Synthesis of Tetrahydrohonokiol Derivates and Their Evaluation for Cytotoxic Activity against CCRF-CEM Leukemia, U251 Glioblastoma and HCT-116 Colon Cancer Cells



Abstract
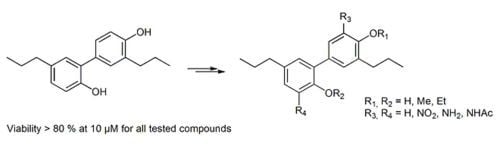
:
1. Introduction
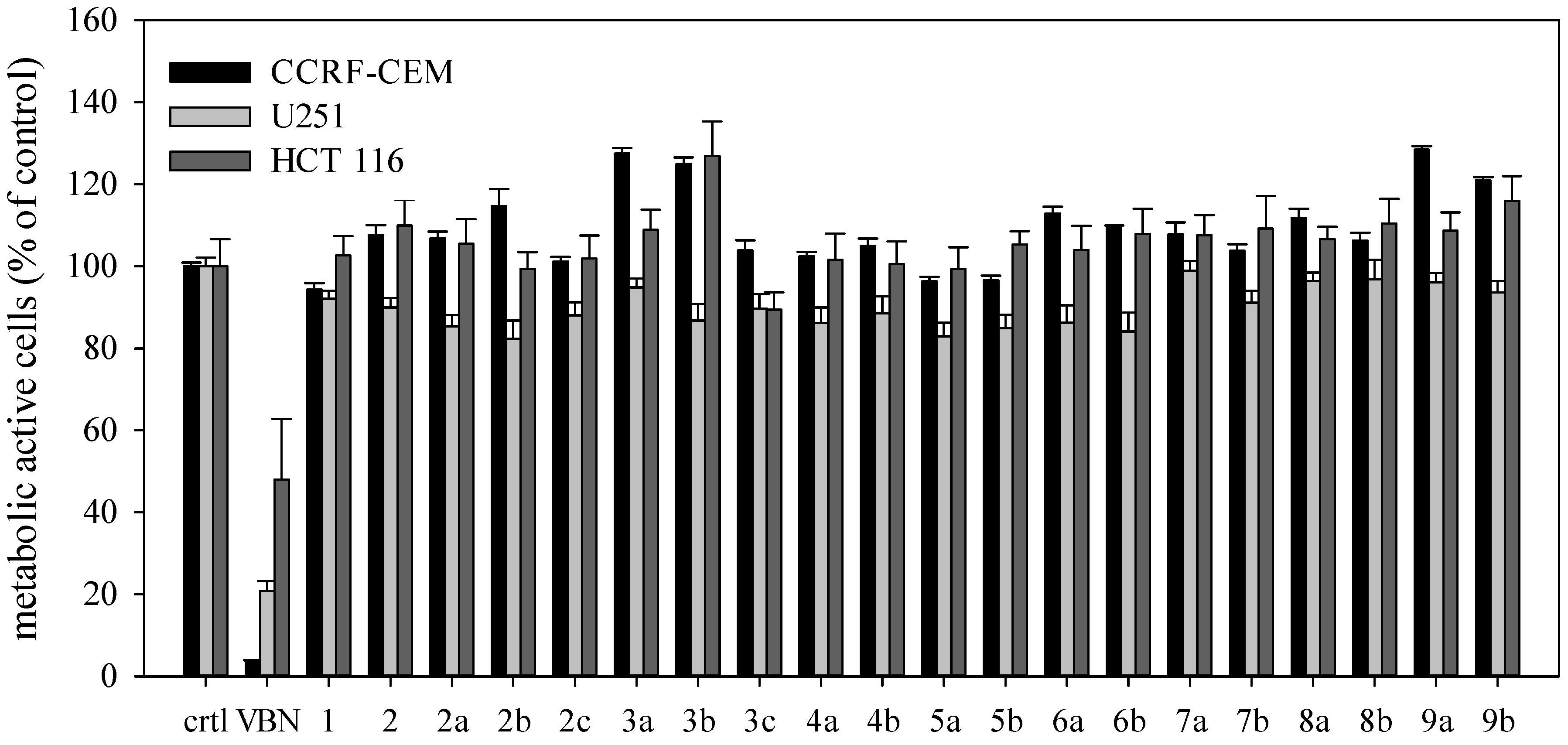
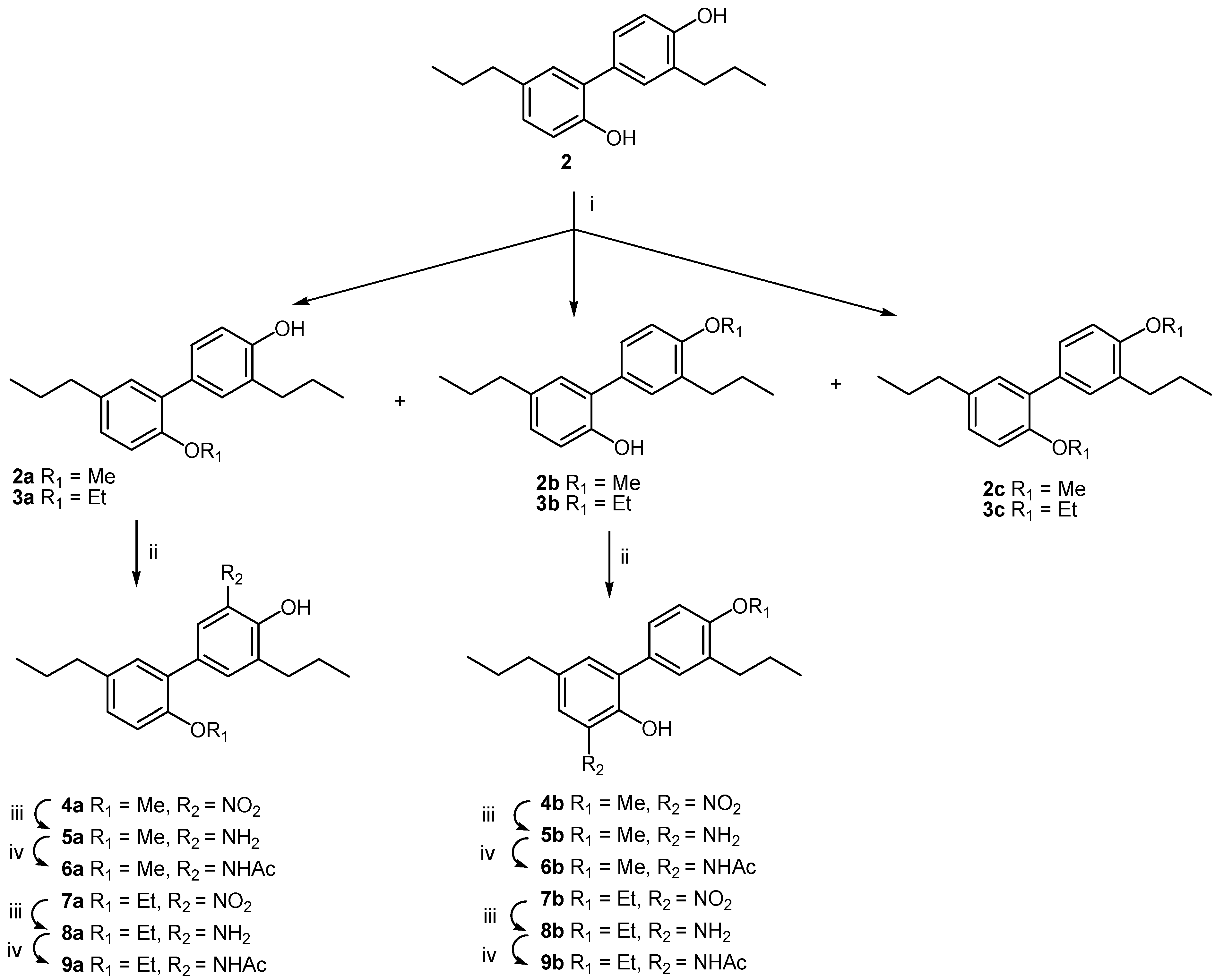
2. Results and Discussion


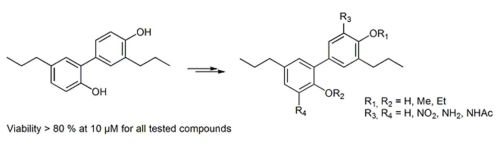{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 | R3 | R4 |
|---|---|---|---|---|
| 2 | -H | -H | -H | -H |
| 2a | -CH3 | -H | -H | -H |
| 2b | -H | -CH3 | -H | -H |
| 2c | -CH3 | -CH3 | -H | -H |
| 3a | -C2H5 | -H | -H | -H |
| 3b | -H | -C2H5 | -H | -H |
| 3c | -C2H5 | -C2H5 | -H | -H |
| 4a | -CH3 | -H | -H | -NO2 |
| 4b | -H | -CH3 | -NO2 | -H |
| 5a | -CH3 | -H | -H | -NH2 |
| 5b | -H | -CH3 | -NH2 | -H |
| 6a | -CH3 | -H | -H | -NHCOCH3 |
| 6b | -H | -CH3 | -NHCOCH3 | -H |
| 7a | -C2H5 | -H | -H | -NO2 |
| 7b | -H | -C2H5 | -NO2 | -H |
| 8a | -C2H5 | -H | -H | -NH2 |
| 8b | -H | -C2H5 | -NH2 | -H |
| 9a | -C2H5 | -H | -H | -NHCOCH3 |
| 9b | -H | -C2H5 | -NHCOCH3 | -H |


3. Experimental
3.1. General
3.1.1. General Procedure for Alkylation (Methylation and Ethylation, Respectively) of Tetrahydrohonokiol (2)
3.1.1.1. 2-Methoxy-3',5-dipropylbiphenyl-2-ol (2a), 4'-methoxy-3',5-dipropylbiphenyl-2-ol (2b) and 2,4'-dimethoxy-3',5-dipropylbiphenyl-2-ol (2c)
3.1.1.2. 2-Ethoxy-3',5-dipropylbiphenyl-2-ol (3a), 4'-ethoxy-3',5-dipropylbiphenyl-2-ol (3b) and 2,4'-diethoxy-3',5-dipropylbiphenyl-2-ol (3c)
3.1.2. General Procedure for Nitration of Monoalkyl Tetrahydrohonokiols 2a, 2b, 3a and 3b
3.1.2.1. 2-Methoxy-5'-nitro-3',5-dipropylbiphenyl-4'-ol (4a)
3.1.2.2. 4'-Methoxy-3-nitro-3',5-dipropylbiphenyl-2-ol (4b)
3.1.2.3. 2-Ethoxy-5'-nitro-3',5-dipropylbiphenyl-4'-ol (7a)
3.1.2.4. 4'-Ethoxy-3-nitro-3',5-dipropylbiphenyl-2-ol (7b)
3.1.3. General Procedure for Reduction of Nitro Tetrahydrohonokiols 4a, 4b, 7a and 7b
3.1.3.1. 5'-Amino-2-methoxy-3',5-dipropylbiphenyl-4'-ol (5a)
3.1.3.2. 2-Amino-4'-methoxy-3',5-dipropylbiphenyl-2-ol (5b)
