Pseudo-Four Component Synthesis of Mono- and Di-Benzylated-1,2,3-Triazoles Derived from Aniline

Abstract
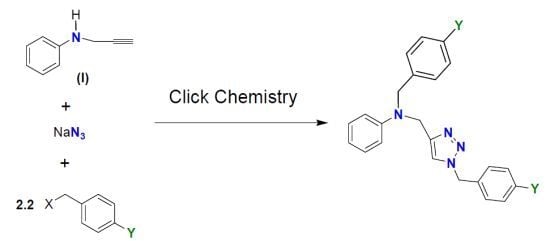
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization


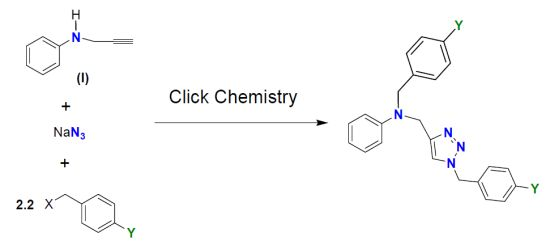{kind=link}
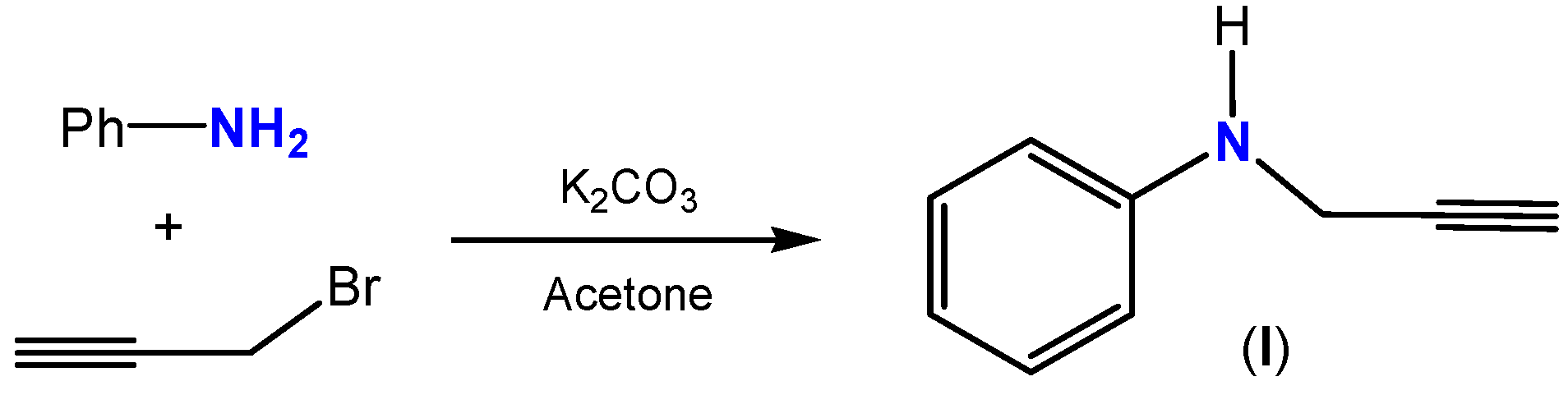{kind=link}
{kind=link}
 Reagents and conditions: Compound I (1.14 mmol), Cu(OAc)2·H2O (5 mol%), 1,10-phenanthroline (5 mol%), sodium ascorbate (1.14 mmol), sodium azide (1.40 mmol), p-substituted benzyl halogenide (1.40 mmol), stirring at room temperature for 18 h in EtOH/H2O (4:1).
Reagents and conditions: Compound I (1.14 mmol), Cu(OAc)2·H2O (5 mol%), 1,10-phenanthroline (5 mol%), sodium ascorbate (1.14 mmol), sodium azide (1.40 mmol), p-substituted benzyl halogenide (1.40 mmol), stirring at room temperature for 18 h in EtOH/H2O (4:1).
| Entry | X | Y | Product | Yield (%) | Product | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | Cl | H | 1a | 42 | 2a | 35 |
| 2 | Cl | F | 1b | 40 | 2b | 31 |
| 3 | Cl | Cl | 1c | 39 | 2c | 35 |
| 4 | Br | Br | 1d | 42 | 2d | 29 |
| 5 | Br | I | 1e | 25 | 2e | 42 |
 Reagents and conditions: Compound II (1.14 mmol), Cu(OAc)2·H2O (5 mol%), 1,10-phenanthroline (5 mol%), sodium ascorbate (1.14 mmol), sodium azide (1.40 mmol), p-substituted benzyl halogenide (2.80 mmol), stirring at room temperature for 24 h in EtOH/H2O (4:1).
