Gold Nanoparticles Decorated with Mannose-6-phosphate Analogues

Abstract
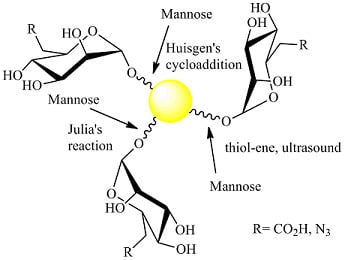
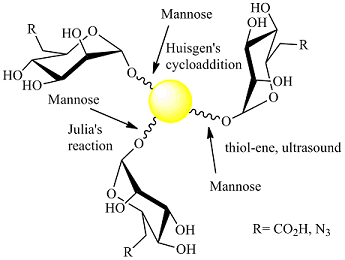
:
1. Introduction
2. Results and Discussion
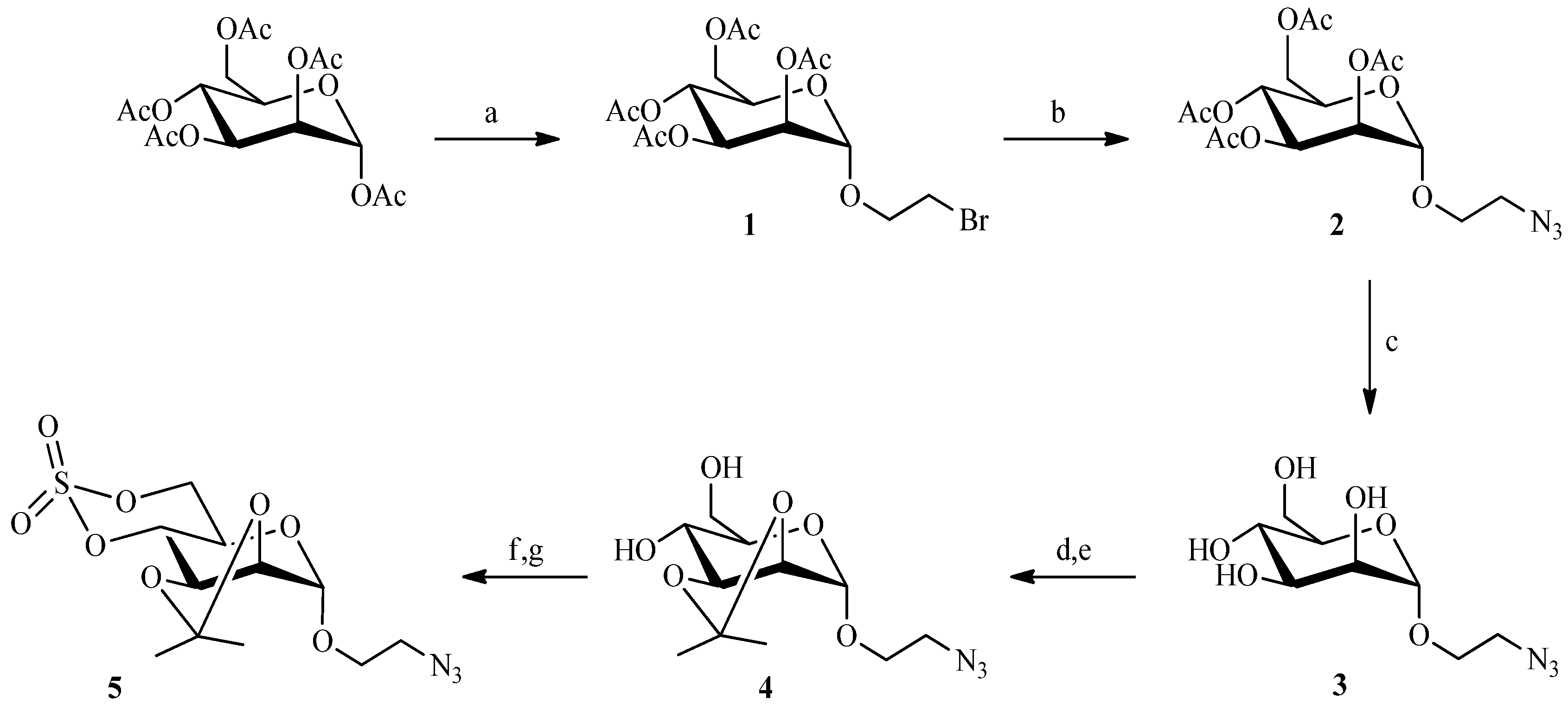
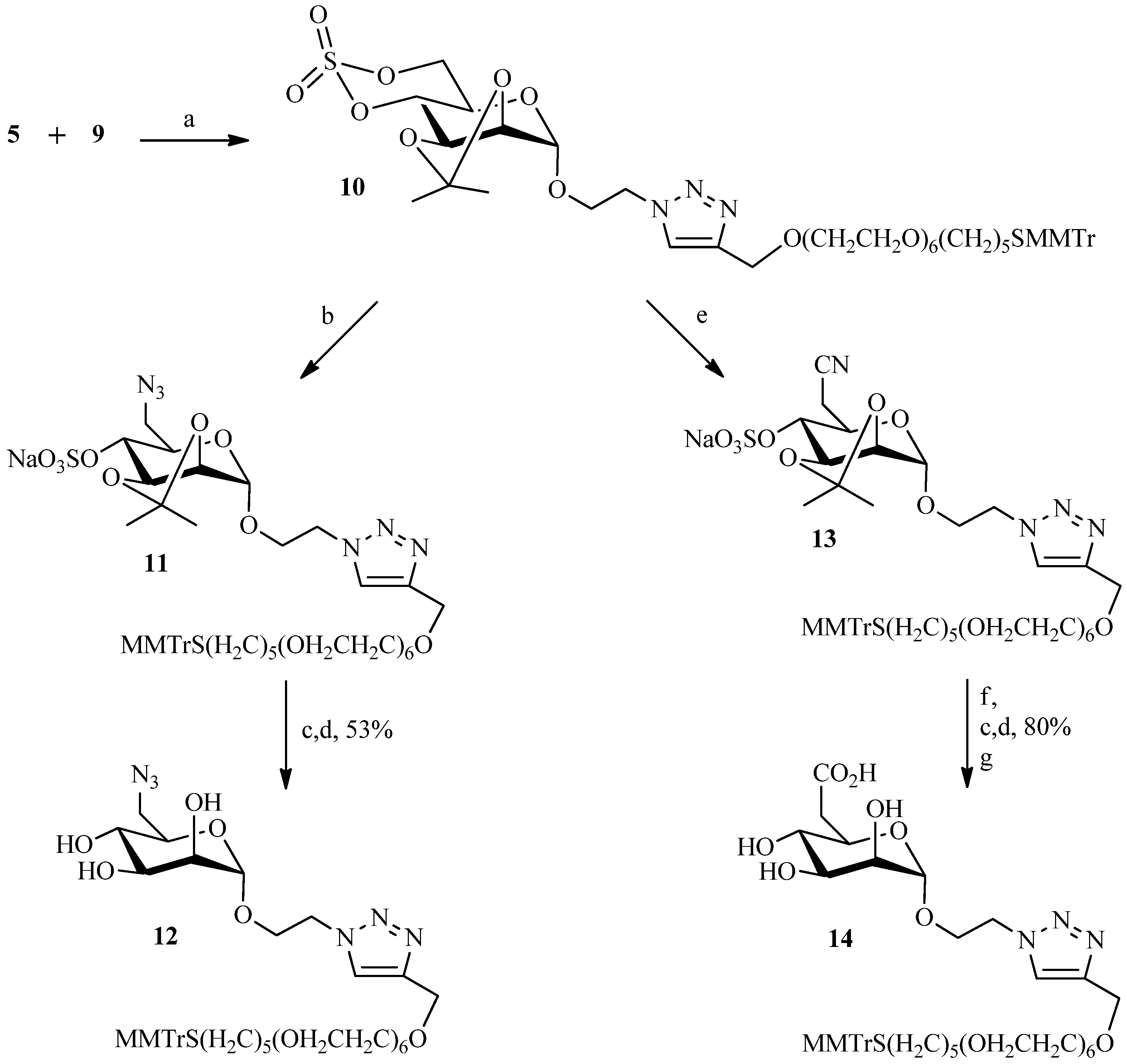
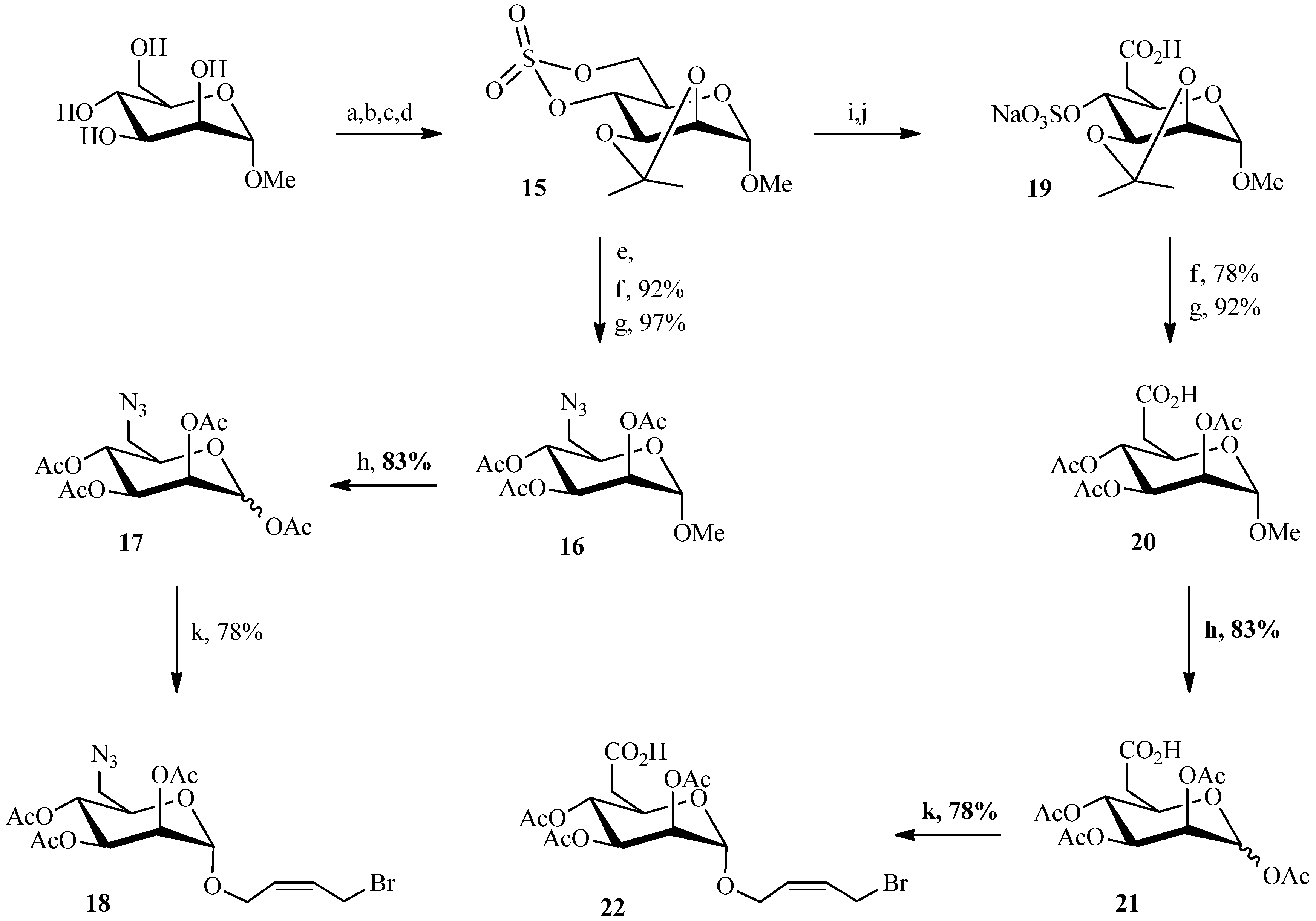
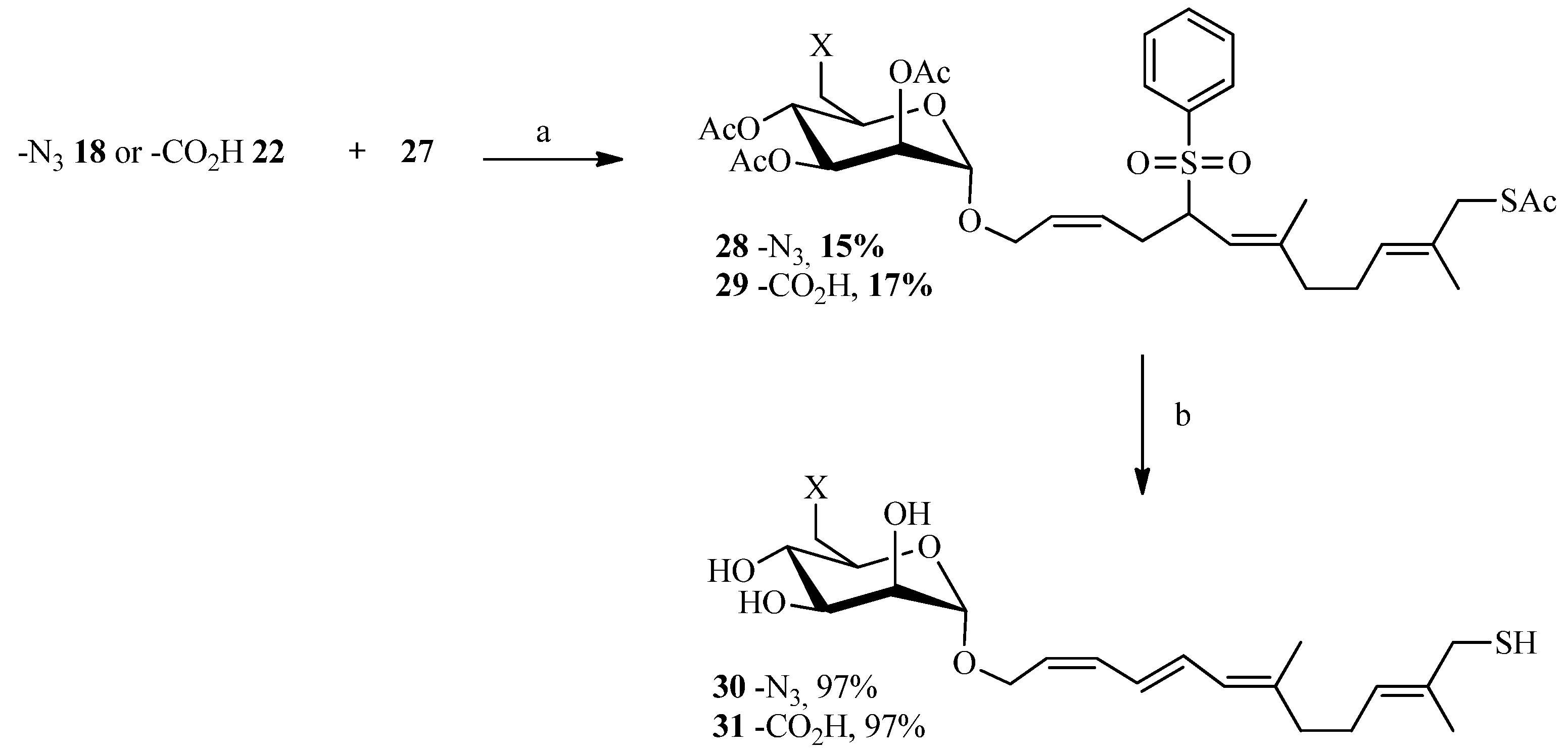
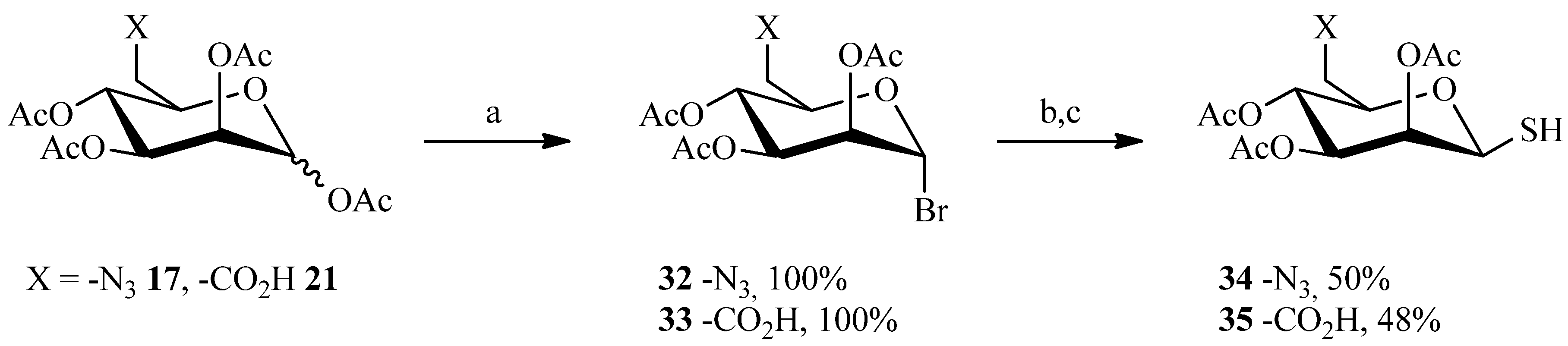
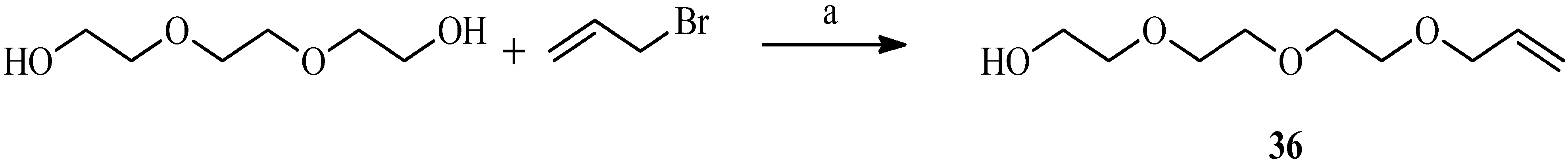
2.1. 1,3Dipolar Cycloaddition

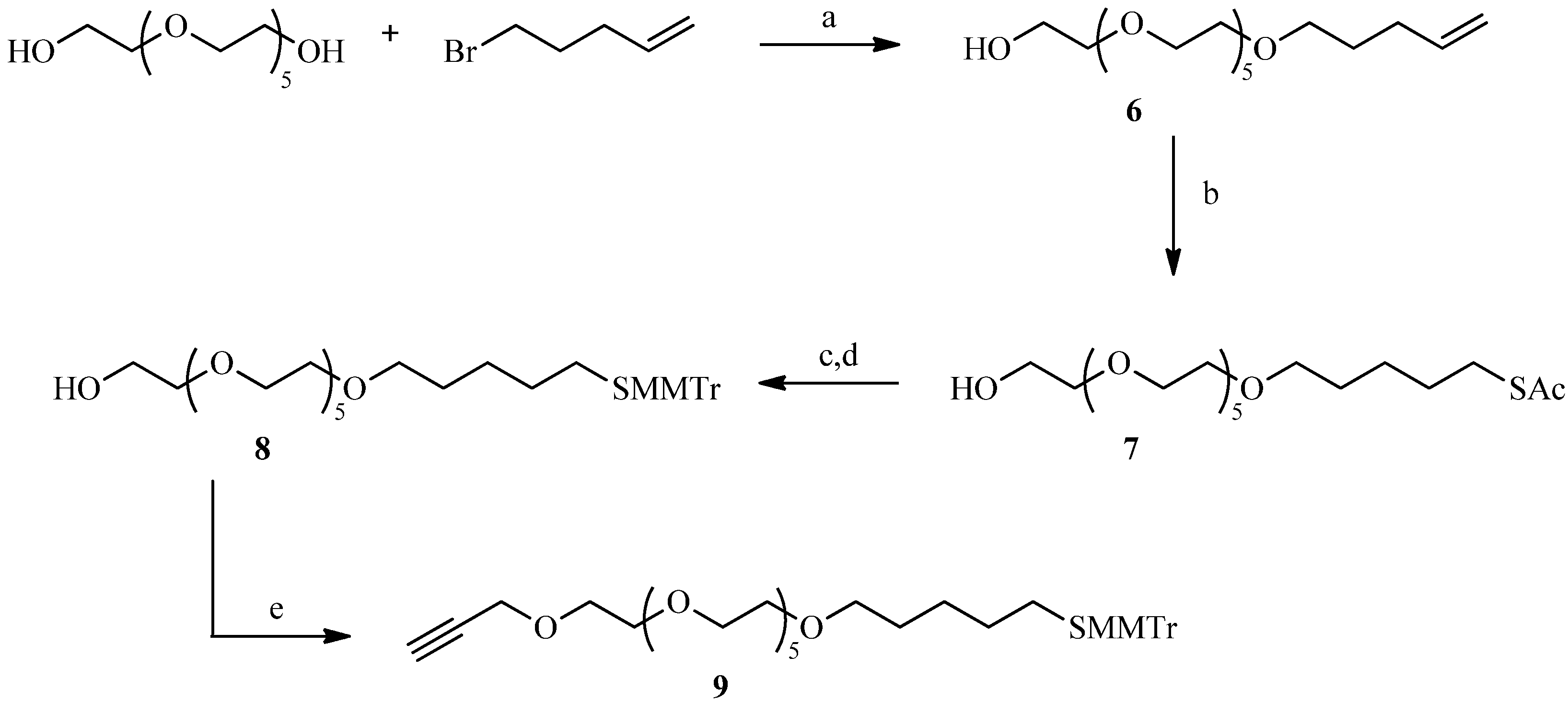

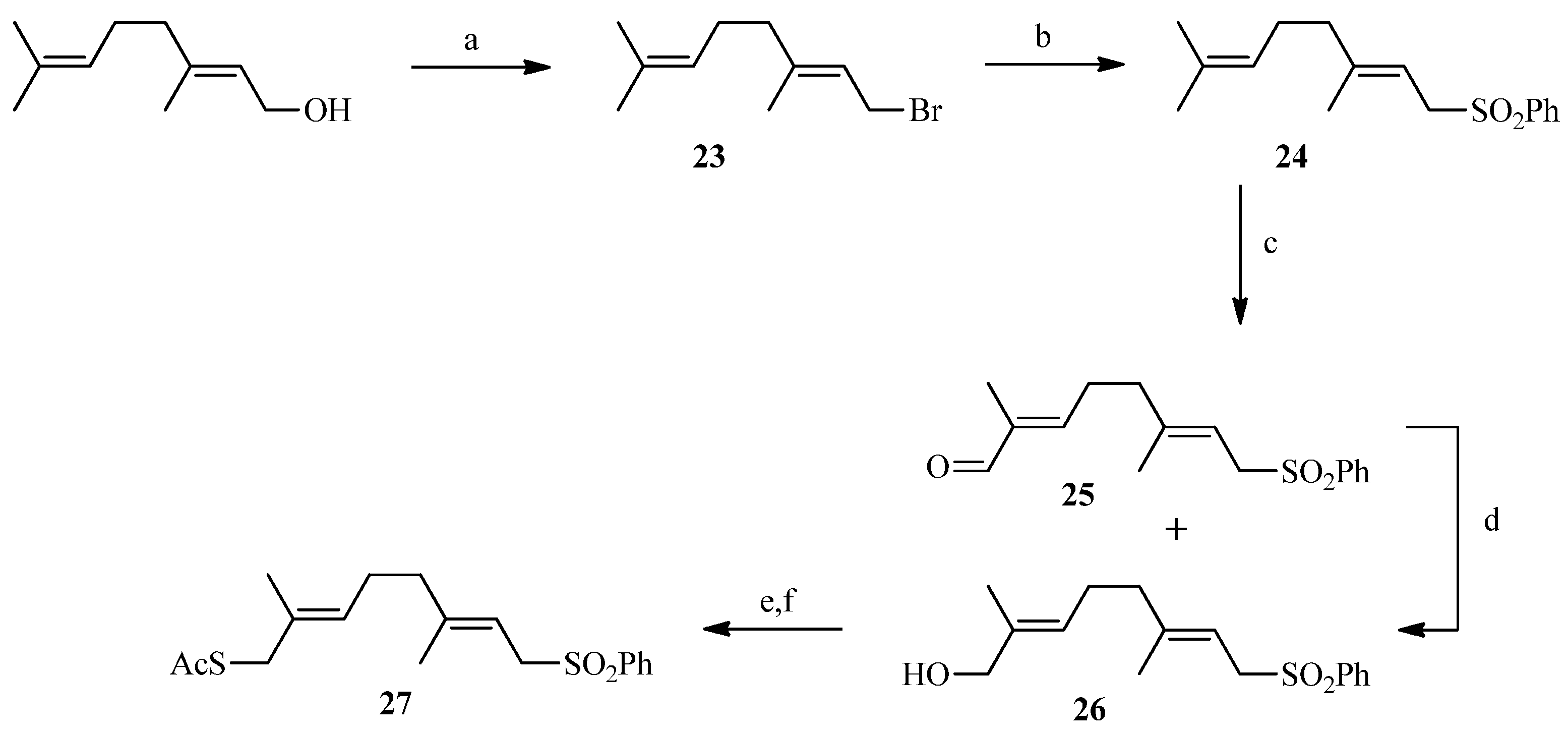
2.2. Julia Reaction



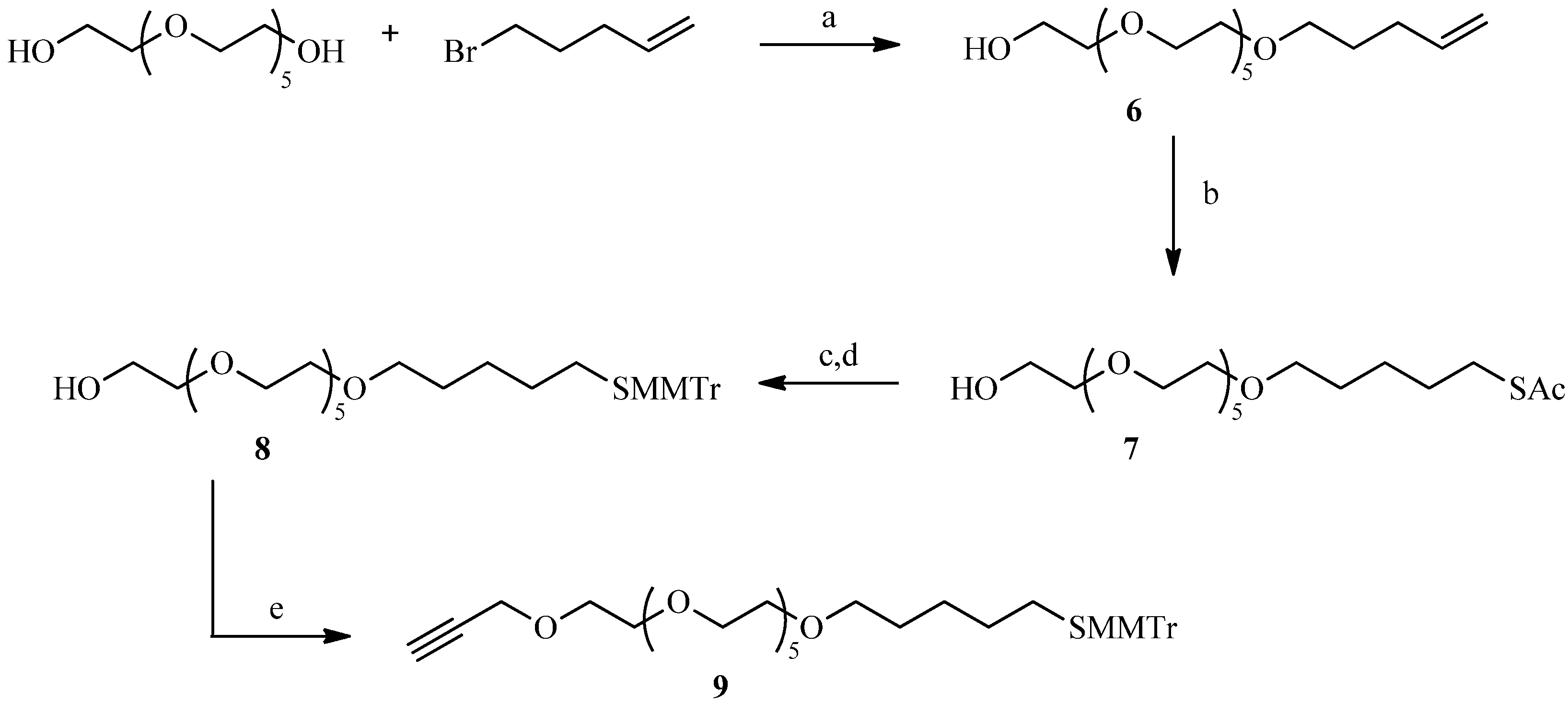
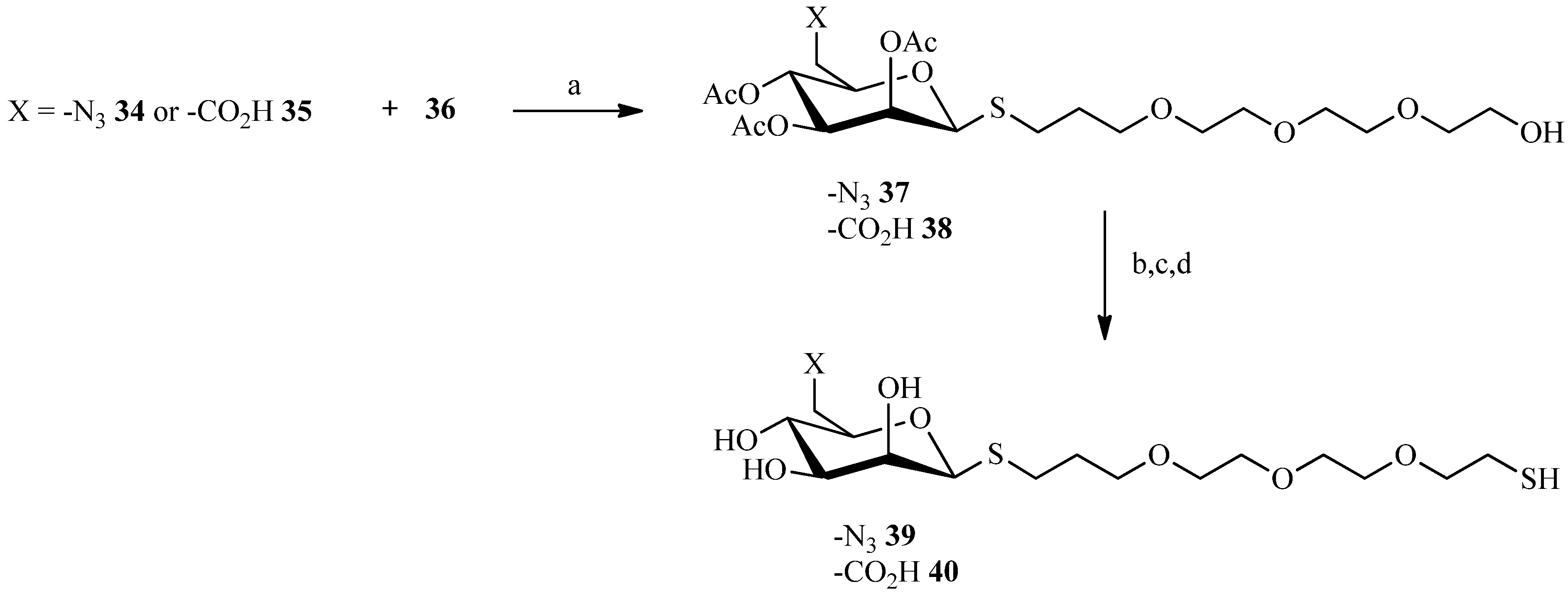
2.3. Thiol-ene Reaction




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Reflux, THF, 24 h | UV, THF, 5 h | Sonication, THF, 4 h | Sonication, Dioxane, 3 h |
|---|---|---|---|---|
| 39 | 76% | 50% | 72% | 79% |
| 40 | 78% | 60% | 75% | 80% |
2.4. Gold Nanoparticles
| Nanoparticles | TEM [a] | DLS [b] |
|---|---|---|
| Azide AuNPs (Huisgen) | 10 | 18–20 |
| Azide AuNPs (Julia) | 10 | 14–16 |
| Azide AuNPs (thiol-ene) | 10 | 12–13 |
| Carboxylic acid AuNPs (Huisgen) | 10 | 19–20 |
| Carboxylic acid AuNPs (Julia) | 10 | 15–16 |
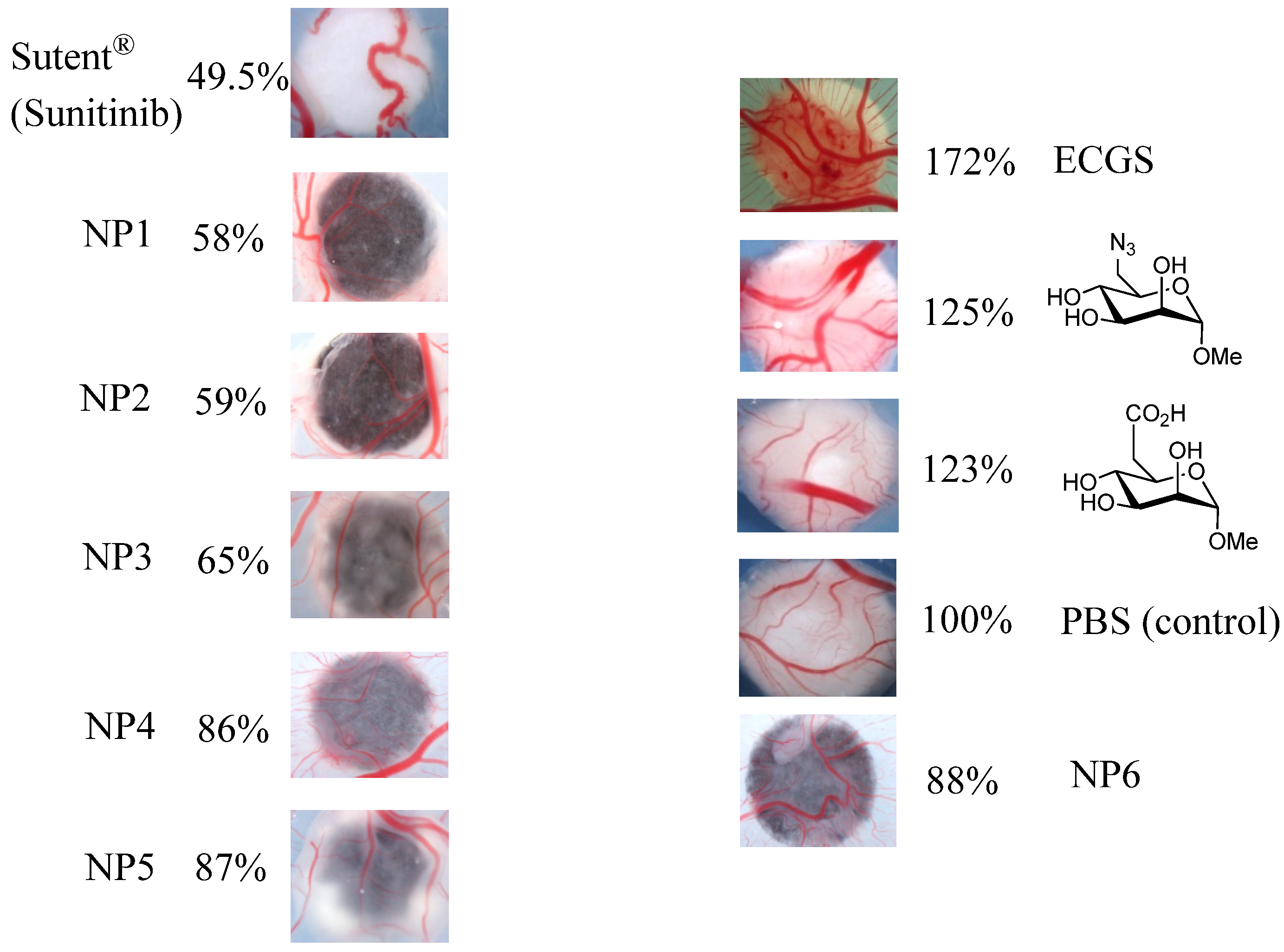
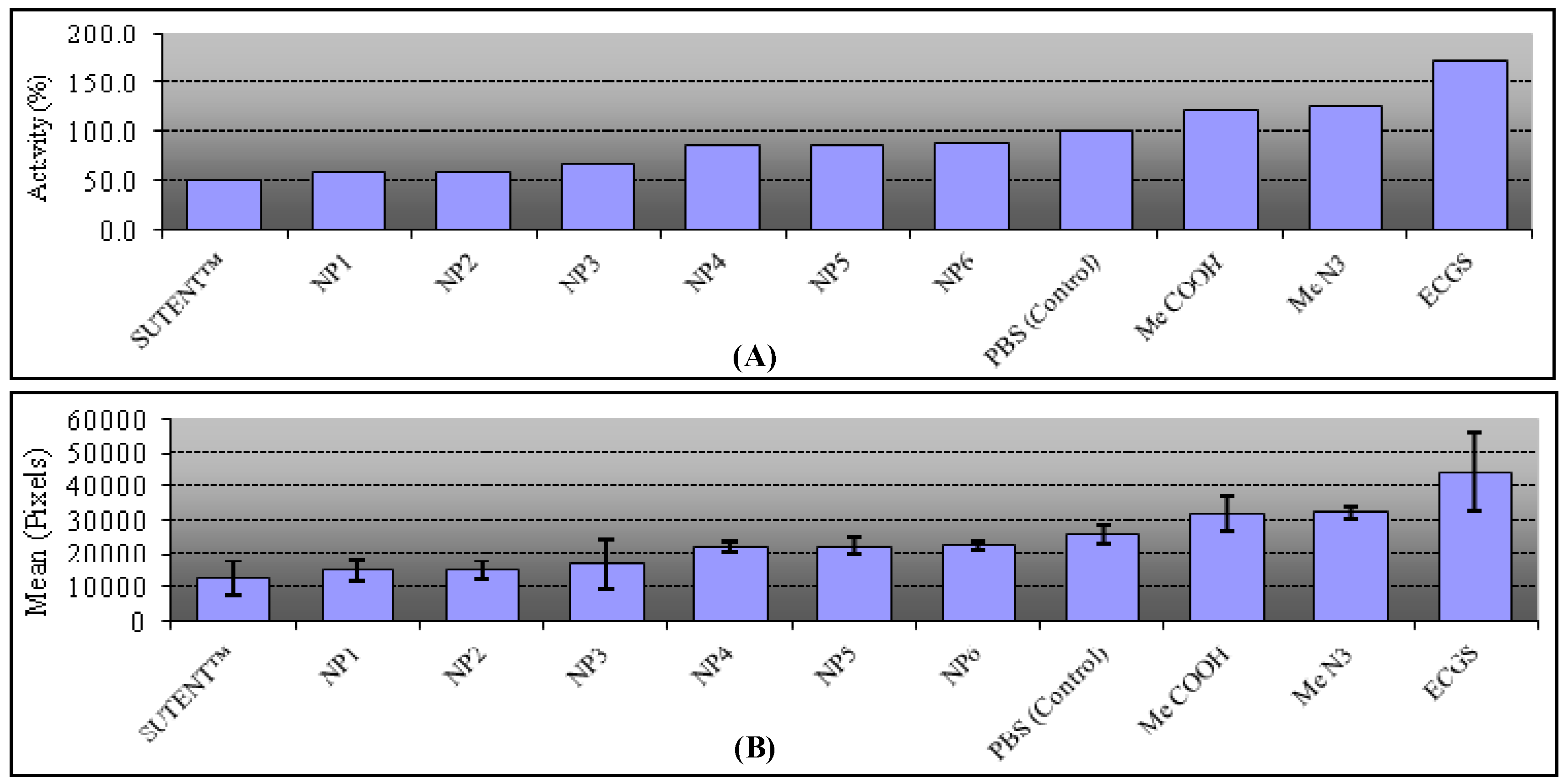
2.5. Biological Assays

3. Experimental
3.1. General Information
 = +42.1 (c = 0.5 in chloroform); 1H-NMR (CDCl3): δ = 2.00, 2.05, 2.11, 2.16 (4s, 12H, 4CH3); 3.52 (t, J = 6.0 Hz, 2H, CH2Br); 3.93 (m, 2H, CH2CH2Br); 4.13 (m, 2H, H5 and H6a); 4.27 (dd, 1H, J = 5.8 Hz, J = 12.6 Hz, H6b); 4.88 (d, J = 1.6 Hz, H1); 5.27 (dd, 1H, J = 2.0 Hz, J = 3.2 Hz, H2); 5.29 (t, 1H, J = 1.6 Hz, H4); 5.35 ppm (dd, 1H, J = 3.6 Hz, J = 10.0 Hz, H3);13C-NMR (CDCl3): δ = 20.67, 20.70, 20.75, 20.87 (4CH3); 29.60 (CH2CH2Br); 62.41 (C6); 66.00 (C4); 68.48 (CH2Br); 68.93 (C5); 69.02 (C3); 69.42 (C2); 97.75 (C1); 169.76, 169.86, 170.03, 170.62 ppm (4CO); MS (ESI) m/z: 477.01, 478.95 [M+Na]+. = +39.0 (c = 0.6 in chloroform); 1H-NMR (CDCl3): δ = 2.0, 2.05, 2.11, 2.16 (4s, 12H, 4CH3); 3.47 (m, 2H, CH2N3); 3.67 (m, 1H, CH2CH2N3); 3.87 (m, 1H, CH2CH2N3); 4.05 (ddd, 1H, J = 2.4 Hz, J = 5.2 Hz, J = 9.7 Hz, H5); 4.13 (dd, 1H, J = 2.6 Hz, J = 12.2 Hz, H6a); 4.29 (dd, 1H, J = 5.2 Hz, J = 12.4 Hz, H6b); 4.87 (d, 1H, J = 1.6 Hz, H1); 5.30 (t, 1H, J = 10.0 Hz, H4); 5.28 (dd, 1H, J = 2.0 Hz, J = 3.2 Hz, H2); 5.36 ppm (dd, 1H, J = 3.2 Hz, J = 10.0 Hz, H3); 13C-NMR (CDCl3) : δ = 20.63, 20.68, 20.71, 20.84 (4CH3); 50.32 (CH2N3); 62.42 (C6); 65.96 (C4); 67.02 (CH2CH2N3); 68.82 (C5 and C3); 69.36 (C2); 97.71 (C1); 169.73, 169.78, 169.98, 170.59 ppm (4CO); MS (ESI) m/z: 440.12 [M+Na]+. = + 54.9 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 3.41 (t, 2H, J = 5.0 Hz, CH2N3); 3.60 (m, 3H, H3, H5 and CH2CH2N3); 3.71 (m, 2H, H4 and H6a); 3.85 (m, 2H, H2 and H6b); 3.92 (m, 1H, CH2CH2N3); 4.81 ppm (d, 1H, J = 1.2 Hz, H1); MS (ESI) m/z: 272.11 [M+Na]+, 288.02 [M+K]+, 521.19 [2M+Na]+. = + 54.9 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.68 (m, 2H, CH2CH2CH=CH2); 2.09 (m, 2H, CH2CH=CH2); 3.46 (t, 2H, J = 6.6 Hz, CH2CH2CH2CH=CH2); 3.56–3.73 (m, 24 h, 12CH2O); 4.99 (m, 2H, CH=CH2); 5.81 ppm (m, 2H, CH=CH2); 