Structural Characterization of Intrinsically Disordered Proteins by NMR Spectroscopy

Abstract
:1. Introduction
2. Resonance Assignment
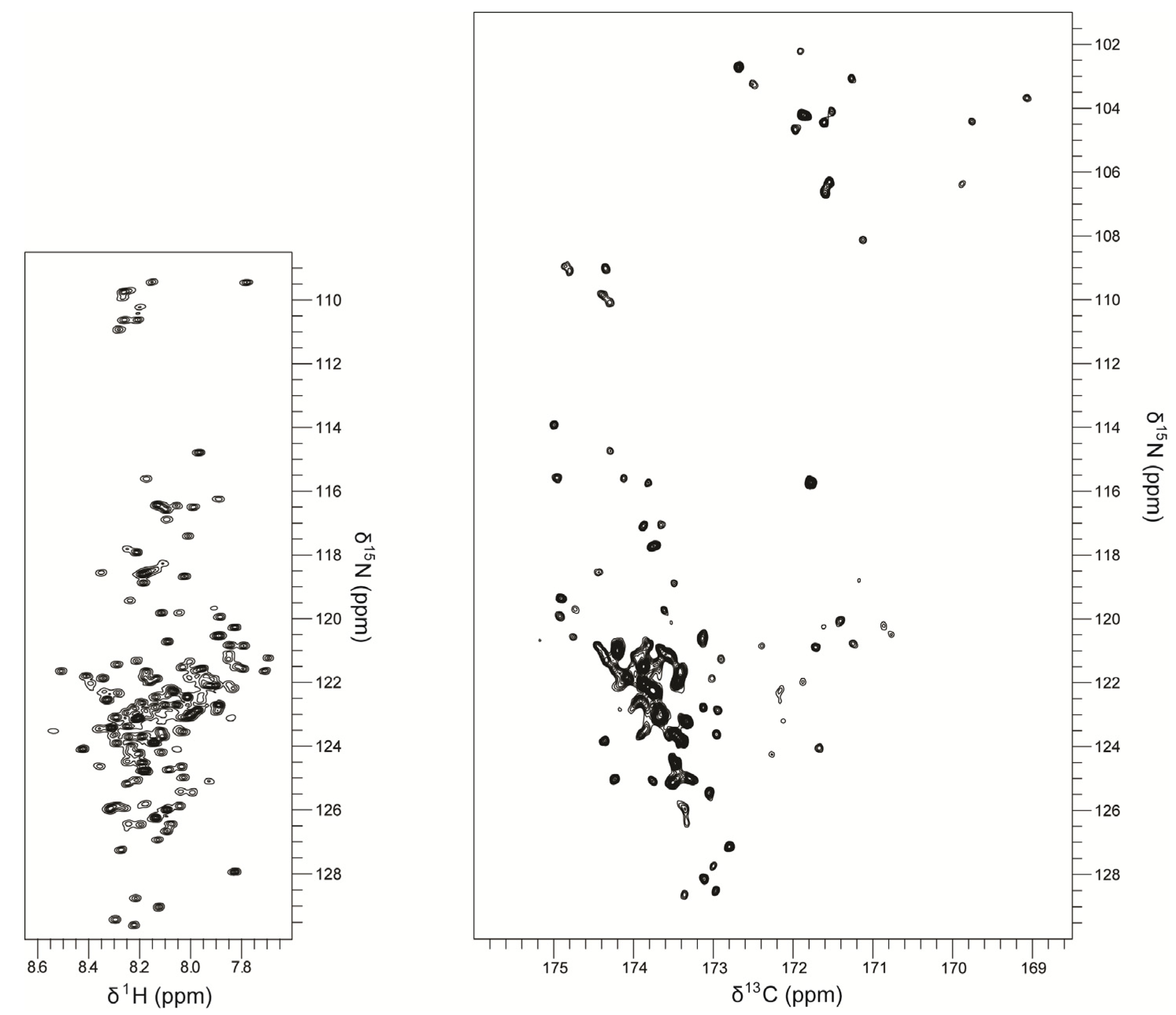
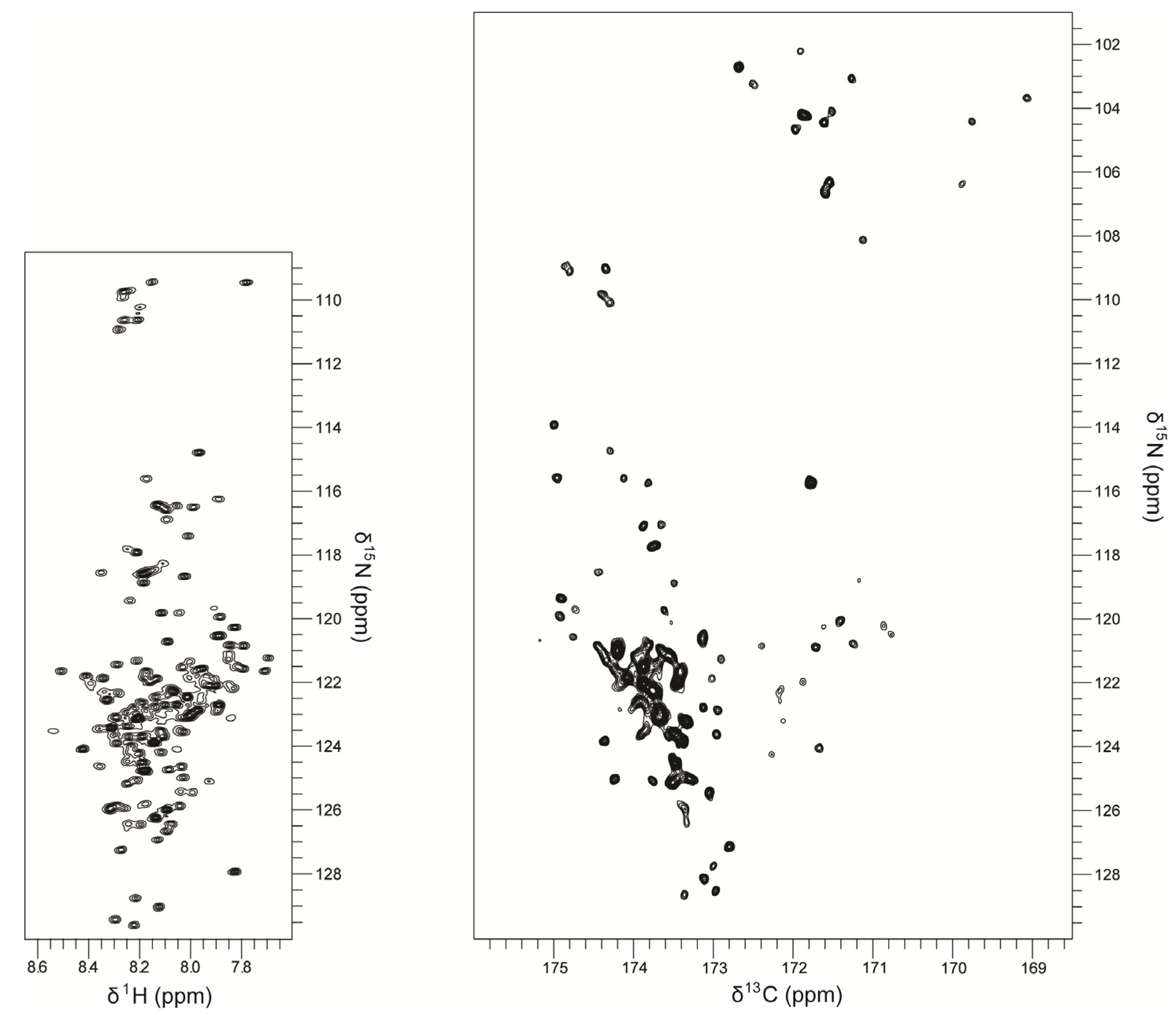
3. Structure and Dynamics Information from NMR Experiments
3.1. Structure Propensities—Local Structure
3.1.1. Chemical Shifts
3.1.2. Scalar Coupling
3.2. Combined Long and Short Range Contact Information
3.2.1. Residual Dipolar Coupling (RDC)
3.2.2. Nuclear Overhauser Effect (NOE)
3.3. Long-Range Contacts
3.3.1. Pulsed-Field Gradient (PFG) NMR
3.3.2. Paramagnetic Relaxation Enhancements (PREs)
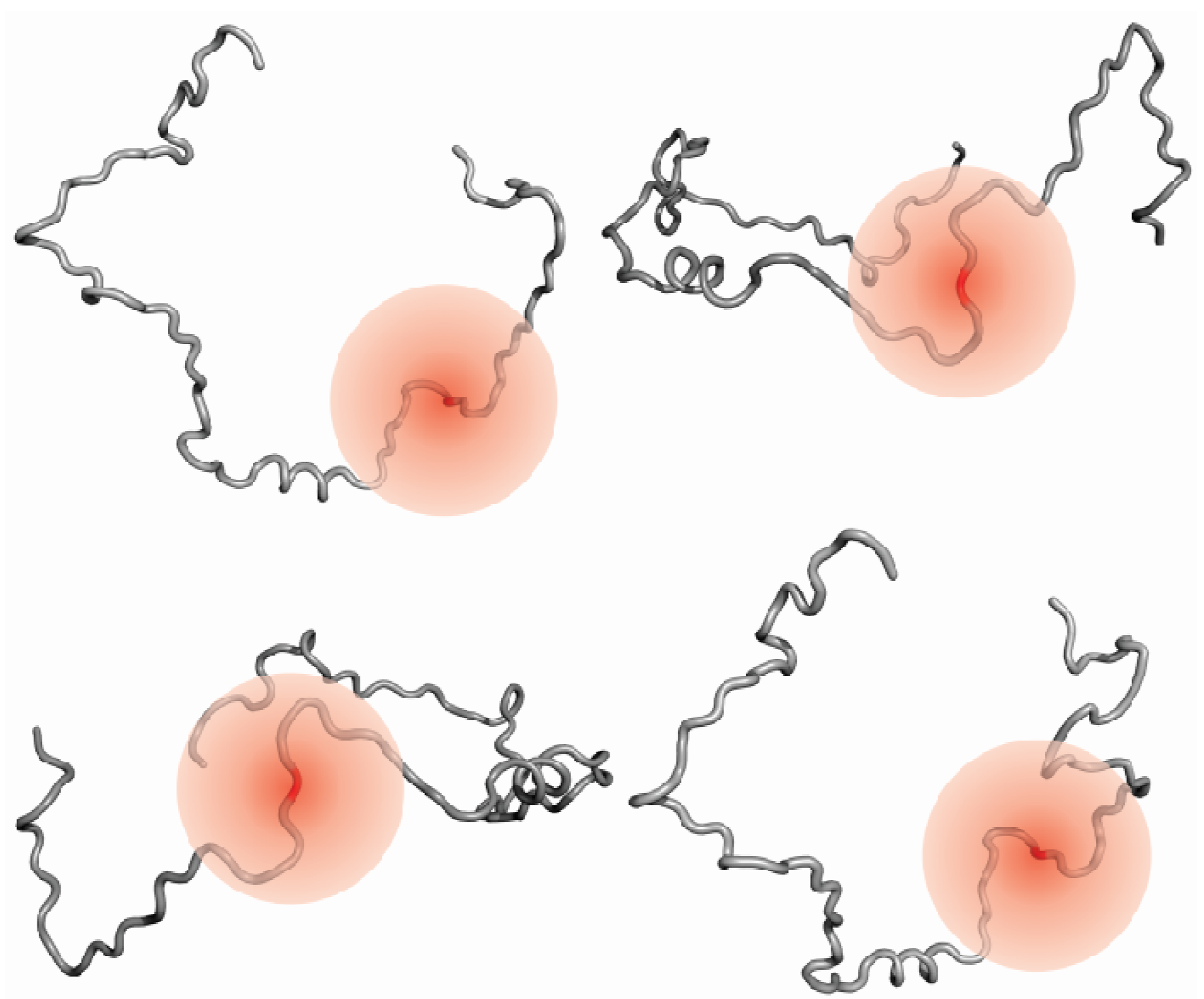
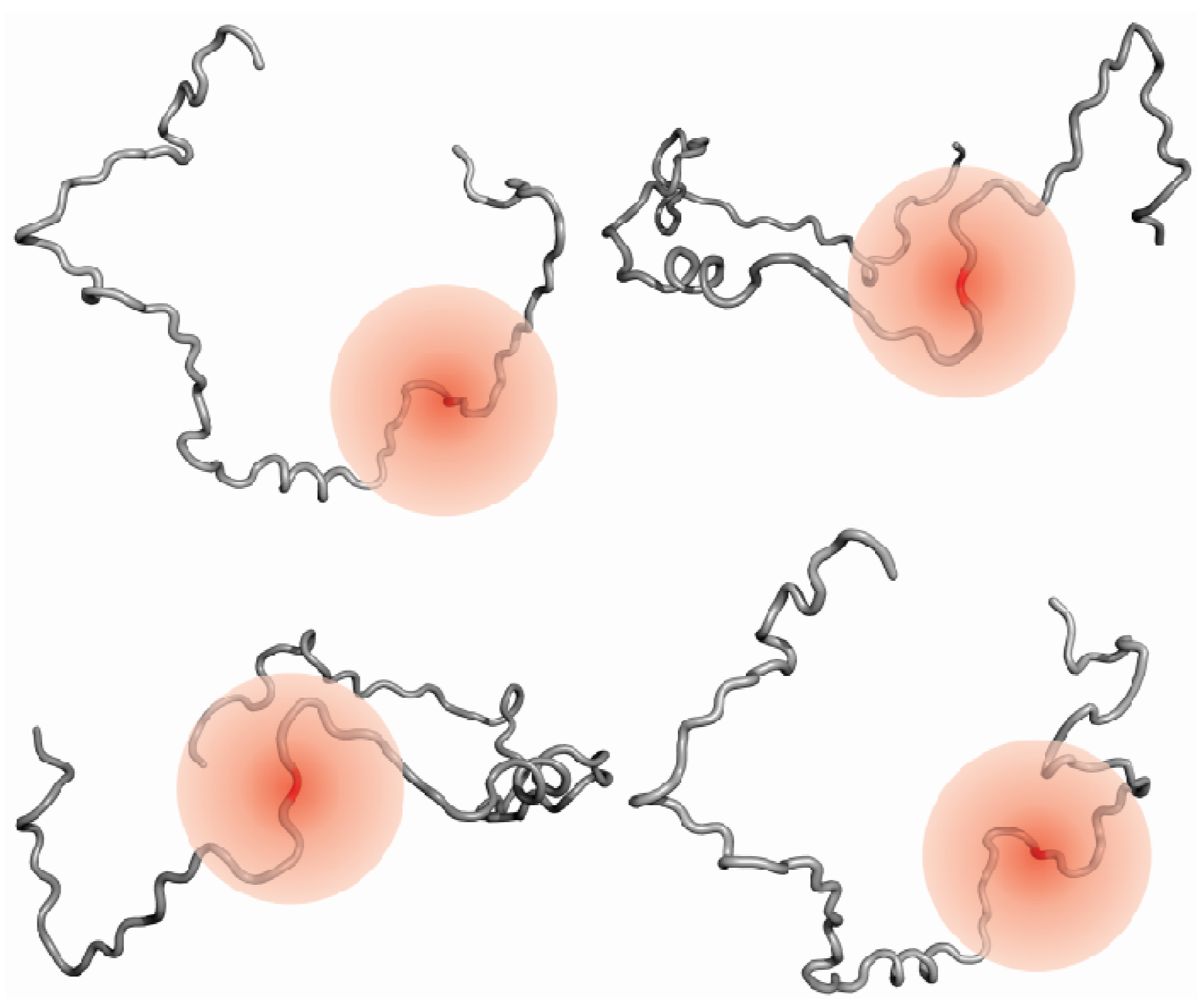
3.4. Information on Dynamics
3.4.1. 15N Relaxation
3.4.2. Heteronuclear NOEs
3.4.3. Relaxation Dispersion
4. 13C -Detected NMR for IDP Studies
4.1. Introduction: How 13C -Detection Can Help
4.2. Sequential Protein Backbone Assignment with 13C-detected Experiments

4.3. 13C-detected Amino-Acid-Selective (CAS) NMR Experiments
4.4. Probing Arginine Side-Chains
4.5. Studying Protein Dynamics Based on 13C-Detected Experiments
4.5.1. 13C-Detected 15N NMR Spin Relaxation
4.5.2. RDCs and J Couplings by 13C-Detected NMR Spectroscopy

{kind=link}
{kind=link}
{kind=link}
| Experiment Type | Use/Aim | Correlations Observed | J-Coupling Supression Used | [Protein] Used | Reference |
|---|---|---|---|---|---|
| 3D COCON | Backbone assignment | C'i-C'i-Ni+1, C'i-1,-C'i-Ni+1, C'i+1 -C'i -Ni+1 | IPAP | NA | [24] |
| 3D CBCACON | Backbone assignment | Cαi-C'i-Ni+1, Cßi-C'i-Ni+1 | IPAP | NA | [24] |
| 2D CON | Backbone assignment | C'i-Ni+1 | IPAP, SE-DIPAP | NA | [24] |
| 3D (H)CBCACON | Backbone assignment | Cαi-1-C'i-1-Ni, Cßi-1-C'i-1-Ni | IPAP | 0.7 mM | [97] |
| 3D (H)CBCANCO | Backbone assignment | Cαi-C'i-1-Ni, Cßi-C'i-1-Ni, Cßi-C'i-Ni+1, Cαi-C'i-Ni+1 | IPAP | 0.7 mM | [97] |
| 3D (H)NCANCO | Backbone assignment | Ni-Ni-C'i-1, Ni+1-Ni-Ci-1, Ni-1-Ni-C'i-1 | IPAP | 0.7 mM | [97] |
| 3D CANCO | Backbone assignment | Cαi-C'i-Ni+1, Cαi+1-C'i-Ni+1 | IPAP | 1–1.8 mM | [98] |
| 3D CACON | Backbone assignment | Cαi-C'i-Ni+1 | IPAP | 1–1.8 mM | [98] |
| 13Cζ-15Nε HSQC | Probing Arg side-chains | Cζi-Nεi | IPAP, CPD | 0.8–2.5 mM | [99] |
| 2D CON-type | Relaxation measurements RDC’s | C'i-Ni+1 | [102,104] | ||
| IPAP | 0.5 mM |
4.5.3. Amide Proton Solvent Exchange
5. In-Cell NMR Spectroscopy
| Cell line | Delivery | Protein studied | Studies | References |
|---|---|---|---|---|
| HeLa (human) COS7 (Chlorocebus) | Peptide tag | Ub, FKBP12, GB1 | Protein–drug interaction, enzymatic cleavage, H/D-exchange | [117] |
| 293-F (human) | Pore-forming toxin | thymosin ß4 | N-terminal acetylation | [118] |
| HeLa cells | α-synuclein | Conformation | [119] | |
| Xenopus laevis | Microinjection | Viral SV40 large T antigen regulatory region | Protein phosphorylation | [120] |
| Tau protein | Interaction with microtubules, protein phosphorylation | [112] | ||
| GB1 | Macromolecular crowding | [121] | ||
| Human embryonic kidney (HEK293T) | Overexpression | Human SOD1 | Monitor folding | [122] |
| Insect cells | Overexpression | G B1 | Chemical shift assignment | [123] |
| Pichia pastoris (budding yeast) | Overexpression | Ubiquitin | Assessment of critical parameters for the cell type, metabolism effect | [124] |
| Escherichia coli | Overexpression | NmerA | Proof of concept | [125] |
| Atox1 | Cis-platin transport | [126] | ||
| Ubiquitin | Protein-protein interaction | [127] | ||
| FKBP-FRB | Drug screening | [117] | ||
| Human SOD1 | Monitor folding | [128] | ||
| TTHA1718 | Structure determination | [115] | ||
| ProtL | Folding | [129] | ||
| Chymiotrypsin inhibitor 2 (CI2) | Correlation spectra | [107] | ||
| α-synuclein | 13C-direct detection | [130] |
5.1. Protein Delivery Methods for In-Cell NMR
5.2. Monitoring Post-Translational Modifications with In-Cell NMR
5.3. Protein Interactions
5.4. 13C direct Detection In-Cell
6. Conclusion and Outlook
Acknowledgments