Reagents and conditions: Compound II (1.14 mmol), Cu(OAc)2·H2O (5 mol%), 1,10-phenanthroline (5 mol%), sodium ascorbate (1.14 mmol), sodium azide (1.40 mmol), p-substituted benzyl halogenide (2.80 mmol), stirring at room temperature for 24 h in EtOH/H2O (4:1).
| Entry | X | Y | Product | Yield (%) |
|---|---|---|---|---|
| 1 | Cl | H | 1a | 71 |
| 2 | Cl | F | 1b | 76 |
| 3 | Cl | Cl | 1c | 80 |
| 4 | Br | Br | 1d | 79 |
| 5 | Br | I | 1e | 86 |

3. Experimental
3.1. General Methods
3.2. Typical Procedure for the Synthesis of Mono- and Dibenzylated 1,2,3-triazoles from Aniline
3.2.1. Benzyl Chloride
3.2.2. 4-Fluorobenzyl Chloride
3.2.3. 4-Chlorobenzyl Chloride
3.2.4. 4-Bromobenzyl Chloride
3.2.5. 4-Iodobenzyl Bromide
3.3. Typical Procedure for the Selective Synthesis of Dibenzylated 1,2,3-triazoles Derived from Aniline
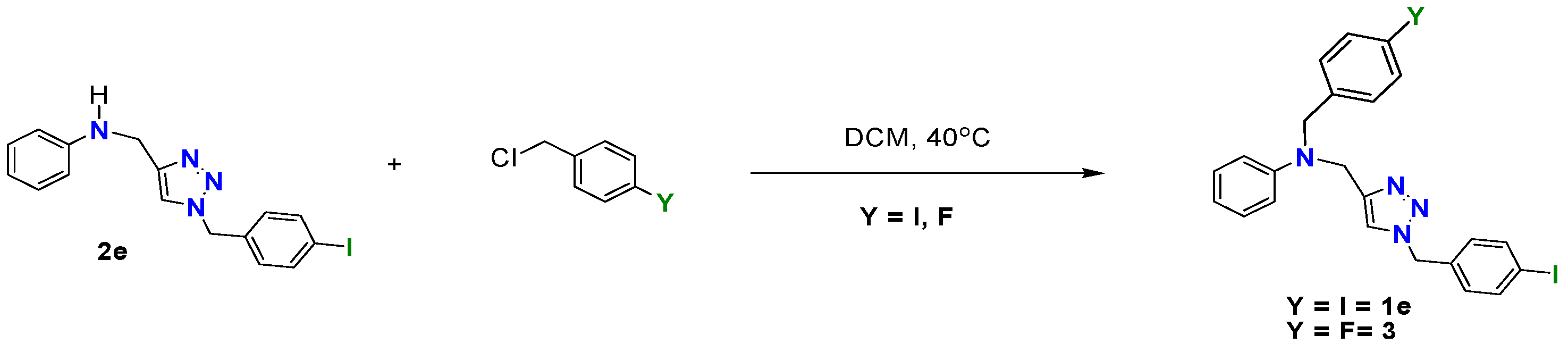
3.4. Reaction of N-((1-(4-Iodobenzyl)-1H-1,2,3-triazol-4-yl-)methyl)benzenamine (2e) with p-Halogenated Benzyl Chlorides
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Armstrong, R.W.; Combs, A.P.; Tempest, T.A.; Brown, S.D.; Keating, T.A. Multiple-component condensation strategies for combinatorial library synthesis. Acc. Chem. Res. 1996, 29, 123–131. [Google Scholar] [CrossRef]
- Dax, S.L.; McNally, J.J.; Youngman, M.A. Multi-component methodologies in solid phase organic synthesis. Curr. Med. Chem. 1999, 6, 255–270. [Google Scholar]
- Hulme, C.; Gore, V. Multi-component reactions: Emerging chemistry in drug discovery. Curr. Med. Chem. 2003, 10, 51–80. [Google Scholar] [CrossRef]
- Zhang, M.; Fang, X.; Neumann, H.; Beller, M. General and regioselective synthesis of pirroles via Ruthenium catalized multicomponent reactions. J. Am. Chem. Soc. 2013, 135, 11384–11388. [Google Scholar] [CrossRef]
- Zhu, J.; Bienaymé, H. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Guillena, G.; Ramón, D.J.; Yus, M. Organocatalytic enantioselective multicomponent reactions (OEMCRs). Tetrahedron-Assymetr. 2007, 18, 693–700. [Google Scholar] [CrossRef]
- Brauch, S.; van Berkel, S.S.; Westermann, B. High-order multicomponent reactions: Beyond four reactants. Chem. Soc. Rev. 2013, 42, 4948–4962. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Liu, J.-M.; Wang, X.; Cheng, T.M.; Li, R.T. Multicomponent reactions leading to symmetric and assymetric multi-substituted 1,4-dihydropyridines on montmorillonite. Tetrahedron 2013, 69, 5242–5247. [Google Scholar] [CrossRef]
