3.1. Chemistry
All chemicals were reagent grade and used as purchased. All reactions were performed under an inert atmosphere of dry argon or nitrogen using distilled dry solvent. 1H (400 MHz) NMR spectra were recorded on a Bruker AVⅢ 400 MHz spectrometer. The chemical shifts were reported in (ppm) using the 7.26 signal of CDCl3 (1H-NMR) and the 2.50 signal of DMSO-d6 (1H-NMR) as internal standards and the 39.50 signal of DMSO-d6 (13C-NMR) as internal standards. ESI Mass spectra (MS) were obtained on a Waters Micromass Platform LCZ Mass Spectrometer. Melting points were recorded on YRT-3 melting point apparatus (Tianjin Reliant Instrument Co., Ltd., Tianjin, China) and were reported without correction.
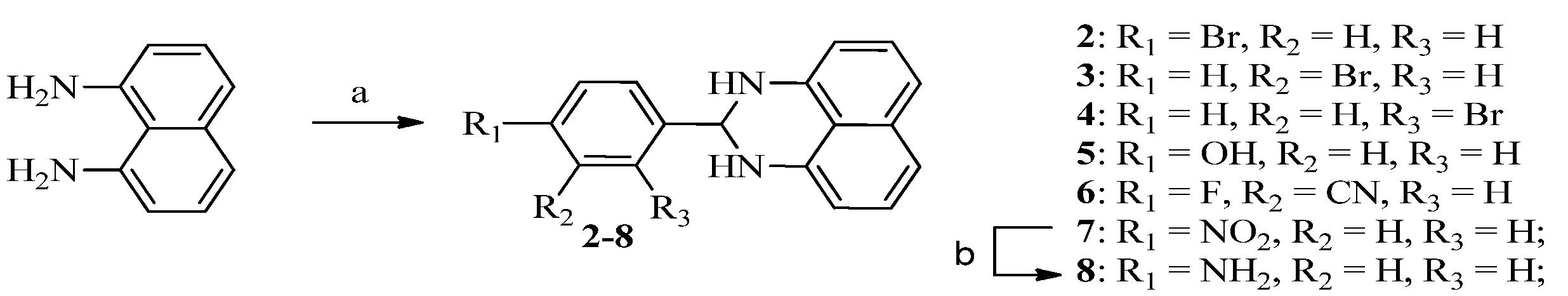
3.1.1. Procedure for the Preparation of Compounds 2–7
To a stirred solution of 4-bromobenzaldehyde (421.8 mg, 2.28 mmol) in methanol (5 mL) was added a solution of naphthalene-1,8-diamine (300 mg, 1.90 mmol) in methanol (5 mL), followed by Zn(OAc)2·H2O (3.5 mg, 0.016 mmol). Then the mixture was stirred at room temperature for 16 h. The reaction mixture was filtered. The filter cake was washed with methanol, dried to get compound 2 (277 mg, 45%) as a brown solid, mp 138.9–142.3 °C. 1H-NMR (CDCl3) δ: 4.68 (brs, 2H), 5.45 (s, 1H), 6.54 (dd, J = 1.6 Hz, 6.4 Hz, 2H), 7.25(m, 4H), 7.52 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 8.4 Hz, 2H); MS (ESI): m/z calcd. for C17H14BrN2[M+H]+ 325.0/327.0, found: 325.2/327.5.
2-(3-Bromophenyl)-2,3-dihydro-1H-perimidine (3). Yield = 48%, mp 161.7–163.3 °C; 1H-NMR (CDCl3) δ: 4.51 (brs, 2H), 5.44 (s, 1H), 6.54 (dd, J = 1.6 Hz, 6.8 Hz, 2H), 7.23–7.34 (m, 5H), 7.55–7.59 (m, 2H), 7.83 (s, 1H); MS (ESI): m/z calcd. for C17H14BrN2 [M+H]+ 325.0/327.0, found: 325.5/327.5.
2-(2-Bromophenyl)-2,3-dihydro-1H-perimidine (4). Yield = 46%, mp 130.4–131.0 °C;1H-NMR (CDCl3) δ: 4.62 (s, 2H), 5.94 (s, 1H), 6.58 (d, J = 7.2 Hz, 2H), 7.23–7.30 (m, 5H), 7.39 (t, J = 7.6 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.84 (dd, J = 1.2 Hz, 7.6 Hz, 1H); MS (ESI): m/z calcd. for C17H14BrN2 [M+H]+ 325.0/327.0, found: 325.4/327.7.
4-(2,3-Dihydro-1H-perimidin-2-yl)phenol (5). Yield = 52%, mp 148.1–152.2 °C; 1H-NMR (DMSO-d6) δ: 5.25 (s, 1H), 6.48 (d, J = 7.2 Hz, 2H), 6.62 (s, 2H), 6.82 (d, J = 8.0 Hz, 2H), 6.98 (d, J = 8.4 Hz, 2H), 7.14 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.41(d, J = 8.4 Hz, 2H), 9.54 (s, 1H); MS (ESI): m/z calcd. for C17H15N2O[M+H]+ 263.1, found: 263.2.
5-(2,3-Dihydro-1H-perimidin-2-yl)-2-fluorobenzonitrile (6). Yield = 55%, mp 204.4–208.4 °C; 1H-NMR (DMSO-d6) δ: 5.44 (s, 1H), 6.50 (d, J = 7.6 Hz, 2H), 6.90 (s, 2H), 7.01(d, J = 7.6 Hz, 2H), 7.17 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.60 (t, J = 9.2 Hz, 1H), 7.99 (dt, J = 2.4 Hz, 6.8 Hz, 1H), 8.14 (dd, J = 2.4 Hz, 6.4 Hz, 1H); MS (ESI): m/z calcd. forC18H13FN3[M+H]+ 290.1, found: 290.3.
2-(4-Nitrophenyl)-2,3-dihydro-1H-perimidine (7). Yield = 54%; 1H-NMR (DMSO-d6) δ: 5.50 (s, 1H), 6.52 (d, J = 7.2 Hz, 2H), 6.99 (s, 2H), 7.01 (d, J = 8.4 Hz, 2H), 7.18 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.85 (d, J = 8.8 Hz, 2H), 8.28 (d, J = 8.8 Hz, 2H); MS (ESI): m/z calcd. for C17H14N3O2 [M+H]+ 292.1, found: 292.5.
3.1.2. Procedure for the Preparation of Compound 8
The mixture of compound 7 (100 mg, 0.34 mmol), iron (38.5 mg, 0.69 mmol) and NH4Cl (55.2 mg, 1.03 mmol) in the solution of ethanol (2 mL) and water (1 mL) was heated at 90 °C for 3 h. After filtration, the filter cake was washed with EtOAc, concentrated the filtrate, and dried to afford compound 8 (80 mg, 89%), mp 166.4–171.9 °C. 1H-NMR (DMSO-d6) δ: 5.17 (s, 1H), 6.47 (d, J = 8.0 Hz, 2H), 6.53 (s, 2H), 6.62 (d, J = 8.0 Hz, 2H), 6.96 (d, J = 8.0 Hz, 2H), 7.13 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H); MS (ESI): m/z calcd. for C17H16N3[M+H]+ 262.1, found: 262.1.