3.1.3.3. 5'-Amino-2-ethoxy-3',5-dipropylbiphenyl-4'-ol (8a)
3.1.3.4. 2-Amino-4'-ethoxy-3',5-dipropylbiphenyl-2-ol (8b)
3.1.4. General Procedure for Acetylation of Amino Tetrahydrohonokiols 5a, 5b, 8a and 8b
3.1.4.1. 5'-Acetamido-2-methoxy-3',5-dipropylbiphenyl-4'-ol (6a)
3.1.4.2. 3-Acetamido-4'-methoxy-3',5-dipropylbiphenyl-2-ol (6b)
3.1.4.3. 5'-Acetamido-2-ethoxy-3',5-dipropylbiphenyl-4'-ol (9a)
3.1.4.4. 3-Acetamido-4'-ethoxy-3',5-dipropylbiphenyl-2-ol (9b)
3.2. Cell Culture
3.3. XTT Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Tang, W.; Eisenbrand, G. Magnolia spp. In Chinese Drugs of Plant Origin; Springer: Berlin, Germany, 1992; pp. 639–646. [Google Scholar]
- Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; People’s Medical Publishing House: Beijing, China, 2005. [Google Scholar]
- Maruyama, Y.; Kuribara, H. Overview of the pharmacological features of honokiol. CNS Drug Rev. 2000, 6, 3–44. [Google Scholar]
- Hamasaki, Y.; Muro, E.; Miyanji, S.; Yamamoto, S.; Kobayashi, I.; Sato, R.; Zaitu, M.; Matsuo, M.; Ichimaru, T.; Tasaki, H.; et al. Inhibition of leukotriene synthesis by honokiol in rat basophilic leukemia cells. Int. Arch. Allergy Immunol. 1996, 110, 278–281. [Google Scholar] [CrossRef]
- Schühly, W.; Hüfner, A.; Pferschy-Wenzig, E.M.; Prettner, E.; Adams, M.; Bodensieck, A.; Kunert, O.; Oluwemimo, A.; Haslinger, E.; Bauer, R. Design and synthesis of ten biphenyl-neolignan derivatives and their in vitro inhibitory potency against cyclooxygenase-1/2 activity and 5-lipoxygenase-mediated LTB4-formation. Bioorg. Med. Chem. 2009, 17, 4459–4465. [Google Scholar] [CrossRef]
- Kuribara, H.; Kishi, E.; Kimura, M.; Weintraub, S.T.; Maruyama, Y. Comparative assessment of the anxiolytic-like activities of honokiol and derivatives. Pharmacol. Biochem. Behavior 2000, 67, 597–691. [Google Scholar] [CrossRef]
- Taferner, B.; Schuehly, W.; Huefner, A.; Baburin, I.; Wiesner, K.; Ecker, G.F.; Hering, S. Modulation of GABAA-Receptors by Honokiol and Derivatives: Subtype Specificity and Structure-Activity Relationship. J. Med. Chem. 2011, 54, 5349–5361. [Google Scholar] [CrossRef]