13C-NMR (CDCl3): δ = 28.66 (CH2CH2CH=CH2); 30.12 (CH2CH=CH2); 61.51–72.58 (13CH2O); 114.59 (CH=CH2); 138.18 ppm (CH=CH2); MS (ESI) m/z: 373.27 [M+Na]+, 389.20 [M+K]+. = + 36.0 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.36 (m, 6H, CH2CH2CH2CH2S); 1.34, 1.49 (2s, 6H, 2CCH3); 2.14 (t, 2H, J = 7.2 Hz, CH2S); 3.38 (t, 2H, J = 6.4 Hz, CH2(CH2)4S); 3.44–3.66 (m, 25 h, H5 and 12CH2O); 3.78 (s, 3H, OCH3); 3.97 (m, 1H, CH2CH2N); 4.15 (m, 1H, CH2CH2N); 4.27 (m, 3H, H2, H3 and H6a); 4.50 (m, 2H, H6b and H4); 4.64 (m, 4 h, CH2N and CH2C=CH); 5.12 (s, 1H, H1); 6.81–7.39 (m, 14 h, CHAr); 8.07 ppm (s, 1H, NCH); 13C-NMR (CD3OD): δ = 26.34, 28.20 (2CCH3); 26.71 (CH2(CH2)2S); 29.62 (CH2CH2S); 30.23 (CH2(CH2)3S); 33.03 (SCH2); 51.19 (CH2N); 55.79 (OCH3); 59.79 (C5); 65.14 (CH2C=CH); 67.42 (CH2CH2N); 70.95, 71.19, 71.49, 71.58, 72.03 (13CH2O); 73.53 (C6); 74.46, 77.23 (C2 and C3); 85.66 (C4); 98.96 (C1); 108.26, 111.66 (SC and C(CH3)2); 114.11, 127.66, 128.86, 130.73, 132.02 (14CHAr); 126.04 (NCH); 138.40, 146.86 (3SCCAr and C=CH); 159.71 ppm (COCH3); MS (ESI) m/z: 1068.62 [M+Na]+, 1080.77 [M+Cl]−. = +17.1 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.37 (m, 6H, CH2CH2CH2CH2S); 1.30, 1.45 (2s, 6H, 2CCH3); 2.17 (t, 2H, J = 7.2 Hz, CH2S); 3.37 (t, 2H, J = 6.6 Hz, CH2(CH2)4S); 3.42–3.67 (m, 27H, H2 and 13CH2O); 3.79 (s, 3H, OCH3); 3.96 (m, 1H, CH2CH2N); 4.12 (m, 1H, CH2CH2N); 4.07 (d, 1H, J = 6.0 Hz, H5); 4.25 (m, 1H, H3); 4.38 (t, 1H, J = 5.8 Hz, H4); 4.70 (m, 6H, H6, CH2N and CH2C=CH); 4.93 (s, 1H, H1); 6.87–7.41 (m, 14 h, CHAr); 9.19 ppm (s, 1H, NCH); 13C-NMR (CD3OD): δ = 27.20, 28.67 (2CCH3); 27.33, 30.16, 30.87 (CH2CH2CH2CH2S); 33.55 (CH2S); 51.66 (C6 and CH2(CH2)4S); 54.55 (CH2N); 56.53 (OCH3); 65.58 (CH2C=CH); 67.89 (CH2CH2N); 70.39–72.33 (12CH2O); 72.13 (C2); 73.88 (C3); 76.31 (C5); 77.37 (C4); 99.59 (C1); 110.79 (SC); 114.87–132.54 (14CHAr); 126.23 (NCH); 138.80–160.13 (3SCCAr and C=CH); 158.40 ppm (COCH3); MS(ESI) m/z:1134.58 [M+Na]+. = −2.1 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.47 (m, 2H, CH2(CH2)2S); 1.60 (m, 2H, CH2(CH2)3S); 1.71 (m, 2H, CH2CH2S); 2.70 (t, 2H, J = 7.2 Hz, CH2S); 3.48 (t, 2H, J = 6.2 Hz, CH2(CH2)4S); 3.19–3.78 (m, 30H, H2-6 and 12CH2O); 3.88 (m, 1H, CH2CH2N); 4.13 (m, 1H, CH2CH2N); 4.63 (m, 4 h, CH2N and CH2C=CH); 4.72 (s, 1H, H1); 8.03 ppm (s, 1H, NCH); 13C-NMR (CD3OD): δ = 26.13 (CH2(CH2)3S); 30.07 (CH2CH2S); 30.36 (CH2(CH2)3S); 39.66 (CH2S); 51.34 (CH2N); 62.85 (C6); 65.05 (CH2C=CH); 66.79 (CH2CH2N); 68.38, 70.81, 71.24, 71.59, 71.93, 72.15, 72.51, 75.01 (C2-5 and 13CH2O); 101.70 (C1); 132.57 (NCH); 161.04 ppm (CH=C); MS (ESI) m/z: 798.62 [M+Na]+. = −9.0 (c = 1.00 in chloroform); 1H-NMR (D2O): δ = 1.35, 1.52 (2s, 6H, 2CCH3); 1.44 (m, 2H, CH2(CH2)2S); 1.60 (m, 2H, CH2(CH2)3S); 1.73 (m, 2H, CH2CH2S); 2.29 (dd, 1H, J = 10.6 Hz, J = 15.0Hz, H6a); 2.80 (dd, 1H, J = 2.0 Hz, J = 15.2 Hz, H6b); 2.89 (t, 2H, J = 8.0 Hz, CH2S); 3.53 (t, 2H, J = 6.6 Hz, CH2(CH2)4S); 3.50–3.69 (m, 25 h, H5 and 12CH2O); 3.75 (s, 3H, OCH3); 3.88 (m, 1H, CH2CH2N); 4.17 (m, 2H, CH2CH2N and H4); 4.19 (d, 1H, J = 5.6 Hz, H2); 4.30 (m, 1H, H3); 4.68 (m, 4 h, CH2N and CH2C=CH); 4.95 (s, 1H, H1); 6.80–7.37 (m, 14 