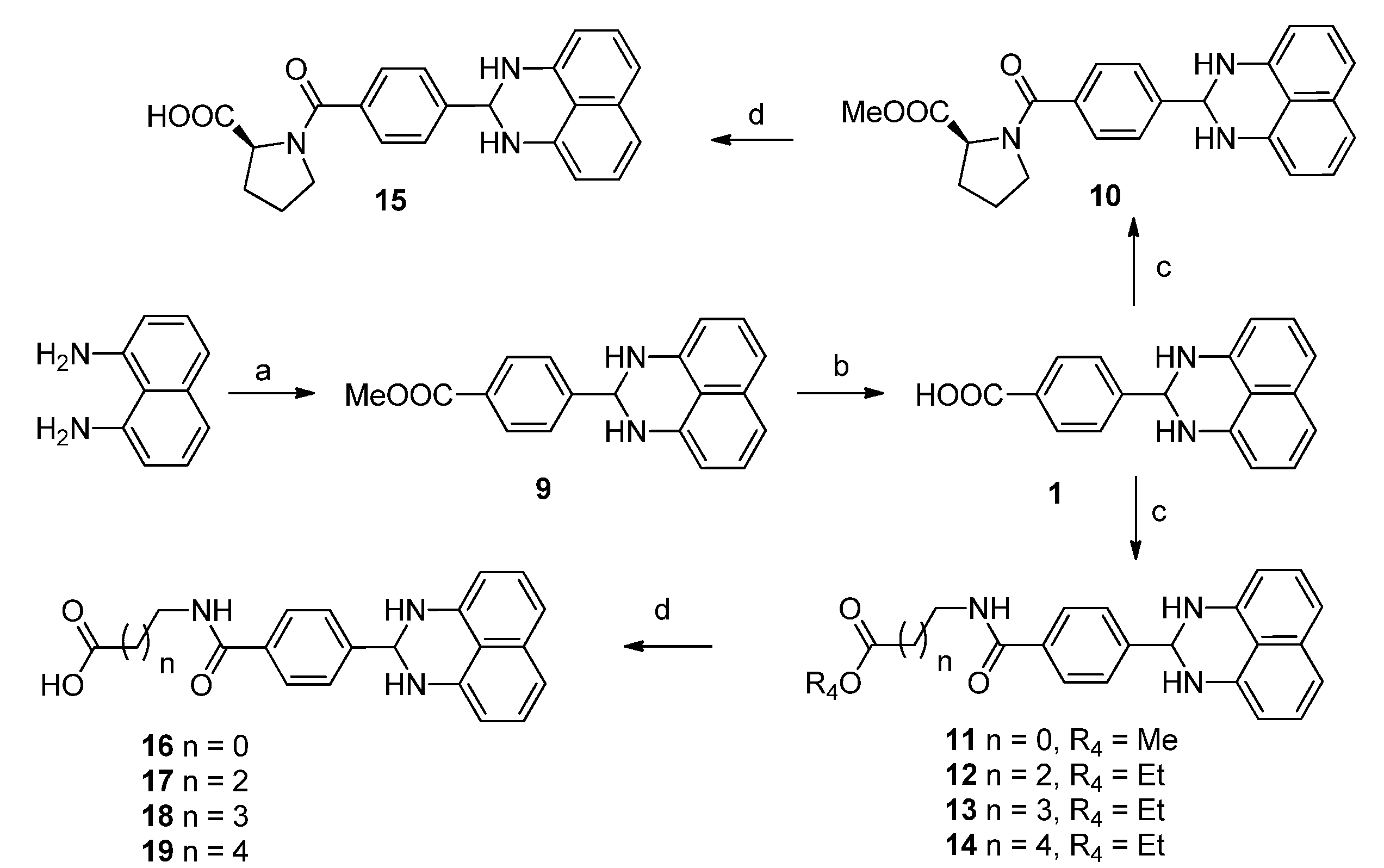
3.1.3. General Procedure for the Preparation of Derivatives 9–19
To a stirred solution of naphthalene-1,8-diamine(500 mg, 3.16 mmol)in methanol (10 mL) was added a solution of 4-formylbenzoic acid methyl ester (621.6 mg, 3.79 mmol) in methanol (5 mL), followed by Zn(OAc)2 (58.2 mg, 0.26 mmol). The mixture was stirred at room temperature for 16 h. After filtration, the filter cake was washed with methanol, dried to give compound 9 (300 mg, 31%), mp 165.0–168.2 °C. 1H-NMR (CDCl3) δ: 3.95 (s, 3H), 4.52 (s, 2H), 5.54 (s, 1H), 6.56 (dd, J = 1.6 Hz, 6.8 Hz, 2H), 7.24–7.30 (m, 4H), 7.72 (d, J = 8.0 Hz, 2H), 8.11 (d, J = 8.0 Hz, 2H); MS (ESI): m/z calcd. for C19H17N2O2 [M+H]+ 305.1, found: 305.2.
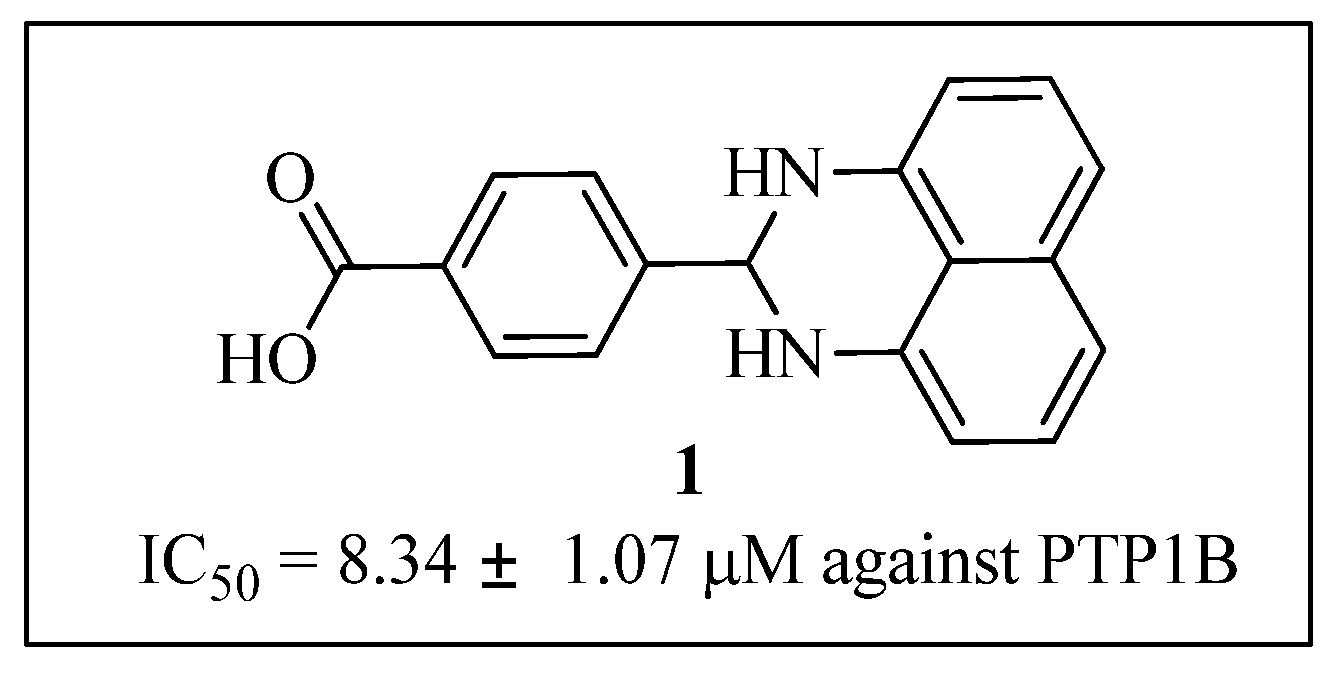
LiOH·H2O (43.5 mg, 0.99 mmol) was added to a solution of compound 9 (100 mg, 0.33 mmol) in THF (1 mL)/H2O (1 mL). The mixture was stirred at room temperature for 3 h. After removal of THF, the water layer was washed with EtOAc, and acidified with HCl (1 M) to pH = 2, filtered and dried to get compound 1 (50 mg, 53%), mp > 265 °C; 1H-NMR (DMSO-d6) δ: 5.45 (s, 1H), 6.51 (d, J = 7.2 Hz, 2H), 6.87 (s, 2H), 7.00 (d, J = 8.0 Hz, 2H), 7.17 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.0 Hz, 2H), 12.93 (brs, 1 H); MS (ESI): m/z calcd. forC18H15N2O2[M+H]+ 291.1, found: 291.0.