Conflicts of Interest
References
- Pancsa, R.; Tompa, P. Structural disorder in eukaryotes. PLoS one 2012, 7, e34687. [Google Scholar] [CrossRef]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009, 19, 31–38. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Rezaei-Ghaleh, N.; Blackledge, M.; Zweckstetter, M. Intrinsically disordered proteins: From sequence and conformational properties toward drug discovery. ChemBioChem 2012, 13, 930–950. [Google Scholar] [CrossRef]
- Borcherds, W.; Kashtanov, S.; Wu, H.; Daughdrill, G.W. Structural divergence is more extensive than sequence divergence for a family of intrinsically disordered proteins. Proteins 2013. [Google Scholar] [CrossRef]
- Tompa, P. On the supertertiary structure of proteins. Nat. Chem. Biol. 2012, 8, 597–600. [Google Scholar] [CrossRef]
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: regulation and disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef]
- Hammoudeh, D.I.; Follis, A.V.; Prochownik, E.V.; Metallo, S.J. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. JACS 2009, 131, 7390–7401. [Google Scholar] [CrossRef]
- Lamberto, G.R.; Binolfi, A.; Orcellet, M.L.; Bertoncini, C.W.; Zweckstetter, M.; Griesinger, C.; Fernandez, C.O. Structural and mechanistic basis behind the inhibitory interaction of PcTS on alpha-synuclein amyloid fibril formation. PNAS 2009, 106, 21057–21062. [Google Scholar] [CrossRef]
- Eliezer, D. Biophysical characterization of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2009, 19, 23–30. [Google Scholar] [CrossRef]
- Ozenne, V.; Bauer, F.; Salmon, L.; Huang, J.R.; Jensen, M.R.; Segard, S.; Bernado, P.; Charavay, C.; Blackledge, M. Flexible-meccano: A tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 2012, 28, 1463–1470. [Google Scholar] [CrossRef]
- Fisher, C.K.; Stultz, C.M. Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2011, 21, 426–431. [Google Scholar] [CrossRef]
- Schneider, R.; Huang, J.R.; Yao, M.; Communie, G.; Ozenne, V.; Mollica, L.; Salmon, L.; Jensen, M.R.; Blackledge, M. Towards a robust description of intrinsic protein disorder using nuclear magnetic resonance spectroscopy. Mol. Biosyst. 2012, 8, 58–68. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Unfolded proteins and protein folding studied by NMR. Chem. Rev. 2004, 104, 3607–3622. [Google Scholar] [CrossRef]
- McCann, J.J.; Zheng, L.; Rohrbeck, D.; Felekyan, S.; Kuhnemuth, R.; Sutton, R.B.; Seidel, C.A.; Bowen, M.E. Supertertiary structure of the synaptic MAGuK scaffold proteins is conserved. PNAS 2012, 109, 15775–15780. [Google Scholar] [CrossRef]
- Zhang, J.; Lewis, S.M.; Kuhlman, B.; Lee, A.L. Supertertiary structure of the MAGUK core from PSD-95. Structure 2013, 21, 402–413. [Google Scholar] [CrossRef]
- Jensen, M.R.; Ruigrok, R.W.; Blackledge, M. Describing intrinsically disordered proteins at atomic resolution by NMR. Curr. Opin. Struct. Biol. 2013, 23, 426–435. [Google Scholar] [CrossRef]
- Sibille, N.; Bernado, P. Structural characterization of intrinsically disordered proteins by the combined use of NMR and SAXS. Biochem. Soc. Trans. 2012, 40, 955–962. [Google Scholar] [CrossRef]
- Felli, I.C.; Pierattelli, R. Recent progress in NMR spectroscopy: toward the study of intrinsically disordered proteins of increasing size and complexity. IUBMB life 2012, 64, 473–481. [Google Scholar] [CrossRef]
- Mantylahti, S.; Hellman, M.; Permi, P. Extension of the HA-detection based approach: (HCA)CON(CA)H and (HCA)NCO(CA)H experiments for the main-chain assignment of intrinsically disordered proteins. J. Biomol. NMR 2011, 49, 99–109. [Google Scholar] [CrossRef]
- Motackova, V.; Novacek, J.; Zawadzka-Kazimierczuk, A.; Kazimierczuk, K.; Zidek, L.; Sanderova, H.; Krasny, L.; Kozminski, W.; Sklenar, V. Strategy for complete NMR assignment of disordered proteins with highly repetitive sequences based on resolution-enhanced 5D experiments. J. Biomol. NMR 2010, 48, 169–177. [Google Scholar] [CrossRef]