h, CHAr); 8.12 ppm (s, 1H, NCH); 13C-NMR (D2O): δ = 23.80 (CH2CH2S); 24.24 (CH2(CH2)2S); 25.45, 26.80 (2CCH3); 28.06 (CH2(CH2)3S); 38.92 (C6); 49.99 (CH2N); 50.89 (CH2S); 55.86 (OCH3); 62.95 (CH2C=CH); 65.47 (CH2CH2N); 66.41 (C5); 66.54, 68.69, 69.07, 69.40, 69.53, 70.70 (13CH2O); 75.21 (C2); 76.00 (C3); 78.30 (C4); 95.99 (C1); 110.39 (SC and C(CH3)2); 114.14–132.02 (14CHAr); 125.55 (NCH); 143.85 (C=CH); 138.38, 146.00, 146.84 (3SCCAr); 159.71 (COCH3);177.75 (CO2H); MS (ESI) m/z: 1037.34 [M+Na]+. = +10.2 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.49 (m, 2H, CH2(CH2)2S); 1.61 (m, 2H, CH2(CH2)3S); 1.80 (m, 2H, CH2CH2S); 2.41 (dd, 1H, J = 10.2 Hz, J = 16.2 Hz, H6a); 2.84 (m, 3H, CH2S and H6b); 3.49 (t, 2H, J = 6.4 Hz, CH2(CH2)4S); 3.40–3.79 (m, 28 h, H2-5 and 12 CH2O); 3.92 (m, 1H, CH2CH2N); 4.22 (m, 1H, CH2CH2N); 4.71 (d, 1H, J = 1.2 Hz, H1); 4.87 (m, 2H, CH2N); 4.92 (m, 2H, CH2C=CH); 8.65 (s, 1H, NCH); 13C-NMR (D2O): δ = 23.79 (CH2CH2S); 24.23 (CH2(CH2)2S); 28.06 (CH2(CH2)3S); 36.51 (C6); 50.67 (CH2N); 50.90 (CH2S); 62.55 (CH2C=CH); 65.29 (CH2N); 67.88, 69.08, 69.54, 70.70 (13CH2O); 52.32, 69.36, 69.82, 70.16 (C2-5); 99.48 (C1); 109.39 (C=CH); 146.74 (NCH); 175.27 ppm (CO2H); MS (ESI) m/z: 765.86 [M-3H+3Na]+. = +54.8 (c = 1.00 in methanol); 1H-NMR (D2O): δ = 3.40 (s, 3H, OCH3); 3.54 (dd, 1H, J = 6.2 Hz, J = 13.3 Hz, H6a); 3.60–3.73 (m, 4 h, H6b, H5, H4 and H3); 3.91 (dd, 1H, J = 3.3 Hz, J = 1.7 Hz, H2); 4.73 (d, 1H, J = 1.6 Hz, H1); 13C-NMR (D2O): δ = 51.4 (C6); 55.2 (OCH3); 67.8 (C5); 70.2 (C2); 70.7 (C3); 71.6 (C4); 101.4 ppm (C1); MS(ESI) m/z:242.31 [M+Na]+, 218.14 [M-H]−. = +65.7 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.97, 2.05, 2.13 (3s, 9H, COCH3); 3.16 (dd, 1H, J = 8.8 Hz, J = 10.8 Hz, H6a); 3.29 (dd, 1H, J = 2.6 Hz, J = 11.0 Hz, H6b); 3.46 (s, 3H, OCH3); 3.78 (td, 1H, J = 2.4 Hz, J = 9.2 Hz, H5); 4.71 (s, 1H, J = 1.2 Hz, H1); 5.09 (t, 1H, J = 9.8 Hz, H4); 5.20 (m, 1H, H2); 5.29 ppm (dd, 1H, J = 3.6 Hz, J = 10.0 Hz, H3); 13C-NMR (CDCl3): δ = 3.85 (C6); 20.60, 20.73, 20.80 (3COCH3); 55.49 (OCH3); 68.60 (C3); 69.52 (C2); 69.90 (C4); 70.07 (C5); 98.44 (C1); 169.77, 169.80, 169.95 ppm (3C=O); MS (ESI) m/z: 368.24 [M+Na]+. = −42.7 (c = 1.01 in chloroform); 1H-NMR (CDCl3): δ = 1.98, 2.03, 2.14, 2.15 (4s, 12H, 4CH3); 3.28 (dd, 1H, J = 5.6 Hz, J = 13.6 Hz, H6a); 3.37 (dd, 1H, J = 2.4 Hz, J = 13.2 Hz, H6b); 3.97 (m, 1H, H5); 5.22 (s, 1H, H2); 5.31 (m, 2H, H3 and H4); 6.06 ppm (d, 1H, J = 1.6 Hz, H1); 13C-NMR (CDCl3): δ = 20.53, 20.56, 20.63, 20.72 (4CH3); 50.55 (C6); 66.30, 68.43 (C3 and C4); 68.14 (C2); 71.70 (C5); 90.19 (C1); 167.96, 169.49, 169.68, 169.92 ppm (4C=O); MS (ESI) m/z: 396.22 [M+Na]+, 408.35 [M-Cl]−. = + 37.7 (c = 1.00 in chloroform); 1H-NMR (acetone-d6): δ = 1.24, 1.41 (2s, 6H, 2CCH3); 2.76 (dd, 1H, J = 9.3 Hz, J = 17.3 Hz, H6a); 3.18 (dd, 1H, J = 2.8 Hz, J = 17.3 Hz, H6b); 3.46 (s, 3H, OCH3); 3.86 (td, 1H, J = 9.6 Hz, J = 2.8 Hz, H5); 4.15 (d, 1H, J = 7.4 Hz, H2); 4.21 (dd, 1H, J = 9.9 Hz, J = 7.0 Hz, H4); 4.44 (m, 1H, H3); 4.93 ppm (s, 1H, H1); 13C-NMR (acetone-d6): δ = 20.60 (C6); 25.5, 27.10 (2CCH3); 54.5 (OCH3); 64.90 (C5); 75.62 (C2); 76.34 (C4); 76.90 (C3); 98.17 (C1); 109.88 (C(CH3)2); 