To a stirred solution of compound 1 (1.0 g, 3.4 mmol) in DMF (10 mL) was added methyl glycinate (0.3 g, 3.4 mmol), followed by EDCI (1.0 g, 5.2 mmol) and DMAP (0.04 g, 0.34 mmol). The mixture was stirred at 40 °C overnight. The reaction was diluted with EtOAc (100 mL), washed with water (200 mL × 3). The combined organic phases were then processed in the usual way and chromatographed (2:1 petroleum ether/EtOAc) to yield compound 11 (0.5 g, 40%), mp 193.3–198.7 °C; 1H-NMR (DMSO-d6) δ: 3.68 (s, 3H), 4.04 (d, J = 6.0 Hz, 2H), 5.44 (s, 1H), 6.51 (d, J = 7.6 Hz, 2H), 6.85 (s, 2H), 7.00 (d, J = 8.0 Hz, 2H), 7.17 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.70 (d, J = 8.0 Hz, 2H), 7.92 (d, J = 8.4 Hz, 2H), 8.99 (t, J = 6.4 Hz, 1H); MS (ESI): m/z calcd. for C21H20N3O3 [M+H]+ 362.2, found: 362.1.
LiOH·H2O (69.8 mg, 1.66 mmol) was added to a solution of compound 11 (200 mg, 0.55 mmol) in THF (2 mL)/H2O (2 mL). The reaction was stirred at room temperature for 3 h. After removal of THF, the water layer was washed with EtOAc, acidified with HCl (1 M) to pH = 2, filtered and dried to afford compound 16 (80 mg, 42%), mp 85.4–90.3 °C;1H-NMR (DMSO-d6) δ: 3.93 (d, J = 6.0 Hz, 2H), 5.42 (s, 1H), 6.50 (d, J = 7.2 Hz, 2H), 6.83 (s, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.17 (t, J = 7.6 Hz, 2H), 7.68 (d, J = 8.4 Hz, 2H), 7.90 (d, J = 8.4 Hz, 2 H), 8.86 (t, J = 6.0 Hz, 1H), 12.60 (brs, 1H); 13C-NMR (DMSO-d6) δ: 41.2, 65.7, 104.4, 112.0, 115.3, 126.8, 127.2, 127.7, 133.9, 134.3, 142.7, 145.1, 166.7, 171.2; MS (ESI): m/z calcd. forC20H18N3O3[M+H]+ 348.1, found: 348.5.
Methyl 1-(4-(2,3-dihydro-1H-perimidin-2-yl)benzoyl)pyrrolidine-2-carboxylate (10). Yield = 41%, mp 171.3–176.0 °C; 1H-NMR (CDCl3) δ:1.93 (m, 1H), 2.04 (m, 2H), 2.34 (m, 1H), 3.55 (m, 1H), 3.64 (m, 1H), 3.80 (s, 3H), 4.53 (s, 2H), 4.69 (dd, J = 4.8 Hz, 8.4 Hz, 1H), 5.51 (s, 1H), 6.55 (d, J = 6.8 Hz, 2H), 7.27 (m, 4H), 7.65 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 8.0 Hz, 2H); MS (ESI): m/z calcd. for C24H24N3O3 [M+H]+ 402.2, found: 401.7.
Ethyl 4-(4-(2,3-dihydro-1H-perimidin-2-yl)benzamido)butanoate (12). Yield = 35%, mp 128.6–132.5 °C; 1H-NMR (DMSO-d6) δ: 1.16 (t, J = 7.2 Hz, 3H), 1.78 (m, 2H), 2.35 (t, J = 7.2 Hz, 2H), 3.27 (m, 2H), 4.04 (q, J = 7.2 Hz, 2H), 5.42 (s, 1H), 6.49 (d, J = 8.0 Hz, 2H), 6.83 (s, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 7.87 (d, J = 8.0 Hz, 2H), 8.50 (t, J = 5.2 Hz, 1H); MS (ESI): m/z calcd. forC24H26N3O3 [M+H]+ 404.2, found: 404.0.
Ethyl 5-(4-(2,3-dihydro-1H-perimidin-2-yl)benzamido)pentanoate (13). Yield = 52%, mp 141.8–143.1 °C; 1H-NMR (DMSO-d6) δ: 1.17 (t, J = 7.2 Hz, 3H), 1.54 (m, 4H), 2.32 (t, J = 6.8 Hz, 2H), 3.26 (m, 2H), 4.04 (q, J = 7.2 Hz, 2H), 5.41 (s, 1H), 6.49 (d, J = 7.6 Hz, 2H), 6.83 (s, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 7.86 (d, J = 8.0 Hz, 2H), 8.48 (t, J = 5.2 Hz, 1H); MS (ESI): m/z calcd. for C25H28N3O3 [M+H]+ 418.2, found: 418.1.
Ethyl 6-(4-(2,3-dihydro-1H-perimidin-2-yl)benzamido)hexanoate (14), Yield = 48%, mp 145.8-147.4 °C; 1H-NMR (DMSO-d6) δ: 1.16 (t, J = 7.2 Hz, 3H), 1.23-1.34 (m, 2H), 1.48-1.57 (m, 4H), 2.28 (t, J = 7.2 Hz, 2H), 3.23(m, 2H), 4.04 (q, J = 7.2 Hz, 2H), 5.42 (s, 1H), 6.49 (d, J = 7.6 Hz, 2H), 6.83(s, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 7.86 (d, J = 8.0 Hz, 2H), 8.46 (t, J = 5.2 Hz, 1H); MS (ESI): m/z calcd. for C26H30N3O3 [M+H]+ 432.2, found: 432.2.
1-(4-(2,3-Dihydro-1H-perimidin-2-yl)benzoyl)pyrrolidine-2-carboxylic acid (15). Yield = 42%, mp 233.1–234.3 °C; 1H-NMR (DMSO-d6) δ: 1.91 (m, 4H), 2.29 (m, 2H), 4.42 (m, 1H), 5.42 (s, 1H), 6.51 (d, J = 8.0 Hz, 2H), 6.84 (s, 2H), 7.00 (d, J = 8.0 Hz, 2H), 7.16 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.58 (d, J = 7.6 Hz, 2H), 7.67 (d, J = 8.0 Hz, 2H); MS (ESI): m/z calcd. for C23H22N3O3 [M+H]+ 388.2, found: 388.2.