- Zawadzka-Kazimierczuk, A.; Kozminski, W.; Sanderova, H.; Krasny, L. High dimensional and high resolution pulse sequences for backbone resonance assignment of intrinsically disordered proteins. J. Biomol. NMR 2012, 52, 329–337. [Google Scholar] [CrossRef]
- Bermel, W.; Bertini, I.; Felli, I.C.; Lee, Y.M.; Luchinat, C.; Pierattelli, R. Protonless NMR experiments for sequence-specific assignment of backbone nuclei in unfolded proteins. JACS 2006, 128, 3918–3919. [Google Scholar] [CrossRef]
- Narayanan, R.L.; Durr, U.H.; Bibow, S.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Automatic assignment of the intrinsically disordered protein Tau with 441-residues. JACS 2010, 132, 11906–11907. [Google Scholar] [CrossRef]
- Yao, J.; Dyson, H.J.; Wright, P.E. Chemical shift dispersion and secondary structure prediction in unfolded and partly folded proteins. FEBS Lett. 1997, 419, 285–289. [Google Scholar] [CrossRef]
- Otten, R.; Wood, K.; Mulder, F.A. Comprehensive determination of 3JHNHα for unfolded proteins using 13C'-resolved spin-echo difference spectroscopy. J. Biomol. NMR 2009, 45, 343–349. [Google Scholar] [CrossRef]
- Bermel, W.; Bruix, M.; Felli, I.C.; Kumar, M.V.V.; Pierattelli, R.; Serrano, S. Improving the chemical shift dispersion of multidimensional NMR spectra of intrinsically disordered proteins. J. Biomol. NMR 2013, 55, 231–237. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef]
- Lacy, E.R.; Filippov, I.; Lewis, W.S.; Otieno, S.; Xiao, L.; Weiss, S.; Hengst, L.; Kriwacki, R.W. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat. Struct. Mol. Biol. 2004, 11, 358–364. [Google Scholar] [CrossRef]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. PNAS 2008, 105, 5762–5767. [Google Scholar] [CrossRef]
- Mittag, T.; Marsh, J.; Grishaev, A.; Orlicky, S.; Lin, H.; Sicheri, F.; Tyers, M.; Forman-Kay, J.D. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure 2010, 18, 494–506. [Google Scholar] [CrossRef]
- Allison, J.R.; Varnai, P.; Dobson, C.M.; Vendruscolo, M. Determination of the free energy landscape of alpha-synuclein using spin label nuclear magnetic resonance measurements. JACS 2009, 131, 18314–18326. [Google Scholar] [CrossRef]
- De Simone, A.; Cavalli, A.; Hsu, S.T.; Vranken, W.; Vendruscolo, M. Accurate random coil chemical shifts from an analysis of loop regions in native states of proteins. JACS 2009, 131, 16332–16333. [Google Scholar]
- Tamiola, K.; Acar, B.; Mulder, F.A. Sequence-specific random coil chemical shifts of intrinsically disordered proteins. JACS 2010, 132, 18000–18003. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 1994, 4, 171–180. [Google Scholar]
- Braun, D.; Wider, G.; Wuthrich, K. Sequence-Corrected 15N Random Coil Chemical-Shifts. JACS 1994, 116, 8466–8469. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Poulsen, F.M. Sequence correction of random coil chemical shifts: correlation between neighbor correction factors and changes in the Ramachandran distribution. J. Biomol. NMR 2011, 50, 157–165. [Google Scholar] [CrossRef]
- Schwarzinger, S.; Kroon, G.J.; Foss, T.R.; Chung, J.; Wright, P.E.; Dyson, H.J. Sequence-dependent correction of random coil NMR chemical shifts. JACS 2001, 123, 2970–2978. [Google Scholar] [CrossRef]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar]
- Wang, L.; Eghbalnia, H.R.; Markley, J.L. Nearest-neighbor effects on backbone alpha and beta carbon chemical shifts in proteins. J. Biomol. NMR 2007, 39, 247–257. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Norholm, A.B.; Hendus-Altenburger, R.; Pedersen, S.F.; Poulsen, F.M.; Kragelund, B.B. Temperature-dependent structural changes in intrinsically disordered proteins: Formation of alpha-helices or loss of polyproline II? Protein Sci. 2010, 19, 1555–1564. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Poulsen, F.M. Disordered proteins studied by chemical shifts. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 60, 42–51. [Google Scholar] [CrossRef]
