schistosoma japonicum" Molecules 18, no. 8: 9163-9178. https://doi.org/10.3390/molecules18089163