4-(4-(2,3-Dihydro-1H-perimidin-2-yl)benzamido)butanoic acid (17). Yield = 55%, mp 99.7–103.3 °C; 1H-NMR (DMSO-d6) δ: 1.75 (m, 2H), 2.27 (m, 2H), 3.27 (m, 2H), 5.42 (s, 1H), 6.49 (d, J = 7.2 Hz, 2H), 6.83(s, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.65 (d, J = 8.4 Hz, 2H), 7.87 (d, J = 8.0 Hz, 2H), 8.51 (t, J = 5.2 Hz, 1H); 13C-NMR (DMSO-d6) δ: 24.5, 31.2, 38.7, 65.7, 104.4, 112.4, 115.3, 126.8, 127.1, 127.6, 134.3, 134.6, 142.7, 144.8, 165.6, 174.2; MS (ESI): m/z calcd. for C22H22N3O3 [M+H]+ 376.2, found: 376.1.
5-(4-(2,3-Dihydro-1H-perimidin-2-yl)benzamido)pentanoic acid (18). Yield = 47%, mp 102.8–107.4 °C; 1H-NMR (DMSO-d6) δ: 1.55 (m, 4H), 2.22–2.28 (m, 2H), 3.25 (m, 2H), 5.42 (s, 1H), 6.50 (d, J = 7.2 Hz, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 7.87 (d, J = 8.0 Hz, 2H), 8.49 (t, J = 5.2 Hz, 1H); 13C-NMR (DMSO-d6) δ: 22.0, 28.6, 33.3, 38.8, 65.8, 104.7, 112.6, 115.6, 126.8, 127.0, 127.7, 134.3, 134.7, 142.4, 144.4, 165.7, 174.4; MS (ESI): m/z calcd. for C23H24N3O3 [M+H]+ 390.2, found: 390.1.
6-(4-(2,3-Dihydro-1H-perimidin-2-yl)benzamido)hexanoic acid (19). Yield = 50%, mp 90.8–91.9 °C; 1H-NMR (DMSO-d6) δ: 1.30 (m, 2H), 1.48–1.56 (m, 4H), 2.21 (t, J = 7.2 Hz, 2H), 3.24 (m, 2H), 5.41 (s, 1H), 6.49 (d, J = 8.0 Hz, 2H), 6.83 (s, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 7.86 (d, J = 8.4 Hz, 2H), 8.46 (t, J = 5.2 Hz, 1H);13C-NMR (DMSO-d6) δ: 24.2, 26.0, 28.8, 33.6, 39.0, 65.7, 104.4, 112.5, 115.3, 126.8, 127.0, 127.6, 134.3, 134.7, 142.7, 144.7, 165.7, 174.4; MS (ESI): m/z calcd. forC24H26N3O3 [M+H]+ 404.2, found: 404.5.
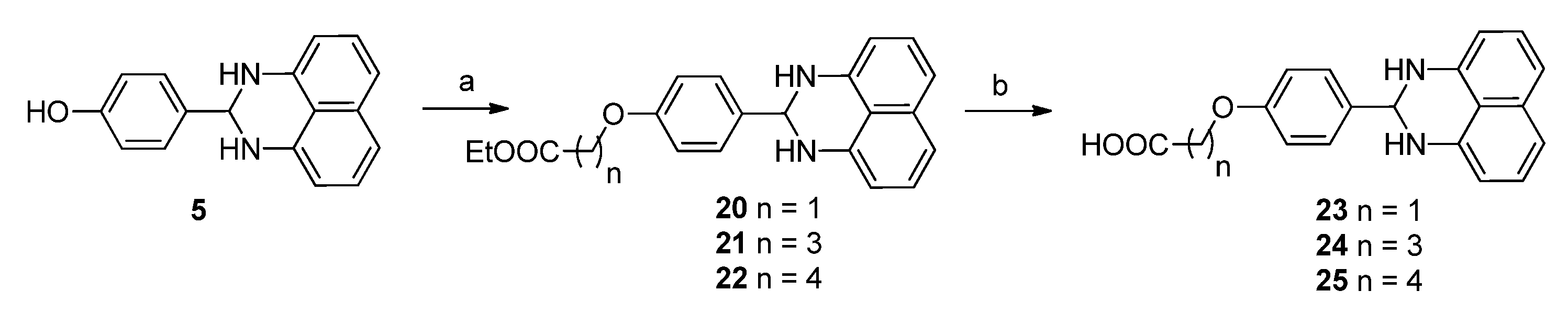
3.1.4. General Procedure for the Preparation of Derivatives 21–25
To a stirred solution of compound 5 (50 mg, 0.19 mmol) in DMF (2 mL) was added ethyl 4-bromobutyrate (44.6 mg, 0.23 mmol) and cesium carbonate (74.9 mg, 0.23 mmol). Then the resulting mixture was stirred at 40 °C overnight. The reaction was diluted with EtOAc (100 mL), washed with water (100 mL × 3). The organic phase was processed in the usual way and chromatographed (1:1 petroleum ether/EtOAc) to yield compound 21 (41 mg, 65%), mp 76.2–78.6 °C; 1H-NMR (DMSO-d6) δ: 1.18 (t, J = 7.2 Hz, 3H), 1.96 (t, J = 6.4 Hz , 2H), 2.45 (m, 2H), 4.00 (t, J = 6.4 Hz, 2H), 4.07 (q, J = 7.2 Hz, 2H), 5.29 (s, 1H), 6.47 (d, J = 7.2 Hz, 2H), 6.66 (s, 2H), 6.96 (d, J = 8.0 Hz, 4H), 7.13 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H); MS (ESI): m/z calcd. for C23H25N2O3 [M+H]+ 377.2, found: 377.6.
The mixture of LiOH·H2O (67.0 mg, 1.60 mmol) and compound 21 (200 mg, 0.53 mmol) in THF (2 mL) and H2O (2 mL) was stirred at room temperature for 3 h. After removal of THF, the water layer was washed with EtOAc, acidified with HCl (1 M) to pH 2, filtered and dried to yield compound 24 (100 mg, 54%), mp 126.3–130.9 °C; 1H-NMR (DMSO-d6) δ: 1.94 (m, 2H), 2.39 (t, J = 7.2 Hz, 2H), 4.02 (t, J = 7.2 Hz, 2H), 5.36 (s, 1H), 6.60 (d, J = 7.2 Hz, 2H), 7.00 (d, J = 8.8 Hz, 2H), 7.09 (d, J = 8.0 Hz, 2H), 7.20 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H); MS (ESI): m/z calcd. for C21H21N2O3 [M+H]+ 349.2, found: 349.3. The following compounds were similarly prepared:
Ethyl 5-[4-(2,3-dihydro-1H-perimidin-2-yl)phenoxy]pentanoate (22). Yield = 40%, mp 93.8–96.4 °C; 1H-NMR (DMSO-d6) δ: 1.18 (t, J = 7.2 Hz, 3H),1.65–1.72 (m, 4H), 2.37 (t, J = 7.2 Hz, 2H), 3.98 (t, J = 6.0 Hz, 2H), 4.06 (q, J = 7.2 Hz, 2H), 5.29 (s, 1H), 6.47 (d, J = 7.2 Hz, 2H), 6.67 (s, 2H), 6.96 (d, J = 8.0 Hz, 4H), 7.13 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.51 (d, J = 8.4 Hz, 2H); MS (ESI): m/z calcd. for C24H27N2O3 [M+H]+ 391.2, found: 391.3.
2-[4-(2,3-Dihydro-1H-perimidin-2-yl)phenoxy]acetic acid (23). Yield = 35%, mp > 265 °C; 1H-NMR (DMSO-d6) δ: 4.33 (m, 2H), 5.27 (s, 1H), 6.47 (d, J = 7.6 Hz, 2H), 6.66 (s, 2H), 6.89 (d, J = 8.4 Hz, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.14 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.47 (d, J = 8.8 Hz, 2H); 13C-NMR (DMSO-d6) δ: 66.1, 67.0, 104.2, 114.3, 115.1, 126.8, 128.2, 128.8, 135.0, 143.3, 152.2, 158.8, 160.9; MS (ESI): m/z calcd. for C19H17N2O3 [M+H]+ 321.1, found: 321.1.