- Kragelj, J.; Ozenne, V.; Blackledge, M.; Jensen, M.R. Conformational Propensities of Intrinsically Disordered Proteins from NMR Chemical Shifts. Chemphyschem. 2013. [Google Scholar]
- Karplus, M. Contact Electron-Spin Coupling of Nuclear Magnetic Moments. J. Chem. Phys. 1959, 30, 11–15. [Google Scholar] [CrossRef]
- Smith, L.J.; Bolin, K.A.; Schwalbe, H.; MacArthur, M.W.; Thornton, J.M.; Dobson, C.M. Analysis of main chain torsion angles in proteins: prediction of NMR coupling constants for native and random coil conformations. J. Mol. Biol. 1996, 255, 494–506. [Google Scholar] [CrossRef]
- Permi, P.; Kilpelainen, I.; Annila, A. Determination of backbone angle psi in proteins using a TROSY-based alpha/beta-HN(CO)CA-J experiment. J. Magn. Reson. 2000, 146, 55–59. [Google Scholar]
- Permi, P. Determination of three-bond scalar coupling between 13C' and 1Hα in proteins using an iHN(CA),CO(alpha/beta-J-COHA) experiment. J. Magn. Reson. 2003, 163, 114–120. [Google Scholar] [CrossRef]
- Petit, A.; Vincent, S.J.; Zwahlen, C. Cosine modulated HSQC: a rapid determination of 3JHNHα scalar couplings in 15N-labeled proteins. J. Magn. Reson. 2002, 156, 313–317. [Google Scholar] [CrossRef]
- Ponstingl, H.; Otting, G. Rapid measurement of scalar three-bond 1HN-1H alpha spin coupling constants in 15N-labelled proteins. J. Biomol. NMR 1998, 12, 319–324. [Google Scholar] [CrossRef]
- Lendel, C.; Damberg, P. 3D J-resolved NMR spectroscopy for unstructured polypeptides: fast measurement of 3JHNHα coupling constants with outstanding spectral resolution. J. Biomol. NMR 2009, 44, 35–42. [Google Scholar] [CrossRef]
- Plaxco, K.W.; Morton, C.J.; Grimshaw, S.B.; Jones, J.A.; Pitkeathly, M.; Campbell, I.D.; Dobson, C.M. The effects of guanidine hydrochloride on the 'random coil' conformations and NMR chemical shifts of the peptide series GGXGG. J. Biomol. NMR 1997, 10, 221–230. [Google Scholar] [CrossRef]
- Tjandra, N.; Bax, A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 1997, 278, 1111–1114. [Google Scholar] [CrossRef]
- Jensen, M.R.; Markwick, P.R.; Meier, S.; Griesinger, C.; Zweckstetter, M.; Grzesiek, S.; Bernado, P.; Blackledge, M. Quantitative determination of the conformational properties of partially folded and intrinsically disordered proteins using NMR dipolar couplings. Structure 2009, 17, 1169–1185. [Google Scholar] [CrossRef]
- Bouvignies, G.; Markwick, P.R.; Blackledge, M. Simultaneous definition of high resolution protein structure and backbone conformational dynamics using NMR residual dipolar couplings. Chemphyschem. 2007, 8, 1901–1909. [Google Scholar] [CrossRef]
- Fischer, M.W.; Losonczi, J.A.; Weaver, J.L.; Prestegard, J.H. Domain orientation and dynamics in multidomain proteins from residual dipolar couplings. Biochemistry 1999, 38, 9013–9022. [Google Scholar] [CrossRef]
- Prestegard, J.H.; Bougault, C.M.; Kishore, A.I. Residual dipolar couplings in structure determination of biomolecules. Chem. Rev. 2004, 104, 3519–3540. [Google Scholar] [CrossRef]
- Mohana-Borges, R.; Goto, N.K.; Kroon, G.J.; Dyson, H.J.; Wright, P.E. Structural characterization of unfolded states of apomyoglobin using residual dipolar couplings. J. Mol. Biol. 2004, 340, 1131–1142. [Google Scholar] [CrossRef]
- Jensen, M.R.; Houben, K.; Lescop, E.; Blanchard, L.; Ruigrok, R.W.; Blackledge, M. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: application to the molecular recognition element of Sendai virus nucleoprotein. JACS 2008, 130, 8055–61. [Google Scholar]
- Salmon, L.; Nodet, G.; Ozenne, V.; Yin, G.; Jensen, M.R.; Zweckstetter, M.; Blackledge, M. NMR characterization of long-range order in intrinsically disordered proteins. JACS 2010, 132, 8407–8418. [Google Scholar] [CrossRef]
- Tjandra, N.; Omichinski, J.G.; Gronenborn, A.M.; Clore, G.M.; Bax, A. Use of dipolar 1H-15N and 1H-13C couplings in the structure determination of magnetically oriented macromolecules in solution. Nat. Struct. Biol. 1997, 4, 732–738. [Google Scholar] [CrossRef]