- Schuehly, W.; Viveros Paredes, J.M.; Kleyer, J.; Huefner, A.; Anavi-Goffer, S.; Raduner, S.; Altmann, K.-H.; Gertsch, J. Mechanisms of osteoclastogenesis inhibition by a new class of biphenyl-type cannabinoid CB2 receptor inverse agonists. Chem. Biol. 2011, 18, 1053–1064. [Google Scholar] [CrossRef]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPrado, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef]
- Costantino, L.; Barlocco, D. Privileged structures as leads in medicinal chemistry. Curr. Med. Chem. 2006, 13, 65–85. [Google Scholar] [CrossRef]
- Kong, Z.-L.; Tzeng, S.-C.; Liu, Y.-C. Cytotoxic neolignans: An SAR study. Bioorg. Med. Chem. Lett. 2005, 15, 163–166. [Google Scholar] [CrossRef]
- Fried, L.E.; Arbiser, J.L. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid. Redox Signal. 2009, 11, 1139–1148. [Google Scholar] [CrossRef]
- Bai, X.; Cerimele, F.; Ushio-Fukai, M.; Waqas, M.; Campbell, P.M.; Govindarajan, B.; Der, C.J.; Battle, T.; Frank, D.A.; Ye, K.; et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J. Biol. Chem. 2003, 278, 35501–35507. [Google Scholar] [CrossRef]
- Ahn, K.W.; Sethi, G.; Shishodia, S.; Sung, B.; Arbiser, J.L.; Aggarwal, B.B. Honokiol potentiates apoptosis, suppresses osteoclastogenesis and inhibits invasion through modulation of nuclear factor-κB activation pathway. Mol. Cancer Res. 2006, 4, 621–633. [Google Scholar] [CrossRef]
- Shigemura, K.; Arbiser, J.L.; Sun, S.-Y.; Zayzafoon, M.; Johnstone, P.A.S.; Fujisawa, M.; Gotoh, A.; Weksler, B.; Zhau, H.E.; Chung, L.W.K. Honokiol, a natural plant product, inhibits the bone metastatic growth of human prostate cancer cells. Cancer 2007, 109, 1279–1289. [Google Scholar] [CrossRef]
- Lin, J.-W.; Chen, J.-T.; Hong, C.-Y.; Lin, Y.-L.; Wang, K.-T.; Yao, C.-J.; Lai, G.-M.; Chen, R.-M. Honokiol traverses the blood-brain barrier and induces apoptosis of neuroblastoma cells via an intrinsic bax-mitochondrion-cytochrome c-caspase protease pathway. Neuro-Oncol. 2012, 14, 302–314. [Google Scholar] [CrossRef]