5-[4-(2,3-Dihydro-1H-perimidin-2-yl)phenoxy]pentanoic acid (25). Yield = 50%, mp 165.9–167.8 °C; 1H-NMR (DMSO-d6) δ: 1.63–1.75 (m, 4H), 2.29 (t, J = 7.2 Hz, 2H), 3.99 (t, J = 6.4 Hz, 2H), 5.32 (s, 1H), 6.52 (d, J = 7.2 Hz, 2H), 7.01 (m, 4H), 7.16 (t, J = 7.6 Hz, 2H), 7.51 (d, J = 8.8 Hz, 2H); MS (ESI): m/z calcd. for C22H23N2O3 [M+H]+ 363.2, found: 363.3.
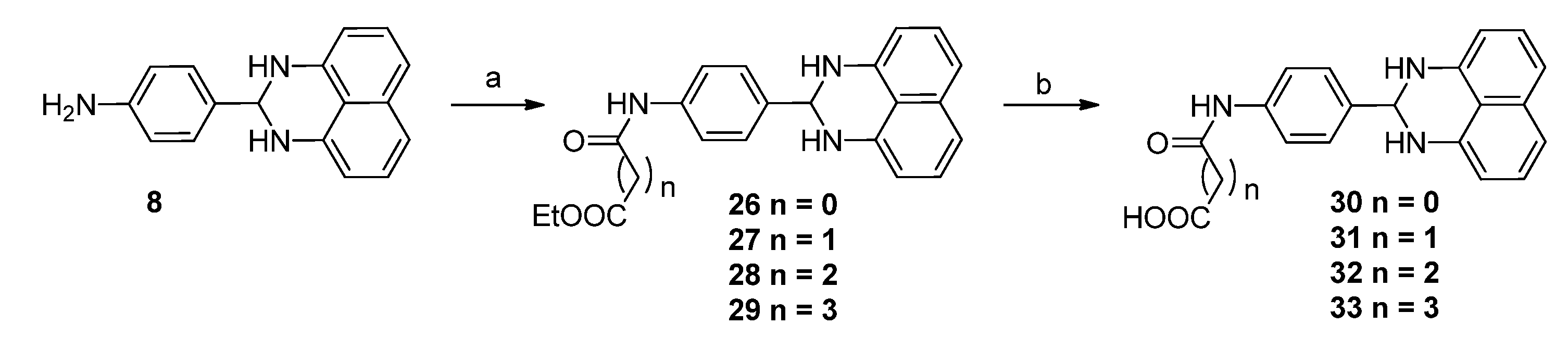
3.1.5. General Procedure for the Preparation of Derivatives 26–33
A mixture of compound 8 (50 mg, 0.19 mmol), succinic acid monoethyl ester (27.7 mg, 0.19 mmol), EDCI (55.2 mg, 28.7 mmol) and DMAP (2.3 mg, 0.019 mmol) in DMF (2 mL) was stirred at 40 °C overnight. The reaction mixture was diluted with EtOAc (100 mL), washed with water (50 mL × 3), the organic layer was then processed in the usual way and chromatographed (1:1 petroleum ether/EtOAc) to yield compound 28 (30 mg, 41%), mp 165.8–170.9 °C; 1H-NMR (DMSO-d6) δ: 1.18 (t, J = 7.2 Hz, 3H), 2.57–2.63 (m, 4H), 4.06 (q, J = 7.2 Hz, 2H), 5.29 (s, 1H), 6.48 (d, J = 8.0 Hz, 2H), 6.67 (s, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.14 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.51 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H); MS (ESI): m/z calcd. for C23H24N3O3 [M+H]+ 390.2, found: 390.0.
A mixture of compound 28 (30 mg, 0.077 mmol) and LiOH·H2O (9.7 mg, 0.23 mmol) in THF (2 mL) and H2O (2 mL) was stirred at room temperature for 3 h. After removal of THF, the water layer was washed with EtOAc, acidified with HCl (1 M) to pH 2, filtered and dried to get compound 32 (15 mg, 54%), mp > 265 °C; 1H-NMR (DMSO-d6) δ: 2.50–2.56 (m, 4H), 5.29 (s, 1H), 6.48 (d, J = 8.0 Hz, 2H), 6.67 (s, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.14 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 10.14 (s, 1H), 12.10 (brs, 1H); MS (ESI): m/z calcd. for C21H20N3O3 [M+H]+ 362.2, found: 362.2. The following compounds were similarly prepared:
Ethyl 2-{[4-(2,3-dihydro-1H-perimidin-2-yl)phenyl]amino}-2-oxoacetate (26). Yield = 41%, mp 132.3–135.5 °C; 1H-NMR (DMSO-d6) δ: 1.32 (t, J = 7.2 Hz, 3H), 4.31 (q, J = 7.2 Hz, 2H), 5.32 (s, 1H), 6.48 (d, J = 7.6 Hz, 2H), 6.73 (s, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.14 (dd, J =7.6 Hz, 8.0 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.8 Hz, 2H), 10.86 (s, 1H); 13C-NMR (DMSO-d6) δ:13.8, 62.4, 65.9, 104.3, 112.4, 115.2, 120.3, 126.8, 128.3, 134.4, 137.6, 138.1, 143.0, 155.6, 160.7; MS (ESI): m/z calcd. for C21H20N3O3 [M+H]+ 362.1, found: 362.0.
Ethyl 3-{[4-(2,3-dihydro-1H-perimidin-2-yl)phenyl]amino}-3-oxopropanoate (27). Yield = 51%, mp 158.2–160.7 °C; 1H-NMR (DMSO-d6) δ: 1.21 (t, J = 7.2 Hz, 3H), 3.47 (s, 2H), 4.12 (q, J = 7.2 Hz, 2H), 5.30 (s, 1H), 6.48 (d, J = 8.0 Hz, 2H), 6.69 (s, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.14 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.53 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H); MS (ESI): m/z calcd. for C22H22N3O3 [M+H]+ 376.2, found: 375.6.
Ethyl 5-{[4-(2,3-dihydro-1H-perimidin-2-yl]phenyl)amino}-5-oxopentanoate (29). Yield = 35%, mp 139.9–142.5 °C; 1H-NMR (DMSO-d6) δ: 1.19 (t, J = 7.2 Hz, 3H), 1.84 (m, 2H), 2.36 (m, 4H), 4.06 (q, J = 7.2 Hz, 2H), 5.29 (s, 1H), 6.48 (d, J = 7.2 Hz, 2H), 6.67 (s, 2H) , 6.97 (d, J = 8.0 Hz, 2H), 7.14 (t, J = 7.6 Hz, 2H), 7.51 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 9.97 (s, 1H); MS (ESI): m/z calcd. for C24H26N3O3 [M+H]+ 404.3, found: 404.1.
2-{[4-(2,3-Dihydro-1H-perimidin-2-yl)phenyl]amino}-2-oxoacetic acid (30). Yield = 35%, mp > 265 °C; 1HNMR (400MHz, DMSO-d6) δ: 5.32 (s, 1H), 6.48 (d, J = 7.2 Hz, 2H), 6.73(s, 2H), 6.97 (d, J = 8.4 Hz, 2H), 7.14 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.4 Hz, 2H), 10.79 (s, 1H).