- Vendruscolo, M. Determination of conformationally heterogeneous states of proteins. Curr. Opin. Struct. Biol. 2007, 17, 15–20. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Dobson, C.M. Towards complete descriptions of the free-energy landscapes of proteins. Philos. Trans. R. Soc. London Ser. A 2005, 363, 433–450; Discussion 450–452. [Google Scholar] [CrossRef]
- Krzeminski, M.; Marsh, J.A.; Neale, C.; Choy, W.Y.; Forman-Kay, J.D. Characterization of disordered proteins with ENSEMBLE. Bioinformatics 2013, 29, 398–399. [Google Scholar] [CrossRef]
- Eliezer, D.; Barre, P.; Kobaslija, M.; Chan, D.; Li, X.; Heend, L. Residual structure in the repeat domain of tau: echoes of microtubule binding and paired helical filament formation. Biochemistry 2005, 44, 1026–1036. [Google Scholar] [CrossRef]
- Crowhurst, K.A.; Forman-Kay, J.D. Aromatic and methyl NOEs highlight hydrophobic clustering in the unfolded state of an SH3 domain. Biochemistry 2003, 42, 8687–8695. [Google Scholar] [CrossRef]
- Ekiel, I.; Abrahamson, M. Folding-related dimerization of human cystatin C. J. Biol. Chem. 1996, 271, 1314–1321. [Google Scholar] [CrossRef]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef]
- Waudby, C.A.; Mantle, M.D.; Cabrita, L.D.; Gladden, L.F.; Dobson, C.M.; Christodoulou, J. Rapid distinction of intracellular and extracellular proteins using NMR diffusion measurements. JACS 2012, 134, 11312–11315. [Google Scholar]
- Gillespie, J.R.; Shortle, D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. I. Paramagnetic relaxation enhancement by nitroxide spin labels. J. Mol. Biol. 1997, 268, 158–169. [Google Scholar] [CrossRef]
- Lietzow, M.A.; Jamin, M.; Dyson, H.J.; Wright, P.E. Mapping long-range contacts in a highly unfolded protein. J. Mol. Biol. 2002, 322, 655–662. [Google Scholar] [CrossRef]
- Iwahara, J.; Tang, C.; Marius Clore, G. Practical aspects of 1H transverse paramagnetic relaxation enhancement measurements on macromolecules. J. Magn. Reson. 2007, 184, 185–195. [Google Scholar] [CrossRef]
- Clore, G.M.; Tang, C.; Iwahara, J. Elucidating transient macromolecular interactions using paramagnetic relaxation enhancement. Curr. Opin. Struct. Biol. 2007, 17, 603–616. [Google Scholar] [CrossRef]
- Pintacuda, G.; Otting, G. Identification of protein surfaces by NMR measurements with a pramagnetic Gd(III) chelate. JACS 2002, 124, 372–373. [Google Scholar] [CrossRef]
- Su, X.C.; Otting, G. Paramagnetic labelling of proteins and oligonucleotides for NMR. J. Biomol. NMR 2010, 46, 101–112. [Google Scholar] [CrossRef]
- Akoury, E.; Gajda, M.; Pickhardt, M.; Biernat, J.; Soraya, P.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Inhibition of tau filament formation by conformational modulation. JACS 2013, 135, 2853–28562. [Google Scholar] [CrossRef]
- Bibow, S.; Ozenne, V.; Biernat, J.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. JACS 2011, 133, 15842–15845. [Google Scholar] [CrossRef]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodoulou, J.; Vendruscolo, M.; Dobson, C.M. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. JACS 2005, 127, 476–477. [Google Scholar] [CrossRef]
- Zangger, K.; Respondek, M.; Gobl, C.; Hohlweg, W.; Rasmussen, K.; Grampp, G.; Madl, T. Positioning of micelle-bound peptides by paramagnetic relaxation enhancements. J. Phys. Chem. B 2009, 113, 4400–4406. [Google Scholar]
- Grossauer, J.; Kosol, S.; Schrank, E.; Zangger, K. The peptide hormone ghrelin binds to membrane-mimetics via its octanoyl chain and an adjacent phenylalanine. Bioorg. Med. Chem. 2010, 18, 5483–5488. [Google Scholar] [CrossRef]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef]
- Palmer, A.G., 3rd. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004, 104, 3623–3640. [Google Scholar] [CrossRef]
- Kim, S.; Wu, K.P.; Baum, J. Fast hydrogen exchange affects 15N relaxation measurements in intrinsically disordered proteins. J. Biomol. NMR 2013, 55, 249–256. [Google Scholar] [CrossRef]