- Wang, X.; Duan, X.; Yang, G.; Zhang, X.; Deng, L.; Zheng, H.; Deng, C.; Wen, J.; Wang, N.; Peng, C.; et al. Honokiol crosses BBB and BCSFB, and inhibits brain tumor growth in rat 9L intracerebral glioscarcoma model and human U251 xenograft glioma model. PloS One 2011, 6, e18490. [Google Scholar]
- Steinmann, P.; Walters, D.K.; Arlt, M.J.E.; Banke, I.J.; Ziegler, U.; Langsam, B.; Arbiser, J.; Muff, R.; Born, W.; Fuchs, B. Antimetastatic activity of honokiol in osteosarcoma. Cancer 2012, 118, 2117–2127. [Google Scholar] [CrossRef]
- Breinbauer, R.; Manger, M.; Scheck, M.; Waldmann, H. Natural product guided compound library development. Curr. Med. Chem. 2002, 9, 2129–2145. [Google Scholar] [CrossRef]
- Kretschmer, N.; Blunder, M.; Kunert, O.; Rechberger, G.N.; Bauer, R.; Schuehly, W. Cytotoxic Furanogermacranolides from the Flowers of Helianthus angustifolius. Planta Med. 2011, 77, 1912–1915. [Google Scholar] [CrossRef]
- Efferth, T.; Davey, M.; Olbrich, A.; Rücker, G.; Gebhart, E.; Davey, R. Activity of drugs from traditional Chinese medicine toward sensitive and MDR1- or MRP1-overexpressing multidrug-resistant human CCRF-CEM leukemia cells. Blood Cell. Mol. Dis. 2002, 28, 160–168. [Google Scholar] [CrossRef]
- Fujita, M.; Itokawa, H.; Sashida, Y. Honokiol, a New Phenolic Compound isolated from the Bark of Magnolia obovata THUNB. Chem. Pharm. Bull. 1972, 20, 212–213. [Google Scholar] [CrossRef]
- Johnson, T.W.; Corey, E.J. Enantiospecific synthesis of the proposed structure of the antitubercular marine diterpenoid pseudopteroxazole; revision of stereochemistry. J. Am. Chem. Soc. 2001, 123, 4475–4479. [Google Scholar] [CrossRef]
- Widdowson, K.L.; Elliott, J.D.; Veber, D.F.; Nie, H.; Rutledge, M.C.; McCleland, B.W.; Xiang, J.N.; Jurewicz, A.J.; Hertzberg, R.P.; Foley, J.J.; et al. Evaluation of potent and selective small-molecule antagonists for the CXCR2 chemokine receptor. J. Med. Chem. 2004, 47, 1319–1321. [Google Scholar] [CrossRef]