3-{[4-(2,3-Dihydro-1H-perimidin-2-yl)phenyl]amino}-3-oxopropanoic acid (31). Yield = 52%, mp 221.3–223.4 °C; 1H-NMR (DMSO-d6) δ: 3.37 (s, 2H), 5.32 (s, 1H), 6.51 (d, J = 7.2 Hz, 2H), 7.00 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 10.24 (s, 1H); 13C-NMR (DMSO-d6) δ: 43.9, 66.3, 105.1, 112.7, 115.9, 118.4, 118.7, 126.8, 128.5, 134.3, 139.3, 142.3, 164.6, 169.2; MS (ESI): m/z calcd. for C20H18N3O3 [M+H]+ 348.1, found: 348.0.
5-{[4-(2,3-Dihydro-1H-perimidin-2-yl)phenyl]amino}-5-oxopentanoic acid (33). Yield = 56%, mp > 265 °C; 1H-NMR (DMSO-d6) δ: 1.80 (m, 2H), 2.26–2.45 (m, 4H), 5.29 (s, 1H), 6.48 (d, J = 7.2 Hz, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.14 (dd, J = 7.6 Hz, 8.0 Hz, 2H),7.50 (d, J = 8.8 Hz, 2H), 7.62 (d, J = 8.8 Hz, 2H), 10.00 (s, 1H); MS (ESI): m/z calcd. for C22H22N3O3 [M+H]+ 376.2, found: 376.1.
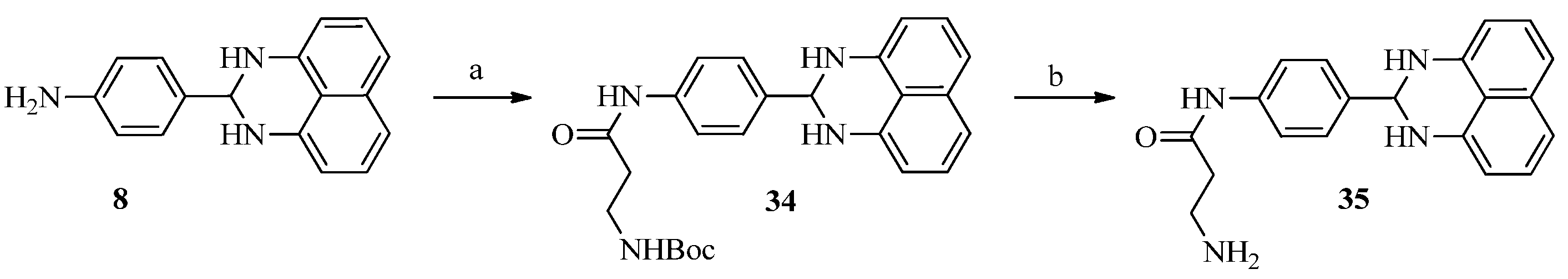
3.1.6. Procedure for the Preparation of Compound 34 and 35
A mixture of compound 8 (100 mg, 0.38 mmol), 3-tert-butoxycarbonylaminopropionic acid (72 mg, 0.38 mmol), EDCI (110 mg, 57 mmol) and DMAP (4.6 mg, 0.038 mmol) in DMF (4 mL) was stirred at 40 °C overnight. The reaction mixture was diluted with EtOAc (100 mL), washed with water (50 mL × 3), the organic layer was then processed in the usual way and chromatographed (2:1 petroleum ether/EtOAc) to yield compound 34 (72 mg, 43%), mp 111.1–113.7 °C; 1H-NMR (DMSO-d6) δ: 1.38 (s, 9H), 2.48 (m, 2H), 3.21 (m, 2H), 5.29 (s, 1H), 6.48 (d, J = 7.2 Hz, 2H), 6.68 (s, 2H), 6.90 (m, 1H), 6.97 (d, J = 8.0 Hz, 2H), 7.14 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.51 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 10.01 (s, 1H); MS (ESI): m/z calcd. for C25H29N4O3 [M+H]+ 433.2, found: 433.1.
Trifluoroacetic acid (1 mL) was added slowly to the solution of compound 34 (50 mg, 0.12 mmol) in DCM (5 mL) at 0 °C. After stirred at room temperature for 5 h, the solution was concentrated to yield compound 35 (26 mg, 67%), mp 239.7–241.2 °C; 1H-NMR (DMSO-d6) δ: 2.72 (m, 2H), 3.12 (m, 2H), 5.31 (s, 1H), 6.48 (d, J = 7.2 Hz, 2H), 6.69 (s, 2H), 6.97 (d, J = 7.6 Hz, 2H), 7.14 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.53 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.8 Hz, 2H), 7.80 (brs, 2H), 10.25 (s, 1H); MS (ESI): m/z calcd. for C20H21N4O [M+H]+ 333.2, found: 333.0.
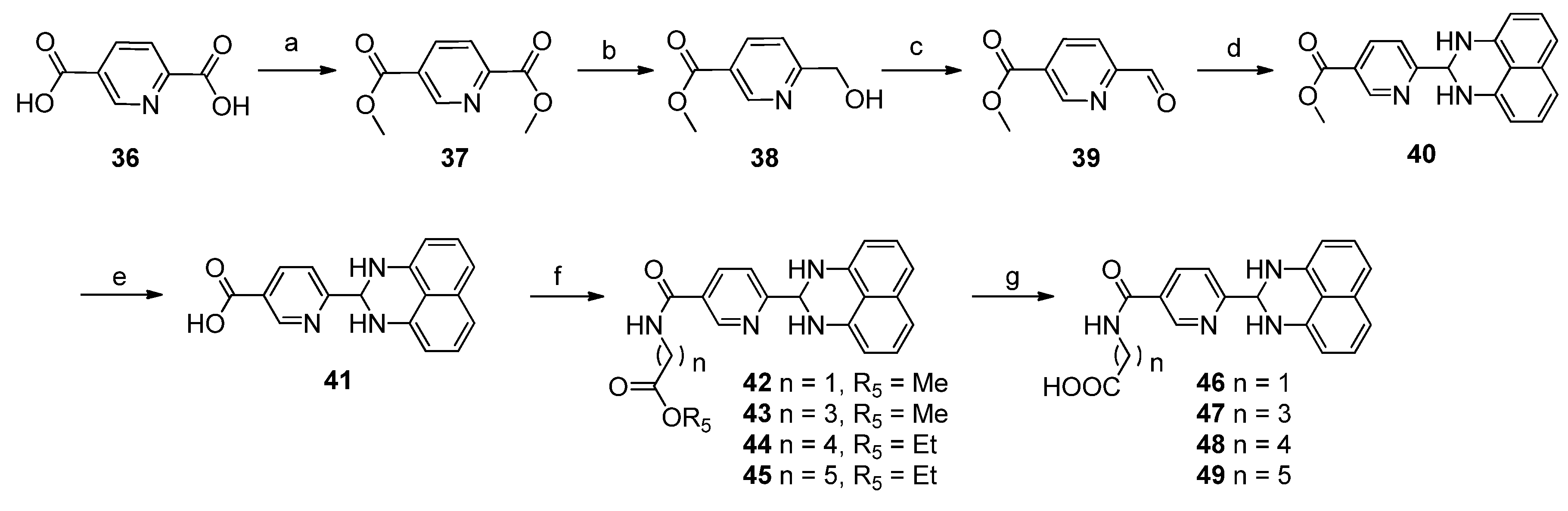
3.1.7. Procedure for the Preparation of Compounds 40–49
SOCl2 (28.5 mL, 0.24 mol) was added slowly to a stirred solution of compound 36 (10 g, 59.9 mmol) in methanol (100 mL) at 0 °C. After the addition, the solution was stirred at 80 °C for 4 h and then concentrated via rotary evaporator to get compound 37 (11.4 g, 98%). NaBH4 (2.4 g, 64 mmol) was added dropwise to the mixture of compound 37 (5 g, 25.6 mmol) and CaCl2 (11.4 g, 102.6 mmol) in THF (25 mL)/EtOH (25 mL) at 0 °C. After completion, the reaction was quenched with water. The aqueous phase was extracted with EtOAc. The combined organic phases were then processed in the usual way and chromatographed (1:1 petroleum ether/EtOAc) to yield compound 38 (2.5 g, 58%). Dess-Martin periodinane (3.0 g, 7.2 mmol) was added slowly to the mixture of compound 38 (1.0 g, 6.0 mmol) in DCM (10 mL). The resulting mixture continued to stir at room temperature overnight. The reaction was quenched with water. The aqueous phase was extracted with EtOAc. The combined organic phases were then processed in the usual way and chromatographed (3:1 petroleum ether/EtOAc) to yield compound 39 (0.85 g, 86%). To a stirred solution of compound 39 (0.3 g, 1.82 mmol) in methanol (5 mL) was added a solution of naphthalene-1,8-diamine (0.24 g, 1.52 mmol) in methanol (5 mL). Then Zn(OAc)2 (0.028 g, 0.128 mmol) was added and the mixture was stirred at room temperature for 16 h. The reaction mixture was filtered, the filter cake was washed with methanol, dried to get compound 40 (125 mg, 27%), mp 92.3–94.8 °C. 1H-NMR (DMSO-d6) δ: 3.88 (s, 3H), 5.50 (s, 1H), 6.54 (d, J = 7.6 Hz, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.08 (s, 2H), 7.16 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.67 (d, J = 8.0 Hz, 1H), 8.31 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 9.08 (d, J = 2.0 Hz, 1H); MS (ESI): m/z calcd. for C18H16N3O2 [M+H]+ 306.1, found: 306.2.