- Rezaei-Ghaleh, N.; Giller, K.; Becker, S.; Zweckstetter, M. Effect of zinc binding on beta-amyloid structure and dynamics: implications for Abeta aggregation. Biophys. J. 2011, 101, 1202–1211. [Google Scholar] [CrossRef]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef]
- Korzhnev, D.M.; Religa, T.L.; Banachewicz, W.; Fersht, A.R.; Kay, L.E. A transient and low-populated protein-folding intermediate at atomic resolution. Science 2010, 329, 1312–1316. [Google Scholar] [CrossRef]
- Sugase, K.; Dyson, H.J.; Wright, P.E. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 2007, 447, 1021–1025. [Google Scholar] [CrossRef]
- Palmer, A.G., 3rd; Kroenke, C.D.; Loria, J.P. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Meth. Enzymol. 2001, 339, 204–238. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Kay, L.E. NMR spectroscopy brings invisible protein states into focus. Nat. Chem. Biol. 2009, 5, 808–814. [Google Scholar] [CrossRef]
- Bermel, W.; Felli, I.C.; Kummerle, R.; Pierattelli, R. 13C direct-detection biomolecular NMR. Concept. Magn. Reson. A 2008, 32A, 183–200. [Google Scholar] [CrossRef]
- Bax, A.; Griffey, R.H.; Hawkins, B.L. Correlation of Proton and 15N Chemical-Shifts by Multiple Quantum Nmr. J. Magn. Reson. 1983, 55, 301–315. [Google Scholar]
- Kovacs, H.; Moskau, D.; Spraul, M. Cryogenically cooled probes - a leap in NMR technology. Prog. Nucl. Mag. Res. Sp. 2005, 46, 131–155. [Google Scholar] [CrossRef]
- Serber, Z.; Richter, C.; Moskau, D.; Bohlen, J.M.; Gerfin, T.; Marek, D.; Haberli, M.; Baselgia, L.; Laukien, F.; Stern, A.S.; Hoch, J.C.; Dotsch, V. New carbon-detected protein NMR experiments using CryoProbes. JACS 2000, 122, 3554–3555. [Google Scholar] [CrossRef]
- Bertini, I.; Duma, L.; Felli, I.C.; Fey, M.; Luchinat, C.; Pierattelli, R.; Vasos, P.R. A heteronuclear direct-detection NMR spectroscopy experiment for protein-backbone assignment. Angew. Chem. Int. Edit. 2004, 43, 2257–2259. [Google Scholar] [CrossRef]
- Bermel, W.; Bertini, I.; Duma, L.; Felli, I.C.; Emsley, L.; Pierattelli, R.; Vasos, P.R. Complete assignment of heteronuclear protein resonances by protonless NMR spectroscopy. Angewandte Chemie 2005, 44, 3089–3092. [Google Scholar] [CrossRef]
- Bermel, W.; Bertini, I.; Felli, I.C.; Matzapetakis, M.; Pierattelli, R.; Theil, E.C.; Turano, P. A method for C(alpha) direct-detection in protonless NMR. J. Magn. Reson. 2007, 188, 301–310. [Google Scholar] [CrossRef]
- Bermel, W.; Bertini, I.; Csizmok, V.; Felli, I.C.; Pierattelli, R.; Tompa, P. H-start for exclusively heteronuclear NMR spectroscopy: the case of intrinsically disordered proteins. J. Magn. Reson. 2009, 198, 275–281. [Google Scholar] [CrossRef]
- Bermel, W.; Bertini, I.; Chill, J.; Felli, I.C.; Haba, N.; Kumar, M.V.V.; Pierattelli, R. Exclusively heteronuclear 13C-detected amino-acid-selective NMR experiments for the study of intrinsically disordered proteins (IDPs). Chembiochem. 2012, 13, 2425–2432. [Google Scholar] [CrossRef]
- Werbeck, N.D.; Kirkpatrick, J.; Hansen, D.F. Probing arginine side-chains and their dynamics with carbon-detected NMR spectroscopy: Application to the 42 kDa human histone deacetylase 8 at high pH. Angewandte Chemie 2013, 52, 3145–3147. [Google Scholar] [CrossRef]
- Andre, I.; Linse, S.; Mulder, F.A. Residue-specific pKa determination of lysine and arginine side chains by indirect 15N and 13C NMR spectroscopy: application to apo calmodulin. JACS 2007, 129, 15805–15813. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. The case for intrinsically disordered proteins playing contributory roles in molecular recognition without a stable 3D structure. F1000 Biol. Rep. 2013, 5, 1. [Google Scholar]
- Lawrence, C.W.; Showalter, S.A. Carbon-Detected 15N NMR Spin Relaxation of an Intrinsically Disordered Protein: FCP1 Dynamics Unbound and in Complex with RAP74. J. Phys. Chem. Lett. 2012, 3, 1409–1413. [Google Scholar] [CrossRef]