- Hibasami, H.; Achiwa, Y.; Katsuzaki, H.; Imai, K.; Yoshioka, K.; Nakanishi, K.; Ishii, Y.; Hasegawa, M.; Komiya, T. Honokiol induces apoptosis in human lymphoid leukemia Molt 4B cells. Int. J. Mol. Med. 1998, 2, 671–673. [Google Scholar]
- Battle, T.E.; Arbiser, J.; Frank, D.A. The natural product honokiol induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood 2013, 106, 690–697. [Google Scholar] [CrossRef]
- Lin, J.M.; Prakasha Gowda, A.S.; Sharma, A.K.; Amin, S. In vitro growth inhibition of human cancer cells by novel honokiol analogs. Bioorg. Med. Chem. 2012, 20, 3202–3211. [Google Scholar] [CrossRef]
- Jeong, J.J.; Lee, J.H.; Chang, K.C.; Kim, H.J. Honokiol exerts an anticancer effect in T98G human glioblastoma cells through the induction of apoptosis and the regulation of adhesion molecules. Int. J. Oncol. 2012, 41, 1358–1364. [Google Scholar]
- Chang, K.-H.; Yan, M.-D.; Yao, C.-J.; Lin, P.-C.; Lai, G.-M. Honokiol-induced apoptosis and autophagy in glioblastoma multiforme cells. Oncol. Lett. 2013, 6, 1435–1438. [Google Scholar]
- Ma, L.; Chen, J.; Wang, X.; Liang, X.; Luo, Y.; Zhu, W.; Wang, T.; Peng, M.; Li, S.; Jie, S.; et al. Structural modification of honokiol, a biphenyl occurring in Magnolia officinalis: The evaluation of honokiol analogues as inhibitors of angiogenesis and for their cytotoxicity and structure-activity relationship. J. Med. Chem. 2011, 54, 6469–6481. [Google Scholar]
- Amblard, F.; Govindarajan, B.; Levkove, B.; Rapp, K.L.; Detorio, M.; Arbiser, J.L.; Schinazi, R. Synthesis, cytotoxicity, and antiviral activities of new neolignans related to honokiol and magnolol. Bioorg. Med. Chem. Lett. 2007, 17, 4428–4431. [Google Scholar] [CrossRef]
- Kaushik, G.; Ramalingam, S.; Subramaniam, D.; Rangarajan, P.; Protti, P.; Rammamoorthy, P.; Anant, S.; Mammen, J.M.V. Honokiol induces cytotoxic and cytostatic effects in malignant melanoma cancer cells. Am. J. Surg. 2012, 204, 868–873. [Google Scholar] [CrossRef]