LiOH·H2O (21.6 mg, 0.49 mmol) was added to a solution of compound 40 (50 mg, 0.16 mmol) in THF (2 mL) and H2O (1 mL), then the mixture was stirred at room temperature for 3 h. After removal of the THF, the water layer was washed with EtOAc, acidified with HCl (1 M) to pH 2, filtered and dried to yield compound 41 (20 mg, 43%), mp 100.7–101.3 °C. 1H-NMR (DMSO-d6) δ: 5.50 (s, 1H), 6.55 (d, J = 7.2 Hz, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.16 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 1H), 8.29 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 9.07 (d, J = 1.2 Hz, 1H); 13C-NMR (DMSO-d6) δ: 66.2, 104.9, 112.3, 115.6, 121.0, 126.2, 127.0, 134.2, 138.1, 141.3, 149.4, 164.8, 165.9; MS (ESI): m/z calcd. for C17H14N3O2 [M+H]+ 292.1, found: 292.5.
To a stirred solution of compound 41 (500 mg, 1.72 mmol) in DMF (10 mL) was added methyl glycinate (230 mg, 2.6 mmol), followed by EDCI (500 mg, 2.5 mmol) and DMAP (21 mg, 0.17 mmol). The mixture was stirred at 40 °C overnight. The reaction was diluted with EtOAc (100 mL), washed with water (200 mL × 3). The combined organic phases were then processed in the usual way and chromatographed (2:1 petroleum ether/EtOAc) to yield compound 42 (249 mg, 40%), mp 183.6–184.7 °C. 1H-NMR (DMSO-d6) δ: 3.66 (s, 3H), 4.04 (d, J = 6.0 Hz, 2H), 5.49 (s, 1H), 6.55 (d, J = 7.2 Hz, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.04 (s, 2H), 7.16 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 1H), 8.21 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 9.01 (d, J = 1.6 Hz, 1H), 9.19 (t, J = 5.6 Hz, 1H); MS (ESI): m/z calcd. for C20H19N4O3 [M+H]+ 363.1, found: 363.3.
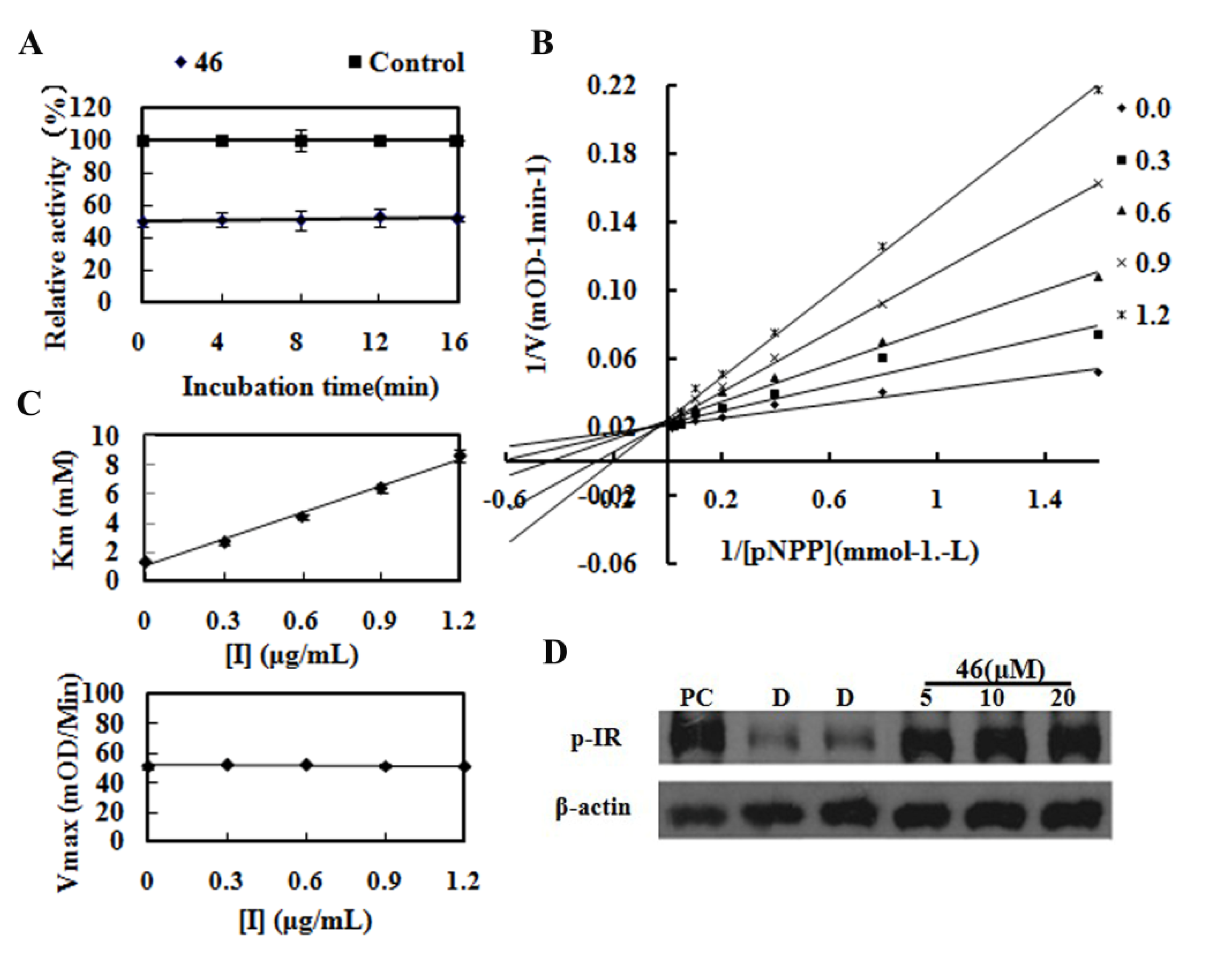
LiOH·H2O (52 mg, 1.2mmol) was added to a solution of compound 42 (150 mg, 0.41 mmol) in THF (2 mL)/H2O (2 mL).The reaction was stirred at room temperature for 3 h. After removal of THF, the water layer was washed with EtOAc, acidified with HCl (1 M) to pH = 2, filtered and dried to get compound 46 (60 mg, 42%), mp 103.5–107.3 °C. 1H-NMR (DMSO-d6) δ: 3.95 (d, J = 6.0 Hz, 2H), 5.48 (s, 1H), 6.55 (d, J = 7.6 Hz, 2H), 6.98 (d, J = 8.4 Hz, 2H), 7.04 (s, 2H), 7.16 (dd, J = 7.6 Hz, 8.4 Hz, 2H), 7.65 (d, J = 8.4 Hz, 1H), 8.22 (dd, J = 2.0 Hz, 8.4 Hz, 1H), 9.01 (d, J = 1.6 Hz, 1H), 9.08 (t, J = 6.0 Hz, 1H), 12.68 (brs, 1H); 13C-NMR (DMSO-d6) δ: 41.2, 66.4, 104.6, 112.3, 115.4, 120.6, 127.0, 128.8, 134.2, 135.8, 141.7, 147.7, 163.7, 164.9, 171.0; MS (ESI): m/z calcd. for C19H17N4O3[M+H]+ 349.1, found: 349.3. The following compounds were similarly prepared:
Methyl 4-[6-(2,3-dihydro-1H-perimidin-2-yl)nicotinamido]butanoate (43). Yield = 52%, mp 100.1–105.4 °C; 1H-NMR (DMSO-d6) δ: 1.78 (m, 2H), 2.37 (t, J = 7.2 Hz, 2H), 3.28 (m, 2H), 3.57 (s, 3H), 5.47 (s, 1H), 6.54 (d, J = 7.2 Hz, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.03 (s, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.62 (d, J = 8.0 Hz, 1H), 8.17 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 8.67 (t, J = 5.2 Hz, 1H), 8.97 (d, J = 1.6 Hz, 1H); MS (ESI): m/z calcd. for C22H23N4O3 [M+H]+ 391.2, found: 391.4.