118.13 ppm (CN); MS (ESI) m/z: 384.23 [M+Na]+, 322.42 [M-Na]−. = +17.23 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.33, 1.53 (2s, 6H, 2CCH3); 2.40 (dd, 1H, J = 9.8 Hz, J = 15.8 Hz, H6a); 3.09 (dd, 1H, J = 2.2 Hz, J = 16.2 Hz, H6b); 3.41 (s, 3H, OCH3); 4.09 (m, 2H, H2 and H5); 4.21 (m, 2H, H3 and H4); 4.81 ppm (s, 1H, H1); 13C-NMR (CD3OD): δ = 26.57, 28.12 (2CCH3); 38.32 (C6); 55.76 (OCH3); 66.91 (C2); 77.19, 78.07, 79.05 (C3, C4 and C5); 99.42 (C1); 110.75 (C(CH3)2); 175.20 ppm (CO2H); MS (ESI) m/z: 387.99 [M+Na]+, 363.12 [M-H]−. = +10.9 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.29, 1.59 (2s, 6H, 2CH=CCH3); 2.00 (m, 4 h, CH2CH2); 2.01, 2.09, 2.14, 2.31 (4s, 12H, 4COCH3); 3.50 (s, 2H, CH2S); 3.78 (m, 1H, CHSO2); 3.97 (m, 4 h, H6 and CH2CHSO2); 4.11 (m, 3H, H5 and CH2O); 4.64 (d, 1H, J = 1.2 Hz, H1); 4.74 (m, 2H, OCH2CH=CH); 5.11 (t, 1H, J = 10.2 Hz, H4); 5.20 (m, 1H, CHCHSO2); 5.30 (m, 2H, H2 and CH=CCH2S); 5.35 (dd, 1H, J = 10.4 Hz, J = 3.6 Hz, H3); 7.53 (m, 2H, CHAr); 7.64 (m, 1H, CHAr); 7.84 ppm (d, 2H, J = 7.2 Hz, CHAr); 13C-NMR (CDCl3): δ = 15.11, 16.09 (2CH=CCH3); 20.58, 20.72, 20.79, 30.44 (4COCH3); 26.15, 39.05 (CH2CH2); 37.96 (CH2S); 55.53 (CHSO2); 55.96, 59.43 (C6 and CHCHSO2); 62.24 (CH2O); 63.25 (C5); 66.10 (C3); 66.82 (C4); 70.31 (C2); 98.34, 98.65 (OCH2CH=CH); 99.35 (C1); 110.57 (CHCHSO2); 127.73 (CH=CCH2S); 128.43 (CHAr); 128.91 (CHAr); 131.96 (CHAr); 133.49 (CHAr); 130.97, 138.61, 145.81 (2C=CH and CSO2); 169.63, 169.73, 170.78 ppm (4C=O); MS (ESI) m/z: 737.01 [M+H]+, 758.98 [M+Na]+. = +9.4 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.29, 1.58 (2s, 6H, 2CH=CCH3); 1.99 (m, 4 h, CH2CH2); 2.04, 2.07, 2.15, 2.31 (4s, 12H, 4COCH3); 3.50 (s, 2H, CH2S); 3.78 (m, 1H, CHSO2); 4.05 (m, 5 h, H5, CH2O and CH2CHSO2); 4.12 (dd, 1H, J = 2.4 Hz, J = 14.4 Hz, H6a); 4.27 (dd, 1H, J = 4.8 Hz, J = 12.4 Hz, H6b); 5.15 (t, 4 h, J = 8.0 Hz, CH=CH, 2CH=CCH3); 5.25 (m, 1H, H2); 5.32 (m, 2H, H3 and H4); 6.08 (d, 1H, J = 1.6 Hz, H1); 7.52 (t, 2H, J = 7.6 Hz, CHAr); 7.63 (m, 1H, CHAr); 7.84 ppm (d, 2H, J = 7.6 Hz, CHAr); 13C-NMR (CDCl3): δ = 15.02, 15.99 (2CH=CCH3); 20.50, 20.60, 20.70 (4COCH3); 26.05, 38.95 (CH2CH2); 37.86 (CH2S); 55.85 (CSO2); 61.92 (C6, CH2O and CH2CHSO2); 65.33, 68.57 (C3 and C4); 68.16 (C2); 70.42 (C5); 90.41 (C1); 110.49, 126.98 (2CH=CCH3); 110.81 (CH=CH); 127.44–133.42 (5CHAr); 138.48, 145.70, 146.84 (2C=CH and CSO2); 169.36, 169.56, 168.80, 170.44 (4C=O); 195.37 ppm (CO2H); MS (ESI) m/z: 740.05 [M+H]+, 761.99 [M+Na]+. = +4.4 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.30, 1.55 (2s, 6H, 2CH3); 1.99 (m, 4 h, CH2CH2); 3.50 (s, 2H, CH2S); 3.76 (m, 1H, CH=CH); 4.03 (m, 5 h, H5, CH2O and CH=CH); 4.12 (dd, 1H, J = 2.4 Hz, J = 14.4 Hz, H6a); 4.27 (dd, 1H, J = 4.8 Hz, J = 12.4 Hz, H6b); 5.15 (t, 4 h, J = 8.0 Hz, 2CH=CH, 2CH=C); 5.25 (m, 1H, H2); 5.32 (m, 2H, H3 and H4); 6.08 ppm (d, 1H, J = 1.6 Hz, H1); 13C-NMR (CDCl3): δ = 15.02, 15.99 (2CH3); 26.05, 38.95 (CH2CH2); 37.86 (CH2S); 55.85 (CH=CH); 61.92 (C6, CH2O andCH=CH); 65.33, 68.57 (C3 and C4); 68.16 (C2); 70.42 (C5); 90.41 (C1); 110.49, 126.98 (2CH=C); 110.81 (2CH=CH); 138.48, 145.70 (2C=CH); 195.37 ppm (CO2H); MS (ESI) m/z: 