- Kim, D.W.; Ko, S.M.; Jeon, Y.J.; Noh, Y.W.; Choi, N.J.; Cho, S.D.; Moon, H.S.; Cho, Y.S.; Shin, J.C.; Park, S.M.; et al. Anti-proliferative effect of honokiol in oral squamous cancer through the regulation of specifity protein 1. Int. J. Oncol. 2013, 43, 1103–1110. [Google Scholar]
- Böhmdorfer, M.; Maier-Salamon, A.; Taferner, B.; Reznicek, G.; Thalhammer, T.; Hering, S.; Huefner, A.; Schuehly, W.; Jäger, W. In vitro metabolism and disposition of honokiol in rat and human livers. J. Pharm. Sci. 2011, 100, 3506–3516. [Google Scholar] [CrossRef]
- Rao, K.V.; Davis, T.L. Constituents of Magnolia grandiflora I: Mono-O-methylhonokiol. Planta Med. 1982, 45, 57–59. [Google Scholar] [CrossRef]
- Scudiero, D.A.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity using human and other tumor cell lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bernaskova, M.; Kretschmer, N.; Schuehly, W.; Huefner, A.; Weis, R.; Bauer, R. Synthesis of Tetrahydrohonokiol Derivates and Their Evaluation for Cytotoxic Activity against CCRF-CEM Leukemia, U251 Glioblastoma and HCT-116 Colon Cancer Cells. Molecules 2014, 19, 1223-1237. https://doi.org/10.3390/molecules19011223
Bernaskova M, Kretschmer N, Schuehly W, Huefner A, Weis R, Bauer R. Synthesis of Tetrahydrohonokiol Derivates and Their Evaluation for Cytotoxic Activity against CCRF-CEM Leukemia, U251 Glioblastoma and HCT-116 Colon Cancer Cells. Molecules. 2014; 19(1):1223-1237. https://doi.org/10.3390/molecules19011223
Chicago/Turabian StyleBernaskova, Marketa, Nadine Kretschmer, Wolfgang Schuehly, Antje Huefner, Robert Weis, and Rudolf Bauer. 2014. "Synthesis of Tetrahydrohonokiol Derivates and Their Evaluation for Cytotoxic Activity against CCRF-CEM Leukemia, U251 Glioblastoma and HCT-116 Colon Cancer Cells" Molecules 19, no. 1: 1223-1237. https://doi.org/10.3390/molecules19011223
APA StyleBernaskova, M., Kretschmer, N., Schuehly, W., Huefner, A., Weis, R., & Bauer, R. (2014). Synthesis of Tetrahydrohonokiol Derivates and Their Evaluation for Cytotoxic Activity against CCRF-CEM Leukemia, U251 Glioblastoma and HCT-116 Colon Cancer Cells. Molecules, 19(1), 1223-1237. https://doi.org/10.3390/molecules19011223