- Balayssac, S.; Bertini, I.; Luchinat, C.; Parigi, G.; Piccioli, M. 13C direct detected NMR increases the detectability of residual dipolar couplings. JACS 2006, 128, 15042–15043. [Google Scholar]
- Bermel, W.; Bertini, I.; Felli, I.C.; Peruzzini, R.; Pierattelli, R. Exclusively heteronuclear NMR experiments to obtain structural and dynamic information on proteins. Chemphyschem. 2010, 11, 689–695. [Google Scholar] [CrossRef]
- Bertini, I.; Felli, I.C.; Gonnelli, L.; Vasantha Kumar, M.V.; Pierattelli, R. High-resolution characterization of intrinsic disorder in proteins: expanding the suite of 13C-detected NMR spectroscopy experiments to determine key observables. Chembiochem. 2011, 12, 2347–2352. [Google Scholar] [CrossRef]
- Cino, E.A.; Karttunen, M.; Choy, W.Y. Effects of molecular crowding on the dynamics of intrinsically disordered proteins. PloS One 2012, 7, e49876. [Google Scholar]
- Li, C.; Charlton, L.M.; Lakkavaram, A.; Seagle, C.; Wang, G.; Young, G.B.; Macdonald, J.M.; Pielak, G.J. Differential dynamical effects of macromolecular crowding on an intrinsically disordered protein and a globular protein: implications for in-cell NMR spectroscopy. JACS 2008, 130, 6310–6311. [Google Scholar] [CrossRef]
- Szasz, C.S.; Alexa, A.; Toth, K.; Rakacs, M.; Langowski, J.; Tompa, P. Protein disorder prevails under crowded conditions. Biochemistry 2011, 50, 5834–5844. [Google Scholar] [CrossRef]
- Johansen, D.; Jeffries, C.M.; Hammouda, B.; Trewhella, J.; Goldenberg, D.P. Effects of macromolecular crowding on an intrinsically disordered protein characterized by small-angle neutron scattering with contrast matching. Biophys. J. 2011, 100, 1120–1108. [Google Scholar] [CrossRef]
- Flaugh, S.L.; Lumb, K.J. Effects of macromolecular crowding on the intrinsically disordered proteins c-Fos and p27(Kip1). Biomacromolecules 2001, 2, 538–540. [Google Scholar] [CrossRef]
- Amata, I.; Maffei, M.; Igea, A.; Gay, M.; Vilaseca, M.; Nebreda, A.R.; Pons, M. Multi-phosphorylation of the Intrinsically Disordered Unique Domain of c-Src Studied by In-Cell and Real-Time NMR Spectroscopy. ChemBioChem 2013. [Google Scholar] [CrossRef]
- Bodart, J.F.; Wieruszeski, J.M.; Amniai, L.; Leroy, A.; Landrieu, I.; Rousseau-Lescuyer, A.; Vilain, J.P.; Lippens, G. NMR observation of Tau in Xenopus oocytes. J. Magn. Reson. 2008, 192, 252–257. [Google Scholar] [CrossRef]
- Burz, D.S.; Dutta, K.; Cowburn, D.; Shekhtman, A. In-cell NMR for protein-protein interactions (STINT-NMR). Nat. Protoc. 2006, 1, 146–152. [Google Scholar] [CrossRef]
- Li, C.; Liu, M. Protein dynamics in living cells studied by in-cell NMR spectroscopy. FEBS Lett. 2013, 587, 1008–1011. [Google Scholar] [CrossRef]
- Sakakibara, D.; Sasaki, A.; Ikeya, T.; Hamatsu, J.; Hanashima, T.; Mishima, M.; Yoshimasu, M.; Hayashi, N.; Mikawa, T.; Walchli, M.; Smith, B.O.; Shirakawa, M.; Guntert, P.; Ito, Y. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature 2009, 458, 102–105. [Google Scholar] [CrossRef]
- Maldonado, A.Y.; Burz, D.S.; Shekhtman, A. In-cell NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc 2011, 59, 197–212. [Google Scholar] [CrossRef]
- Inomata, K.; Ohno, A.; Tochio, H.; Isogai, S.; Tenno, T.; Nakase, I.; Takeuchi, T.; Futaki, S.; Ito, Y.; Hiroaki, H.; Shirakawa, M. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature 2009, 458, 106–109. [Google Scholar] [CrossRef]
- Ogino, S.; Kubo, S.; Umemoto, R.; Huang, S.; Nishida, N.; Shimada, I. Observation of NMR signals from proteins introduced into living mammalian cells by reversible membrane permeabilization using a pore-forming toxin, streptolysin O. JACS 2009, 131, 10834–10835. [Google Scholar]
- Bekei, B.; Rose, H.M.; Herzig, M.; Selenko, P. In-cell NMR in mammalian cells: part 2. Methods Mol. Biol. 2012, 895, 55–66. [Google Scholar] [CrossRef]
- Selenko, P.; Frueh, D.P.; Elsaesser, S.J.; Haas, W.; Gygi, S.P.; Wagner, G. In situ observation of protein phosphorylation by high-resolution NMR spectroscopy. Nat. Struct. Mol. Biol. 2008, 15, 321–329. [Google Scholar] [CrossRef]