471.76 [M+H]+, 493.78 [M+Na]+. = +96.1 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 2.01, 2.10, 2.17 (3s, 9H, 3CH3); 3.21 (dd, 1H, J = 6.6 Hz, J = 11.4 Hz, H6a); 3.35 (dd, 1H, J = 2.9 Hz, J = 11.4 Hz, H6b); 3.94–3.99 (m, 1H, H5); 5.27 (t, 1H, J = 10.0 Hz, H4); 5.41 (dd, 1H, J = 1.4 Hz, J = 3.3 Hz, H2); 5.71 (dd, 1H, J = 3.4 Hz, J = 10.0 Hz, H3); 6.3 ppm (d, 1H, J = 1.1 Hz, H1); 13C-NMR (CDCl3): δ = 2.46 (C6); 20.60, 20.73, 20.78 (3CH3); 67.66 (C3); 69.49 (C4); 72.21 (C2); 73.45 (C5); 82.46 (C1); 169.58, 169.64, 169.72 (3C=O); MS (ESI) m/z: 393.98, 395.40 [M+H]+. = +43.8 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.98 (s, 3H, CH3); 2.03 (m, 5 h, CH2CH2S and CH3); 2.14 (s, 3H, CH3); 3.27 (m, 5 h, H6a, CH2S and CH2(CH2)2S); 3.35 (dd, 1H, J = 6.4 Hz, J = 13.2 Hz, H6b); 3.59–3.72 (m, 12H, 6CH2O); 4.01 (m, 1H, H5); 4.87 (d, 1H, J = 7.4 Hz, H1); 5.22 (t, 1H, J = 10.0 Hz, H4); 5.25 (dd, 1H, J = 1.6 Hz, J = 3.6 Hz, H2); 5.35 ppm (dd, 1H, J = 3.2 Hz, J = 10.0 Hz, H3); 13C-NMR (CDCl3): δ = 20.65, 20.69, 20.83 (3CH3); 27.98 (CH2S); 35.59 (CH2CH2S); 58.73 (CH2(CH2)2S); 51.05 (C6); 61.62–72.51 (CH2O); 67.16 (C4); 68.80 (C3); 69.48 (C2); 69.93 (C5); 97.42 (C1); 169.82, 169.95, 170.08 ppm (3C=O); MS(ESI) m/z: 560.12 [M+Na]+. = +40.0 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.87 (qt, 2H, J = 6.8 Hz, CH2CH2S); 1.95, 2.06, 2.12 (3s, 12H, 4CH3); 2.69 (m, 2H, CH2S); 3.51 (t, 2H, J = 6.0 Hz, CH2(CH2)2S); 3.54–3.69 (m, 12H, 6CH2O); 4.05 (dd, 1H, J = 2.2 Hz, J = 12.2 Hz, H6a); 4.27 (dd, 1H, J = 5.4 Hz, J = 12.2 Hz, H6b); 4.34 (m, 1H, H5); 4.80 (d, 1H, J = 7.4 Hz, H1); 5.23 (m, 2H, H3 et H4); 5.30 ppm (dd, 1H, J = 1.6 Hz, J = 3.2 Hz, H2); 13C-NMR (CDCl3): δ = 19.62, 19.69, 19.91 (3CH3); 27.19 (CH2S); 28.41 (CH2CH2S); 60.69 (CH2(CH2)2S); 61.41 (C6); 65.31 (C3); 67.96 (C5); 68.45 (C4); 68.26–71.53 (7CH2O); 70.14 (C2); 81.64 (C1); 168.72, 168.80, 168.99, 169.63 ppm (3C=O et CO2H); MS (ESI) m/z: 563.76 [M+Na]+. = +40.0 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.95 (m, 2H, CH2CH2SC); 2.88 (t, 2H, J = 6.6 Hz, CH2SH); 3.26 (m, 4 h, CH2SC and CH2(CH2)2SC); 3.40 (dd, 1H, J = 7.0 Hz, J = 13.0 Hz, H6a); 3.55 (m, 2H, H5 and H6a); 3.58–3.69 (m, 10H, H3,4 and 4CH2O); 3.72 (t, 2H, J = 6.4 Hz, CH2CH2SH); 3.80 (dd, 1H, J = 1.6 Hz, J = 3.2 Hz, H2); 4.77 ppm (d, 1H, J = 1.6 Hz, H1); 13C-NMR (CD3OD): δ = 28.02 (CH2SC); 37.42 (CH2CH2SC); 39.51 (CH2SH); 57.92 (CH2(CH2)2SC); 53.02 (C6); 62.55–71.65 (5CH2O); 69.52, 72.01, 72.36 (C3, C4 and C5); 73.86 (C2); 101.87 ppm (C1); MS (ESI) m/z: 428.73 [M+H]+, 460.57 [M+Na]+. = +32.0 (c = 1.05 in chloroform); 1H-NMR (CD3OD): δ = 1.92 (qt, 2H, J = 6.4 Hz, CH2CH2SC); 2.73 (m, 2H, CH2SC); 3.09 (t, 2H, J = 6.6 Hz, CH2(CH2)2SC); 3.53 (t, 2H, J = 6.0 Hz, CH2SH); 3.55–3.62 (m, 10H, 5CH2O); 4.08 (dd, 1H, J = 2.2 Hz, J = 12.2 Hz, H6a); 4.30 (dd, 1H, J = 5.4 Hz, J = 12.2 Hz, H6b); 4.37 (m, 1H, H5); 5.21 (m, 2H, H1, H3 and H4); 5.31 (dd, 1H, J = 1.6 Hz, J = 3.2 Hz, H2); 13C-NMR (CD3OD): δ = 27.22 (CH2SC); 27.82 (CH2CH2SC); 28.48 (CH2(CH2)2SC); 61.41 (C6); 65.32 (C3); 67.96 (C5); 68.44 (C4); 68.28–69.59 (5CH2O and CH2SH); 70.14 (C2); 81.65 (C1);168.90 (CO2H); MS (ESI) m/z: 431.59 [M+H]+, 463.64 [M+Na]+.