Ethyl 5-[6-(2,3-dihydro-1H-perimidin-2-yl)nicotinamido]pentanoate (44). Yield = 60%, mp 109.4–113.5 °C; 1H-NMR (DMSO-d6) δ: 1.16 (t, J = 6.8 Hz, 3H), 1.54 (m, 4H), 2.32 (m, 2H), 3.26 (m, 2H), 4.03 (q, J = 6.8 Hz, 2H), 5.47 (s, 1H), 6.54 (d, J = 7.2 Hz, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.03 (s, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.62 (d, J = 8.4 Hz, 1H), 8.16 (dd, J= 2.0 Hz, 8.0 Hz, 1H), 8.65 (t, J = 5.6 Hz, 1H), 8.96 (d, J = 1.6 Hz, 1H); MS (ESI): m/z calcd. for C24H27N4O3 [M+H]+ 419.2, found: 419.1.
Ethyl 6-[6-(2,3-dihydro-1H-perimidin-2-yl)nicotinamido]hexanoate (45). Yield = 44%, mp 119.3–123.8 °C; 1H-NMR (DMSO-d6) δ: 1.15 (t, J = 7.2 Hz, 3H), 1.23–1.34 (m, 2H), 1.49–1.56 (m, 4H), 2.26–2.30 (m, 2H), 3.22–3.27 (m, 2H), 4.02 (q, J = 7.2 Hz, 2H), 5.47 (s, 1H), 6.45 (d, J = 7.2 Hz, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.03 (s, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.61 (d, J = 8.0 Hz, 1H), 8.16 (d, J = 2.0 Hz, 8.0 Hz, 1H), 8.62 (t, J = 5.6 Hz, 1H), 8.96 (d, J = 1.6 Hz, 1H); MS (ESI): m/z calcd. for C25H29N4O3 [M+H]+ 433.2, found: 433.2.
4-[6-(2,3-Dihydro-1H-perimidin-2-yl)nicotinamido]butanoic acid (47). Yield = 50%, mp 95.3–99.0 °C; 1H-NMR (DMSO-d6) δ: 1.72 (m, 2H), 2.21 (t, J = 6.8 Hz, 2H), 3.26 (m, 2H), 5.47 (s, 1H), 6.54 (d, J = 7.2 Hz, 2H), 6.97 (d, J = 8.4 Hz, 2H), 7.02 (s, 2H), 7.15 (t, J = 7.6 Hz, 2H), 7.61 (d, J = 8.4 Hz, 1H), 8.19 (dd, J = 1.6 Hz, 8.0 Hz, 1H), 8.99 (s, 1H), 9.33 (brs, 1H); 13C-NMR (DMSO-d6) δ: 24.6, 32.6, 39.2, 66.3, 104.6, 112.3, 115.4, 120.4, 126.9, 129.5, 134.2, 135.7, 141.7, 147.6, 163.3, 164.4, 175.0; MS (ESI): m/z calcd. for C21H21N4O3 [M+H]+ 377.2, found: 377.1.
5-[6-(2,3-Dihydro-1H-perimidin-2-yl)nicotinamido]pentanoic acid (48). Yield = 46%, mp 117.1–120.3 °C; 1H-NMR (DMSO-d6) δ: 1.54 (m, 4H), 2.24 (m, 2H), 3.25 (m, 2H), 5.47 (s, 1H), 6.54 (d, J = 7.2 Hz, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.02 (s, 2H), 7.15 (t, J = 7.6 Hz, 2H), 7.62 (d, J = 8.0 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 8.65 (t, J = 5.2 Hz, 1H), 8.97 (s, 1H), 12.03 (brs, 1H); 13C-NMR (DMSO-d6) δ: 22.1, 28.5, 33.4, 39.0, 66.3, 104.7, 112.4, 115.5, 120.6, 127.0, 129.5, 134.3, 135.8, 141.7, 147.6, 163.4, 164.6, 174.5; MS (ESI): m/z calcd. for C22H23N4O3 [M+H]+ 391.2, found: 391.4.
6-[6-(2,3-Dihydro-1H-perimidin-2-yl)nicotinamido]hexanoic acid (49). Yield = 48%, mp 86.7–88.4 °C; 1H-NMR (DMSO-d6) δ: 1.23–1.34 (m, 2H), 1.48–1.57 (m, 4H), 2.20 (t, J = 7.2 Hz, 2H), 3.24 (m, 2H), 5.48 (s, 1H), 6.55 (d, J = 7.2 Hz, 2H), 6.97 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.62 (d, J = 8.0 Hz, 1H), 8.17 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 8.64 (t, J = 5.6 Hz, 1H), 8.96 (d, J = 1.6 Hz, 1H); 13C-NMR (DMSO-d6) δ:24.3, 26.1, 28.7, 33.7, 39.2, 66.3, 104.8, 112.2, 115.6, 120.3, 127.0, 128.6, 134.9, 135.9, 141.6, 147.8, 163.4, 164.5, 174.5; MS (ESI): m/z calcd. for C23H25N4O3 [M+H]+ 405.2, found: 405.3.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

