- Selenko, P.; Serber, Z.; Gadea, B.; Ruderman, J.; Wagner, G. Quantitative NMR analysis of the protein G B1 domain in Xenopus laevis egg extracts and intact oocytes. PNAS 2006, 103, 11904–11909. [Google Scholar]
- Banci, L.; Barbieri, L.; Bertini, I.; Luchinat, E.; Secci, E.; Zhao, Y.; Aricescu, A.R. Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat. Chem. Biol. 2013, 9, 297–299. [Google Scholar] [CrossRef]
- Hamatsu, J.; O'Donovan, D.; Tanaka, T.; Shirai, T.; Hourai, Y.; Mikawa, T.; Ikeya, T.; Mishima, M.; Boucher, W.; Smith, B.O.; et al. High-resolution heteronuclear multidimensional NMR of proteins in living insect cells using a baculovirus protein expression system. JACS 2013, 135, 1688–1691. [Google Scholar] [CrossRef]
- Bertrand, K.; Reverdatto, S.; Burz, D.S.; Zitomer, R.; Shekhtman, A. Structure of proteins in eukaryotic compartments. JACS 2012, 134, 12798–12806. [Google Scholar]
- Serber, Z.; Ledwidge, R.; Miller, S.M.; Dotsch, V. Evaluation of parameters critical to observing proteins inside living Escherichia coli by in-cell NMR spectroscopy. JACS 2001, 123, 8895–8901. [Google Scholar] [CrossRef]
- Arnesano, F.; Banci, L.; Bertini, I.; Felli, I.C.; Losacco, M.; Natile, G. Probing the interaction of cisplatin with the human copper chaperone Atox1 by solution and in-cell NMR spectroscopy. JACS 2011, 133, 18361–18369. [Google Scholar]
- Burz, D.S.; Shekhtman, A. The STINT-NMR method for studying in-cell protein-protein interactions. Curr. Protoc. Protein Sci. 2010. [Google Scholar] [CrossRef]
- Banci, L.; Barbieri, L.; Bertini, I.; Cantini, F.; Luchinat, E. In-cell NMR in E. coli to monitor maturation steps of hSOD1. PLoS one 2011, 6, e23561. [Google Scholar]
- Schlesinger, A.P.; Wang, Y.; Tadeo, X.; Millet, O.; Pielak, G.J. Macromolecular crowding fails to fold a globular protein in cells. JACS 2011, 133, 8082–8085. [Google Scholar] [CrossRef]
- Bertini, I.; Felli, I.C.; Gonnelli, L.; Kumar, M.V. V.; Pierattelli, R. 13C direct-detection biomolecular NMR spectroscopy in living cells. Angewandte Chemie 2011, 50, 2339–2341. [Google Scholar] [CrossRef]
- Bekei, B.; Rose, H.M.; Herzig, M.; Dose, A.; Schwarzer, D.; Selenko, P. In-cell NMR in mammalian cells: part 1. Methods Mol. Biol. 2012, 895, 43–54. [Google Scholar] [CrossRef]
- Theillet, F.X.; Smet-Nocca, C.; Liokatis, S.; Thongwichian, R.; Kosten, J.; Yoon, M.K.; Kriwacki, R.W.; Landrieu, I.; Lippens, G.; Selenko, P. Cell signaling, post-translational protein modifications and NMR spectroscopy. J. Biomol. NMR 2012, 54, 217–236. [Google Scholar] [CrossRef]
- Liokatis, S.; Dose, A.; Schwarzer, D.; Selenko, P. Simultaneous detection of protein phosphorylation and acetylation by high-resolution NMR spectroscopy. JACS 2010, 132, 14704–14705. [Google Scholar] [CrossRef]
- Theillet, F.X.; Liokatis, S.; Jost, J.O.; Bekei, B.; Rose, H.M.; Binolfi, A.; Schwarzer, D.; Selenko, P. Site-specific mapping and time-resolved monitoring of lysine methylation by high-resolution NMR spectroscopy. JACS 2012, 134, 7616–7619. [Google Scholar] [CrossRef]
- Jensen, M.R.; Communie, G.; Ribeiro, E.A., Jr.; Martinez, N.; Desfosses, A.; Salmon, L.; Mollica, L.; Gabel, F.; Jamin, M.; Longhi, S.; et al. Intrinsic disorder in measles virus nucleocapsids. PNAS 2011, 108, 9839–9844. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kosol, S.; Contreras-Martos, S.; Cedeño, C.; Tompa, P. Structural Characterization of Intrinsically Disordered Proteins by NMR Spectroscopy. Molecules 2013, 18, 10802-10828. https://doi.org/10.3390/molecules180910802
Kosol S, Contreras-Martos S, Cedeño C, Tompa P. Structural Characterization of Intrinsically Disordered Proteins by NMR Spectroscopy. Molecules. 2013; 18(9):10802-10828. https://doi.org/10.3390/molecules180910802
Chicago/Turabian StyleKosol, Simone, Sara Contreras-Martos, Cesyen Cedeño, and Peter Tompa. 2013. "Structural Characterization of Intrinsically Disordered Proteins by NMR Spectroscopy" Molecules 18, no. 9: 10802-10828. https://doi.org/10.3390/molecules180910802