= +42.1 (c = 0.5 in chloroform); 1H-NMR (CDCl3): δ = 2.00, 2.05, 2.11, 2.16 (4s, 12H, 4CH3); 3.52 (t, J = 6.0 Hz, 2H, CH2Br); 3.93 (m, 2H, CH2CH2Br); 4.13 (m, 2H, H5 and H6a); 4.27 (dd, 1H, J = 5.8 Hz, J = 12.6 Hz, H6b); 4.88 (d, J = 1.6 Hz, H1); 5.27 (dd, 1H, J = 2.0 Hz, J = 3.2 Hz, H2); 5.29 (t, 1H, J = 1.6 Hz, H4); 5.35 ppm (dd, 1H, J = 3.6 Hz, J = 10.0 Hz, H3);13C-NMR (CDCl3): δ = 20.67, 20.70, 20.75, 20.87 (4CH3); 29.60 (CH2CH2Br); 62.41 (C6); 66.00 (C4); 68.48 (CH2Br); 68.93 (C5); 69.02 (C3); 69.42 (C2); 97.75 (C1); 169.76, 169.86, 170.03, 170.62 ppm (4CO); MS (ESI) m/z: 477.01, 478.95 [M+Na]+. = +39.0 (c = 0.6 in chloroform); 1H-NMR (CDCl3): δ = 2.0, 2.05, 2.11, 2.16 (4s, 12H, 4CH3); 3.47 (m, 2H, CH2N3); 3.67 (m, 1H, CH2CH2N3); 3.87 (m, 1H, CH2CH2N3); 4.05 (ddd, 1H, J = 2.4 Hz, J = 5.2 Hz, J = 9.7 Hz, H5); 4.13 (dd, 1H, J = 2.6 Hz, J = 12.2 Hz, H6a); 4.29 (dd, 1H, J = 5.2 Hz, J = 12.4 Hz, H6b); 4.87 (d, 1H, J = 1.6 Hz, H1); 5.30 (t, 1H, J = 10.0 Hz, H4); 5.28 (dd, 1H, J = 2.0 Hz, J = 3.2 Hz, H2); 5.36 ppm (dd, 1H, J = 3.2 Hz, J = 10.0 Hz, H3); 13C-NMR (CDCl3) : δ = 20.63, 20.68, 20.71, 20.84 (4CH3); 50.32 (CH2N3); 62.42 (C6); 65.96 (C4); 67.02 (CH2CH2N3); 68.82 (C5 and C3); 69.36 (C2); 97.71 (C1); 169.73, 169.78, 169.98, 170.59 ppm (4CO); MS (ESI) m/z: 440.12 [M+Na]+. = + 54.9 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 3.41 (t, 2H, J = 5.0 Hz, CH2N3); 3.60 (m, 3H, H3, H5 and CH2CH2N3); 3.71 (m, 2H, H4 and H6a); 3.85 (m, 2H, H2 and H6b); 3.92 (m, 1H, CH2CH2N3); 4.81 ppm (d, 1H, J = 1.2 Hz, H1); MS (ESI) m/z: 272.11 [M+Na]+, 288.02 [M+K]+, 521.19 [2M+Na]+. = + 54.9 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.68 (m, 2H, CH2CH2CH=CH2); 2.09 (m, 2H, CH2CH=CH2); 3.46 (t, 2H, J = 6.6 Hz, CH2CH2CH2CH=CH2); 3.56–3.73 (m, 24 h, 12CH2O); 4.99 (m, 2H, CH=CH2); 5.81 ppm (m, 2H, CH=CH2); 13C-NMR (CDCl3): δ = 28.66 (CH2CH2CH=CH2); 30.12 (CH2CH=CH2); 61.51–72.58 (13CH2O); 114.59 (CH=CH2); 138.18 ppm (CH=CH2); MS (ESI) m/z: 373.27 [M+Na]+, 389.20 [M+K]+. = + 36.0 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.36 (m, 6H, CH2CH2CH2CH2S); 1.34, 1.49 (2s, 6H, 2CCH3); 2.14 (t, 2H, J = 7.2 Hz, CH2S); 3.38 (t, 2H, J = 6.4 Hz, CH2(CH2)4S); 3.44–3.66 (m, 25 h, H5 and 12CH2O); 3.78 (s, 3H, OCH3); 3.97 (m, 1H, CH2CH2N); 4.15 (m, 1H, CH2CH2N); 4.27 (m, 3H, H2, H3 and H6a); 4.50 (m, 2H, H6b and H4); 4.64 (m, 4 h, CH2N and CH2C=CH); 5.12 (s, 1H, H1); 6.81–7.39 (m, 14 h, CHAr); 8.07 ppm (s, 1H, NCH); 13C-NMR (CD3OD): δ = 26.34, 28.20 (2CCH3); 26.71 (CH2(CH2)2S); 29.62 (CH2CH2S); 30.23 (CH2(CH2)3S); 33.03 (SCH2); 51.19 (CH2N); 55.79 (OCH3); 59.79 (C5); 65.14 (CH2C=CH); 67.42 (CH2CH2N); 70.95, 71.19, 71.49, 71.58, 72.03 (13CH2O); 73.53 (C6); 74.46, 77.23 (C2 and C3); 85.66 (C4); 98.96 (C1); 108.26, 111.66 (SC and C(CH3)2); 114.11, 127.66, 128.86, 130.73, 132.02 (14CHAr); 126.04 (NCH); 138.40, 146.86 (3SCCAr and C=CH); 159.71 ppm (COCH3); MS (ESI) m/z: 1068.62 [M+Na]+, 1080.77 [M+Cl]−. = +17.1 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.37 (m, 6H, CH2CH2CH2CH2S); 1.30, 1.45 (2s, 6H, 2CCH3); 2.17 (t, 2H, J = 7.2 Hz, CH2S); 3.37 (t, 2H, J = 6.6 Hz, CH2(CH2)4S); 3.42–3.67 (m, 27H, H2 and 13CH2O); 3.79 (s, 3H, OCH3); 3.96 (m, 1H, CH2CH2N); 4.12 (m, 1H, CH2CH2N); 4.07 (d, 1H, J = 6.0 Hz, H5); 4.25 (m, 1H, H3); 4.38 (t, 1H, J = 5.8 Hz, H4); 4.70 (m, 6H, H6, CH2N and CH2C=CH); 4.93 (s, 1H, H1); 6.87–7.41 (m, 14 h, CHAr); 9.19 ppm (s, 1H, NCH); 13C-NMR (CD3OD): δ = 27.20, 28.67 (2CCH3); 27.33, 30.16, 30.87 (CH2CH2CH2CH2S); 33.55 (CH2S); 51.66 (C6 and CH2(CH2)4S); 54.55 (CH2N); 56.53 (OCH3); 65.58 (CH2C=CH); 67.89 (CH2CH2N); 70.39–72.33 (12CH2O); 72.13 (C2); 73.88 (C3); 76.31 (C5); 77.37 (C4); 99.59 (C1); 110.79 (SC); 114.87–132.54 (14CHAr); 126.23 (NCH); 138.80–160.13 (3SCCAr and C=CH); 158.40 ppm (COCH3); MS(ESI) m/z:1134.58 [M+Na]+. = −2.1 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.47 (m, 2H, CH2(CH2)2S); 1.60 (m, 2H, CH2(CH2)3S); 1.71 (m, 2H, CH2CH2S); 2.70 (t, 2H, J = 7.2 Hz, CH2S); 3.48 (t, 2H, J = 6.2 Hz, CH2(CH2)4S); 3.19–3.78 (m, 30H, H2-6 and 12CH2O); 3.88 (m, 1H, CH2CH2N); 4.13 (m, 1H, CH2CH2N); 4.63 (m, 4 h, CH2N and CH2C=CH); 4.72 (s, 1H, H1); 8.03 ppm (s, 1H, NCH); 13C-NMR (CD3OD): δ = 26.13 (CH2(CH2)3S); 30.07 (CH2CH2S); 30.36 (CH2(CH2)3S); 39.66 (CH2S); 51.34 (CH2N); 62.85 (C6); 65.05 (CH2C=CH); 66.79 (CH2CH2N); 68.38, 70.81, 71.24, 71.59, 71.93, 72.15, 72.51, 75.01 (C2-5 and 13CH2O); 101.70 (C1); 132.57 (NCH); 161.04 ppm (CH=C); MS (ESI) m/z: 798.62 [M+Na]+. = −9.0 (c = 1.00 in chloroform); 1H-NMR (D2O): δ = 1.35, 1.52 (2s, 6H, 2CCH3); 1.44 (m, 2H, CH2(CH2)2S); 1.60 (m, 2H, CH2(CH2)3S); 1.73 (m, 2H, CH2CH2S); 2.29 (dd, 1H, J = 10.6 Hz, J = 15.0Hz, H6a); 2.80 (dd, 1H, J = 2.0 Hz, J = 15.2 Hz, H6b); 2.89 (t, 2H, J = 8.0 Hz, CH2S); 3.53 (t, 2H, J = 6.6 Hz, CH2(CH2)4S); 3.50–3.69 (m, 25 h, H5 and 12CH2O); 3.75 (s, 3H, OCH3); 3.88 (m, 1H, CH2CH2N); 4.17 (m, 2H, CH2CH2N and H4); 4.19 (d, 1H, J = 5.6 Hz, H2); 4.30 (m, 1H, H3); 4.68 (m, 4 h, CH2N and CH2C=CH); 4.95 (s, 1H, H1); 6.80–7.37 (m, 14 h, CHAr); 8.12 ppm (s, 1H, NCH); 13C-NMR (D2O): δ = 23.80 (CH2CH2S); 24.24 (CH2(CH2)2S); 25.45, 26.80 (2CCH3); 28.06 (CH2(CH2)3S); 38.92 (C6); 49.99 (CH2N); 50.89 (CH2S); 55.86 (OCH3); 62.95 (CH2C=CH); 65.47 (CH2CH2N); 66.41 (C5); 66.54, 68.69, 69.07, 69.40, 69.53, 70.70 (13CH2O); 75.21 (C2); 76.00 (C3); 78.30 (C4); 95.99 (C1); 110.39 (SC and C(CH3)2); 114.14–132.02 (14CHAr); 125.55 (NCH); 143.85 (C=CH); 138.38, 146.00, 146.84 (3SCCAr); 159.71 (COCH3);177.75 (CO2H); MS (ESI) m/z: 1037.34 [M+Na]+. = +10.2 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.49 (m, 2H, CH2(CH2)2S); 1.61 (m, 2H, CH2(CH2)3S); 1.80 (m, 2H, CH2CH2S); 2.41 (dd, 1H, J = 10.2 Hz, J = 16.2 Hz, H6a); 2.84 (m, 3H, CH2S and H6b); 3.49 (t, 2H, J = 6.4 Hz, CH2(CH2)4S); 3.40–3.79 (m, 28 h, H2-5 and 12 CH2O); 3.92 (m, 1H, CH2CH2N); 4.22 (m, 1H, CH2CH2N); 4.71 (d, 1H, J = 1.2 Hz, H1); 4.87 (m, 2H, CH2N); 4.92 (m, 2H, CH2C=CH); 8.65 (s, 1H, NCH); 13C-NMR (D2O): δ = 23.79 (CH2CH2S); 24.23 (CH2(CH2)2S); 28.06 (CH2(CH2)3S); 36.51 (C6); 50.67 (CH2N); 50.90 (CH2S); 62.55 (CH2C=CH); 65.29 (CH2N); 67.88, 69.08, 69.54, 70.70 (13CH2O); 52.32, 69.36, 69.82, 70.16 (C2-5); 99.48 (C1); 109.39 (C=CH); 146.74 (NCH); 175.27 ppm (CO2H); MS (ESI) m/z: 765.86 [M-3H+3Na]+. = +54.8 (c = 1.00 in methanol); 1H-NMR (D2O): δ = 3.40 (s, 3H, OCH3); 3.54 (dd, 1H, J = 6.2 Hz, J = 13.3 Hz, H6a); 3.60–3.73 (m, 4 h, H6b, H5, H4 and H3); 3.91 (dd, 1H, J = 3.3 Hz, J = 1.7 Hz, H2); 4.73 (d, 1H, J = 1.6 Hz, H1); 13C-NMR (D2O): δ = 51.4 (C6); 55.2 (OCH3); 67.8 (C5); 70.2 (C2); 70.7 (C3); 71.6 (C4); 101.4 ppm (C1); MS(ESI) m/z:242.31 [M+Na]+, 218.14 [M-H]−. = +65.7 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.97, 2.05, 2.13 (3s, 9H, COCH3); 3.16 (dd, 1H, J = 8.8 Hz, J = 10.8 Hz, H6a); 3.29 (dd, 1H, J = 2.6 Hz, J = 11.0 Hz, H6b); 3.46 (s, 3H, OCH3); 3.78 (td, 1H, J = 2.4 Hz, J = 9.2 Hz, H5); 4.71 (s, 1H, J = 1.2 Hz, H1); 5.09 (t, 1H, J = 9.8 Hz, H4); 5.20 (m, 1H, H2); 5.29 ppm (dd, 1H, J = 3.6 Hz, J = 10.0 Hz, H3); 13C-NMR (CDCl3): δ = 3.85 (C6); 20.60, 20.73, 20.80 (3COCH3); 55.49 (OCH3); 68.60 (C3); 69.52 (C2); 69.90 (C4); 70.07 (C5); 98.44 (C1); 169.77, 169.80, 169.95 ppm (3C=O); MS (ESI) m/z: 368.24 [M+Na]+. = −42.7 (c = 1.01 in chloroform); 1H-NMR (CDCl3): δ = 1.98, 2.03, 2.14, 2.15 (4s, 12H, 4CH3); 3.28 (dd, 1H, J = 5.6 Hz, J = 13.6 Hz, H6a); 3.37 (dd, 1H, J = 2.4 Hz, J = 13.2 Hz, H6b); 3.97 (m, 1H, H5); 5.22 (s, 1H, H2); 5.31 (m, 2H, H3 and H4); 6.06 ppm (d, 1H, J = 1.6 Hz, H1); 13C-NMR (CDCl3): δ = 20.53, 20.56, 20.63, 20.72 (4CH3); 50.55 (C6); 66.30, 68.43 (C3 and C4); 68.14 (C2); 71.70 (C5); 90.19 (C1); 167.96, 169.49, 169.68, 169.92 ppm (4C=O); MS (ESI) m/z: 396.22 [M+Na]+, 408.35 [M-Cl]−. = + 37.7 (c = 1.00 in chloroform); 1H-NMR (acetone-d6): δ = 1.24, 1.41 (2s, 6H, 2CCH3); 2.76 (dd, 1H, J = 9.3 Hz, J = 17.3 Hz, H6a); 3.18 (dd, 1H, J = 2.8 Hz, J = 17.3 Hz, H6b); 3.46 (s, 3H, OCH3); 3.86 (td, 1H, J = 9.6 Hz, J = 2.8 Hz, H5); 4.15 (d, 1H, J = 7.4 Hz, H2); 4.21 (dd, 1H, J = 9.9 Hz, J = 7.0 Hz, H4); 4.44 (m, 1H, H3); 4.93 ppm (s, 1H, H1); 13C-NMR (acetone-d6): δ = 20.60 (C6); 25.5, 27.10 (2CCH3); 54.5 (OCH3); 64.90 (C5); 75.62 (C2); 76.34 (C4); 76.90 (C3); 98.17 (C1); 109.88 (C(CH3)2); 118.13 ppm (CN); MS (ESI) m/z: 384.23 [M+Na]+, 322.42 [M-Na]−. = +17.23 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.33, 1.53 (2s, 6H, 2CCH3); 2.40 (dd, 1H, J = 9.8 Hz, J = 15.8 Hz, H6a); 3.09 (dd, 1H, J = 2.2 Hz, J = 16.2 Hz, H6b); 3.41 (s, 3H, OCH3); 4.09 (m, 2H, H2 and H5); 4.21 (m, 2H, H3 and H4); 4.81 ppm (s, 1H, H1); 13C-NMR (CD3OD): δ = 26.57, 28.12 (2CCH3); 38.32 (C6); 55.76 (OCH3); 66.91 (C2); 77.19, 78.07, 79.05 (C3, C4 and C5); 99.42 (C1); 110.75 (C(CH3)2); 175.20 ppm (CO2H); MS (ESI) m/z: 387.99 [M+Na]+, 363.12 [M-H]−. = +10.9 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.29, 1.59 (2s, 6H, 2CH=CCH3); 2.00 (m, 4 h, CH2CH2); 2.01, 2.09, 2.14, 2.31 (4s, 12H, 4COCH3); 3.50 (s, 2H, CH2S); 3.78 (m, 1H, CHSO2); 3.97 (m, 4 h, H6 and CH2CHSO2); 4.11 (m, 3H, H5 and CH2O); 4.64 (d, 1H, J = 1.2 Hz, H1); 4.74 (m, 2H, OCH2CH=CH); 5.11 (t, 1H, J = 10.2 Hz, H4); 5.20 (m, 1H, CHCHSO2); 5.30 (m, 2H, H2 and CH=CCH2S); 5.35 (dd, 1H, J = 10.4 Hz, J = 3.6 Hz, H3); 7.53 (m, 2H, CHAr); 7.64 (m, 1H, CHAr); 7.84 ppm (d, 2H, J = 7.2 Hz, CHAr); 13C-NMR (CDCl3): δ = 15.11, 16.09 (2CH=CCH3); 20.58, 20.72, 20.79, 30.44 (4COCH3); 26.15, 39.05 (CH2CH2); 37.96 (CH2S); 55.53 (CHSO2); 55.96, 59.43 (C6 and CHCHSO2); 62.24 (CH2O); 63.25 (C5); 66.10 (C3); 66.82 (C4); 70.31 (C2); 98.34, 98.65 (OCH2CH=CH); 99.35 (C1); 110.57 (CHCHSO2); 127.73 (CH=CCH2S); 128.43 (CHAr); 128.91 (CHAr); 131.96 (CHAr); 133.49 (CHAr); 130.97, 138.61, 145.81 (2C=CH and CSO2); 169.63, 169.73, 170.78 ppm (4C=O); MS (ESI) m/z: 737.01 [M+H]+, 758.98 [M+Na]+. = +9.4 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.29, 1.58 (2s, 6H, 2CH=CCH3); 1.99 (m, 4 h, CH2CH2); 2.04, 2.07, 2.15, 2.31 (4s, 12H, 4COCH3); 3.50 (s, 2H, CH2S); 3.78 (m, 1H, CHSO2); 4.05 (m, 5 h, H5, CH2O and CH2CHSO2); 4.12 (dd, 1H, J = 2.4 Hz, J = 14.4 Hz, H6a); 4.27 (dd, 1H, J = 4.8 Hz, J = 12.4 Hz, H6b); 5.15 (t, 4 h, J = 8.0 Hz, CH=CH, 2CH=CCH3); 5.25 (m, 1H, H2); 5.32 (m, 2H, H3 and H4); 6.08 (d, 1H, J = 1.6 Hz, H1); 7.52 (t, 2H, J = 7.6 Hz, CHAr); 7.63 (m, 1H, CHAr); 7.84 ppm (d, 2H, J = 7.6 Hz, CHAr); 13C-NMR (CDCl3): δ = 15.02, 15.99 (2CH=CCH3); 20.50, 20.60, 20.70 (4COCH3); 26.05, 38.95 (CH2CH2); 37.86 (CH2S); 55.85 (CSO2); 61.92 (C6, CH2O and CH2CHSO2); 65.33, 68.57 (C3 and C4); 68.16 (C2); 70.42 (C5); 90.41 (C1); 110.49, 126.98 (2CH=CCH3); 110.81 (CH=CH); 127.44–133.42 (5CHAr); 138.48, 145.70, 146.84 (2C=CH and CSO2); 169.36, 169.56, 168.80, 170.44 (4C=O); 195.37 ppm (CO2H); MS (ESI) m/z: 740.05 [M+H]+, 761.99 [M+Na]+. = +4.4 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.30, 1.55 (2s, 6H, 2CH3); 1.99 (m, 4 h, CH2CH2); 3.50 (s, 2H, CH2S); 3.76 (m, 1H, CH=CH); 4.03 (m, 5 h, H5, CH2O and CH=CH); 4.12 (dd, 1H, J = 2.4 Hz, J = 14.4 Hz, H6a); 4.27 (dd, 1H, J = 4.8 Hz, J = 12.4 Hz, H6b); 5.15 (t, 4 h, J = 8.0 Hz, 2CH=CH, 2CH=C); 5.25 (m, 1H, H2); 5.32 (m, 2H, H3 and H4); 6.08 ppm (d, 1H, J = 1.6 Hz, H1); 13C-NMR (CDCl3): δ = 15.02, 15.99 (2CH3); 26.05, 38.95 (CH2CH2); 37.86 (CH2S); 55.85 (CH=CH); 61.92 (C6, CH2O andCH=CH); 65.33, 68.57 (C3 and C4); 68.16 (C2); 70.42 (C5); 90.41 (C1); 110.49, 126.98 (2CH=C); 110.81 (2CH=CH); 138.48, 145.70 (2C=CH); 195.37 ppm (CO2H); MS (ESI) m/z: 471.76 [M+H]+, 493.78 [M+Na]+. = +96.1 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 2.01, 2.10, 2.17 (3s, 9H, 3CH3); 3.21 (dd, 1H, J = 6.6 Hz, J = 11.4 Hz, H6a); 3.35 (dd, 1H, J = 2.9 Hz, J = 11.4 Hz, H6b); 3.94–3.99 (m, 1H, H5); 5.27 (t, 1H, J = 10.0 Hz, H4); 5.41 (dd, 1H, J = 1.4 Hz, J = 3.3 Hz, H2); 5.71 (dd, 1H, J = 3.4 Hz, J = 10.0 Hz, H3); 6.3 ppm (d, 1H, J = 1.1 Hz, H1); 13C-NMR (CDCl3): δ = 2.46 (C6); 20.60, 20.73, 20.78 (3CH3); 67.66 (C3); 69.49 (C4); 72.21 (C2); 73.45 (C5); 82.46 (C1); 169.58, 169.64, 169.72 (3C=O); MS (ESI) m/z: 393.98, 395.40 [M+H]+. = +43.8 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.98 (s, 3H, CH3); 2.03 (m, 5 h, CH2CH2S and CH3); 2.14 (s, 3H, CH3); 3.27 (m, 5 h, H6a, CH2S and CH2(CH2)2S); 3.35 (dd, 1H, J = 6.4 Hz, J = 13.2 Hz, H6b); 3.59–3.72 (m, 12H, 6CH2O); 4.01 (m, 1H, H5); 4.87 (d, 1H, J = 7.4 Hz, H1); 5.22 (t, 1H, J = 10.0 Hz, H4); 5.25 (dd, 1H, J = 1.6 Hz, J = 3.6 Hz, H2); 5.35 ppm (dd, 1H, J = 3.2 Hz, J = 10.0 Hz, H3); 13C-NMR (CDCl3): δ = 20.65, 20.69, 20.83 (3CH3); 27.98 (CH2S); 35.59 (CH2CH2S); 58.73 (CH2(CH2)2S); 51.05 (C6); 61.62–72.51 (CH2O); 67.16 (C4); 68.80 (C3); 69.48 (C2); 69.93 (C5); 97.42 (C1); 169.82, 169.95, 170.08 ppm (3C=O); MS(ESI) m/z: 560.12 [M+Na]+. = +40.0 (c = 1.00 in chloroform); 1H-NMR (CDCl3): δ = 1.87 (qt, 2H, J = 6.8 Hz, CH2CH2S); 1.95, 2.06, 2.12 (3s, 12H, 4CH3); 2.69 (m, 2H, CH2S); 3.51 (t, 2H, J = 6.0 Hz, CH2(CH2)2S); 3.54–3.69 (m, 12H, 6CH2O); 4.05 (dd, 1H, J = 2.2 Hz, J = 12.2 Hz, H6a); 4.27 (dd, 1H, J = 5.4 Hz, J = 12.2 Hz, H6b); 4.34 (m, 1H, H5); 4.80 (d, 1H, J = 7.4 Hz, H1); 5.23 (m, 2H, H3 et H4); 5.30 ppm (dd, 1H, J = 1.6 Hz, J = 3.2 Hz, H2); 13C-NMR (CDCl3): δ = 19.62, 19.69, 19.91 (3CH3); 27.19 (CH2S); 28.41 (CH2CH2S); 60.69 (CH2(CH2)2S); 61.41 (C6); 65.31 (C3); 67.96 (C5); 68.45 (C4); 68.26–71.53 (7CH2O); 70.14 (C2); 81.64 (C1); 168.72, 168.80, 168.99, 169.63 ppm (3C=O et CO2H); MS (ESI) m/z: 563.76 [M+Na]+. = +40.0 (c = 1.00 in chloroform); 1H-NMR (CD3OD): δ = 1.95 (m, 2H, CH2CH2SC); 2.88 (t, 2H, J = 6.6 Hz, CH2SH); 3.26 (m, 4 h, CH2SC and CH2(CH2)2SC); 3.40 (dd, 1H, J = 7.0 Hz, J = 13.0 Hz, H6a); 3.55 (m, 2H, H5 and H6a); 3.58–3.69 (m, 10H, H3,4 and 4CH2O); 3.72 (t, 2H, J = 6.4 Hz, CH2CH2SH); 3.80 (dd, 1H, J = 1.6 Hz, J = 3.2 Hz, H2); 4.77 ppm (d, 1H, J = 1.6 Hz, H1); 13C-NMR (CD3OD): δ = 28.02 (CH2SC); 37.42 (CH2CH2SC); 39.51 (CH2SH); 57.92 (CH2(CH2)2SC); 53.02 (C6); 62.55–71.65 (5CH2O); 69.52, 72.01, 72.36 (C3, C4 and C5); 73.86 (C2); 101.87 ppm (C1); MS (ESI) m/z: 428.73 [M+H]+, 460.57 [M+Na]+. = +32.0 (c = 1.05 in chloroform); 1H-NMR (CD3OD): δ = 1.92 (qt, 2H, J = 6.4 Hz, CH2CH2SC); 2.73 (m, 2H, CH2SC); 3.09 (t, 2H, J = 6.6 Hz, CH2(CH2)2SC); 3.53 (t, 2H, J = 6.0 Hz, CH2SH); 3.55–3.62 (m, 10H, 5CH2O); 4.08 (dd, 1H, J = 2.2 Hz, J = 12.2 Hz, H6a); 4.30 (dd, 1H, J = 5.4 Hz, J = 12.2 Hz, H6b); 4.37 (m, 1H, H5); 5.21 (m, 2H, H1, H3 and H4); 5.31 (dd, 1H, J = 1.6 Hz, J = 3.2 Hz, H2); 13C-NMR (CD3OD): δ = 27.22 (CH2SC); 27.82 (CH2CH2SC); 28.48 (CH2(CH2)2SC); 61.41 (C6); 65.32 (C3); 67.96 (C5); 68.44 (C4); 68.28–69.59 (5CH2O and CH2SH); 70.14 (C2); 81.65 (C1);168.90 (CO2H); MS (ESI) m/z: 431.59 [M+H]+, 463.64 [M+Na]+.3.2. Preparation of Citrate-Reduced Gold Nanoparticles
3.3. CAM Assays

4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kornfeld, S. Structure and function of the mannose 6-phosphate insulin-like growth factor-ii receptors. Annu. Rev. Biochem. 1992, 61, 307–330. [Google Scholar] [CrossRef]
- Kang, J.K.; Li, Y.; Leaf, A. Retinoic acid alters the intracellular trafficking of the mannose-6-phosphate insulin-like growth factor II receptor and lysosomal enzymes. Proc. Natl. Acad. Sci. USA 1998, 95, 13671–13676. [Google Scholar]
- Volpert, A.; Jackson, D.; Bouck, N.; Linzer, D.I.H. The insulin-like growth factor II mannose 6-phosphate receptor is required for proliferin-induced angiogenesis. Endocrinology 1996, 137, 3871–3876. [Google Scholar]
- Barragan-Montero, V.; Awwad, A.; Combemale, S.; de Santa Barbara, P.; Jover, B.; Molès, J.-P.; Montero, J.-L. Synthesis of Mannose-6-Phosphate analogues and their utility as angiogenesis regulators. Chem. Med. Chem. 2011, 6, 1771–1774. [Google Scholar] [CrossRef]
- Clavel, C.; de Santa Barbara, P.; Jover, B.; Moles, J.-P.; Montero, J.-L.; Montero, V. Novel Uses of D-Mannopyranose Derivatives. U.S. Patent WO 2009138601 A2, 19 November 2009. [Google Scholar]
- Awwad, A.; Combemale, S.; de Santa Barbara, P.; Jover, B.; Molès, J.-P.; Montero, J.-L.; Barragan-Montero, V. Nouveaux Derives de Mannopyranoside Ayant une Activite Anticancereuse. EP 2499150 A1, 19 September 2012. [Google Scholar]
- De Santa Barbara, P.; Jover, B.; Moles, J.-P.; Montero, J.-L.; Montero, V. Nouvelles Utilisations de Derives de D-Mannopyranose Activateurs de l’angiogenese. EP 2285817 A2, 23 February 2011. [Google Scholar]
- Kitov, P.I.; Bundle, D.R. Carbohydrate-Based Drug Discovery; Wong, C.-H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Volume 2, pp. 541–574. [Google Scholar]
- Kiessling, L.L.; Pontrello, J.K.; Schuster, M.C. Carbohydrate-Based Drug Discovery; Wong, C.-H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Volume 2, pp. 575–608. [Google Scholar]
- Kiessling, L.; Gestwicki, J.E.; Strong, L.E. Synthetic multivalent ligands as probes of signal transduction. Angew. Chem. Int. Ed. 2006, 45, 2348–2368. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, C.R.T. Synthetic multivalent ligands as probes of signal transduction. Acc. Chem. Res. 1995, 28, 321–327. [Google Scholar] [CrossRef]
- Lis, H.; Sharon, N. Lectins: Carbohydrate-specific proteins that mediate cellular recognition. Chem. Rev. 1998, 98, 637–674. [Google Scholar] [CrossRef]
- Lundquist, J.J.; Toone, E.J. The cluster glycoside effect. Chem. Rev. 2002, 102, 555–578. [Google Scholar] [CrossRef]
- De La Fuente, J.M.; Barrientos, A.G.; Rojas, T.C.; Rojo, J.; Cañada, J.; Fernández, A.; Penadés, S. Gold glyconanoparticles as water-soluble polyvalent models to study carbohydrate interactions. Angew. Chem. Int. Ed. 2001, 40, 2257–2261. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; Wiley-VCH: Weinheim, Germany, 1984; Volume 1, pp. 1–176. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Li, N.; Binder, W.H. Click-chemistry for nanoparticle-modification. Mater. Chem. 2011, 21, 16717–16734. [Google Scholar] [CrossRef]
- Bastide, J.; Henri-Rousseau, O. The Chemistry of the Carbon-Carbon Triple Bond; Patai, S., Ed.; Interscience Publishers: London, UK, 1978; pp. 447–552. [Google Scholar]
- Tornøe, W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Davis, M. Chemotherapy of schistosomiasis. V. Cholesteryl and choloyl derivatives of 4-amino-2-methoxyphenyl ethers. J. Chem. Soc. 1962, 1962, 178–181. [Google Scholar] [CrossRef]
- Zemplén, G. Decomposition of reducing disaccharides, VII Determination of the constitution of maltose. Berich. Deutschen Chem. Gesellsch. 1927, 7, 1555–1564. [Google Scholar] [CrossRef]
- Khanjin, N.A.; Montero, J.-L. Synthesis of mannose-6-phosphate analogs: Large-scale preparation of isosteric mannose-6-phosphonate via cyclic sulfate precursor. Tetrahedron Lett. 2002, 43, 4017–4020. [Google Scholar] [CrossRef]
- Kim, B.M.; Sharpless, K.B. Cyclic sulfates containing acid-sensitive groups and chemoselective hydrolysis of sulfate esters. Tetrahedron Lett. 1989, 30, 655–658. [Google Scholar] [CrossRef]
- Gibson, T. Phase-transfer synthesis of mono-alkyl ethers of oligoethylene glycols. J. Org. Chem. 1980, 45, 1095–1098. [Google Scholar] [CrossRef]
- Cunneen, J.I. The addition of thio-compounds to olefins. II. Reactions of thiolacetic and mono-chlorothiolacetic, di-chlorothiolacetic, and tri-chlorothiolacetic acids. J. Chem. Soc. 1947, 1947, 134–141. [Google Scholar] [CrossRef]
- Ciavatta, L.; Ferri, D.; Palombari, R. On the equilibrium Cu2++Cu(s) reversible 2Cu+. J. Inorg. Nucl. Chem. 1980, 42, 593–598. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-Catalyzed regioselective ligation of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar]
- Bock, V.D.; Hiemstra, H.; van Maarseveen, J.H. Cu-I-catalyzed alkyne-azide “click” cycloadditions from a mechanistic and synthetic perspective. Eur. J. Org. Chem. 2006, 2006, 51–68. [Google Scholar]
- Rodionov, V.O.; Fokin, V.V.; Finn, M.G. Mechanism of the ligand-free CuI-catalyzed azide-alkyne cycloaddition reaction. Angew. Chem. Int. Ed. 2005, 44, 2210–2215. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Bertrand, P.; Gesson, J.-P. Click chemistry with O-dimethylpropargylcarbamate for preparation of pH-sensitive functional groups. A case study. J. Org. Chem. 2007, 72, 3596–3599. [Google Scholar]
- Orgueira, H.A.; Fokas, D.; Isome, Y.; Chan, P.C.; Baldino, C.M. Regioselective synthesis of [1,2,3]-triazoles catalyzed by Cu(I) generated in situ from Cu(0) nanosize activated powder and amine hydrochloride salts. Tetrahedron Lett. 2005, 46, 2911–2914. [Google Scholar] [CrossRef]
- Smith, G.F.; Sullivan, V.R. Frank, hexanitrato ammonium cerate as a proposed reference standard in oxidimetry G. Ind. Eng. Chem. Anal. Ed. 1936, 8, 449–451. [Google Scholar] [CrossRef]
- Jarowicki, K.; Kocienski, P. Protecting groups. J. Chem. Soc. Perkin Trans. 2001, 18, 2109–2135. [Google Scholar] [CrossRef]
- Wittig, G.; Geissler, G. Zur reaktionsweise des pentaphenyl-phosphors und einiger derivate. Liebigs Ann. Chem. 1953, 580, 44–57. [Google Scholar] [CrossRef]
- Wittig, G.; Schöllkopf, U. Uber triphenyl-phosphinmethylene als olefinbildende reagenzien. Chem. Ber. 1954, 87, 1318–1330. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Reitz, A.B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions-stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar] [CrossRef]
- Horner, L.; Hoffmann, H.; Wippel, H.G. Phosphororganische Verbindungen, XII. Phosphinoxyde als Olefinierungsreagenzien. Chem. Ber. 1958, 91, 61–63. [Google Scholar] [CrossRef]
- Horner, L.; Hoffmann, H.; Wippel, H.G.; Klahre, G. Phosphororganische verbindungen, XX. phosphinoxyde als olefinierungsreagenzien. Chem. Ber. 1959, 92, 2499–2505. [Google Scholar] [CrossRef]
- Wadsworth, W.S.; Emmons, W.D. Utility of phosphonate carbanions in olefin synthesis. J. Am. Chem. Soc. 1961, 83, 1733–1738. [Google Scholar] [CrossRef]
- Wadsworth, W.S. Synthetic applications ofphosphoryl-stabilized anions. Org. React. 1977, 25, 73–253. [Google Scholar]
- Peterson, D.J. A carbonyl olefination reaction using silyl-subsituted organometallic compounds. J. Org. Chem. 1968, 33, 780–784. [Google Scholar] [CrossRef]
- Van Staden, L.F.; Gravestock, D.; Ager, D.J. New developments in the Peterson olefination reaction. Chem. Soc. Rev. 2002, 31, 195–200. [Google Scholar] [CrossRef]
- Ager, D.J. The Peterson olefination reaction. Org. React. 1990, 38, 1–223. [Google Scholar]
- Johnson, C.R.; Shanklin, J.R.; Kirchhoff, R.A. Olefin synthesis by reductive elimination of β-hydroxysulfoximines-methylenation of carbonyl-compounds. J. Am. Chem. Soc. 1973, 95, 6462–6463. [Google Scholar] [CrossRef]
- Julia, M.; Paris, J.-M. Syntheses using sulfones V(+). Method for general synthesis of double bonds. Tetrahedron Lett. 1973, 14, 4833–4836. [Google Scholar] [CrossRef]
- Baudin, J.B.; Hareau, G.; Julia, S.A.; Ruel, O. A direct synthesis of olefins by reaction of carbonyl-compounds with lithio derivatives of 2-[alkyl-sulfonyl or (2'-alkenyl)-sulfonyl or benzyl-sulfonyl]-benzothiazoles. Tetrahedron Lett. 1991, 32, 1175–1178. [Google Scholar] [CrossRef]
- Baudin, J.B.; Hareau, G.; Julia, S.A.; Lorne, R.; Ruel, O. Stereochemistry of the olefin formation from anti and syn heterocyclic beta-hydroxy-sulfones. Bull. Soc. Chim. Fr. 1993, 130, 336–357. [Google Scholar]
- Blakemore, P.R. The modified Julia olefination: Alkene synthesis via the condensation of metallated heteroarylalkylsulfones with carbonyl compounds. J. Chem. Soc. Perkin Trans. 2002, 2002, 2563–2585. [Google Scholar] [CrossRef]
- Umbreit, M.A.; Sharpless, K.B. Allylic oxidation of olefins by catalytic and stoichiometric selenium dioxide with tert-butyl hydroperoxide. J. Am. Chem. Soc. 1977, 99, 5526–5528. [Google Scholar] [CrossRef]
- Stevens, M.P. Polymer Chemistry, 3rd ed.; Oxford University Press: Oxford, NY, USA, 1999; pp. 265–267. [Google Scholar]
- Mayo, F.R.; Walling, C. The peroxide effect in the addition of reagents to unsaturated compounds and in rearrangement reactions. Chem. Rev. 1940, 27, 351–412. [Google Scholar] [CrossRef]
- Zard, S.Z. Radical Reactions in Organic Synthesis, 1st ed.; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Posner, T. Information on unsaturated compounds II. The addition of mercaptan to unsaturated hydrocarbon. Berich. Deutschen Chem. Gesellsch. 1905, 38, 646–657. [Google Scholar] [CrossRef]
- Katz, E.; Willner, I. Integrated nanoparticle-biomolecule hybrid systems: Synthesis, properties, and applications. Angew. Chem. Int. Ed. 2004, 43, 6042–6108. [Google Scholar] [CrossRef]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of thiol-derivatized gold nanoparticles in a 2-phase liquid-liquid system. J. Chem. Soc. Chem. Commun. 1994, 7, 801–802. [Google Scholar]
- Turkevich, J.; Stevenson, P.C.; Hillier, J.A. A study of the nucleation and growth processes in the synthesis of colloidal gold. Disc. Far. Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Frens, G. Controlled nucleation for regulation of particle-size in monodisperse gold suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Slot, J.W.; Gueuze, H.J. Sizing of protein a-colloidal gold probes for immunoelectron microscopy. J. Cell Biol. 1981, 90, 533–536. [Google Scholar] [CrossRef]
- Chow, M.K.; Zukoski, C.F. Gold sol formation mechanisms-role of colloidal stability. J. Colloid Interf. Sci. 1994, 165, 97–109. [Google Scholar] [CrossRef]
- Kimling, J.; Maier, M.; Okenve, B.; Kotaidis, V.; Ballot, H.; Plech, A. Turkevich method for gold nanoparticle synthesis revisited. J. Phys. Chem. B 2006, 110, 15700–15707. [Google Scholar]
- Ribatti, D. Chick embryo chorioallantoic membrane as a useful tool to study angiogenesis. Int. Rev. Cell Mol. Biol. 2008, 270, 181–224. [Google Scholar] [CrossRef]
- Richardson, M.; Singh, G. Observations on the use of the avian chorioallantoic membrane (CAM) model in investigations into angiogenesis. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2003, 3, 155–185. [Google Scholar] [CrossRef]
- Lokman, N.A.; Elder, A.S.F.; Ricciardelli, C.; Oehler, M.K. Chick chorioallantoic membrane (CAM) Assay as an in vivo model to study the effect of newly identified molecules on ovarian cancer invasion and metastasis. Int. J. Mol. Sci. 2012, 13, 9959–9970. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 3rd ed.; Pergamon Press: Oxford, UK, 1998. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Combemale, S.; Assam-Evoung, J.-N.; Houaidji, S.; Bibi, R.; Barragan-Montero, V. Gold Nanoparticles Decorated with Mannose-6-phosphate Analogues. Molecules 2014, 19, 1120-1149. https://doi.org/10.3390/molecules19011120
Combemale S, Assam-Evoung J-N, Houaidji S, Bibi R, Barragan-Montero V. Gold Nanoparticles Decorated with Mannose-6-phosphate Analogues. Molecules. 2014; 19(1):1120-1149. https://doi.org/10.3390/molecules19011120
Chicago/Turabian StyleCombemale, Stéphanie, Jean-Norbert Assam-Evoung, Sabrina Houaidji, Rashda Bibi, and Véronique Barragan-Montero. 2014. "Gold Nanoparticles Decorated with Mannose-6-phosphate Analogues" Molecules 19, no. 1: 1120-1149. https://doi.org/10.3390/molecules19011120