- Bienaymé, H.; Hulme, C.; Oddon, G.; Schmitt, P. Maximizing synthetic efficiency: Multi-component transformations lead the way. Chem. Eur. J. 2000, 6, 3321–3329. [Google Scholar] [CrossRef]
- Lalli, C.; Bouma, J.M.; Bonne, D.; Masson, J.; Zhu, J. Exploiting the divergent reactivity of α-isocyanoacetate: Multicomponent synthesis of 5-alkoxyolxazoles and related heterocycles. Chem. Eur. J. 2011, 17, 880–889. [Google Scholar] [CrossRef]
- Li, M.; Lv, X.-L.; Wen, L.-R.; Hu, Z.-Q. Direct solvent-free regioselective construction of pyrrolo[1,2-α][1,10]phenanthrolines based on isocyanide-based multicomponent reactions. Org. Lett. 2013, 15, 1262–1265. [Google Scholar] [CrossRef]
- Gu, Y. Multicomponent reactions in unconventional solvents: State of art. Green Chem. 2012, 14, 2091–2128. [Google Scholar] [CrossRef]
- Rajesh, S.P.; Perumal, S.; Menendez, J.C.; Pandian, S.; Murugesan, R. Facile ionic liquid-mediated, three component sequential reactions for the green, region- and diastereoselective synthesis of furocoumarins. Tetrahedron 2012, 68, 5631–5636. [Google Scholar] [CrossRef]
- Singh, M.S.; Chowdhury, S. Recent developments in solvent-free multicomponent reactions: A perfect synergy for eco-compatible organic synthesis. RSC Adv. 2012, 2, 4547–4592. [Google Scholar] [CrossRef]
- Vlaar, T.; Maes, B.U.W.; Ruijter, E.; Orru, R.V.A. Palladium-catalyzed migratory insertion of isocyanides: An emerging platform in cross coupling reactions. Angew. Chem. Int. Ed. 2013, 52, 7084–7097. [Google Scholar] [CrossRef]
- Maghari, S.; Ramezanpour, S.; Balalaie, S.; Darvish, F.; Rominger, F.; Bijanzadeh, H.R. Synthesis of functionalized pseudopeptides through five-component sequential Ugi/Nucleophilic Reaction of N-substituted 2-alkylamides with hydrazides. J. Org. Chem. 2013, 78, 6450–6456. [Google Scholar] [CrossRef]
- Castellano, T.; Neo, A.G.; Marcaccini, S.; Marcos, C.F. Enols as feasible acid components in the Ugi condensation. Org. Lett. 2012, 14, 6218–6221. [Google Scholar] [CrossRef]
- Doemling, A.; Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- La Spisa, F.; Feo, A.; Mossetti, R.; Tron, G.C. An efficient synthesis of symmetric and unsymmetric Bis-(β-aminoamides) via Ugi multicomponent reaction. Org. Lett. 2012, 14, 6044–6047. [Google Scholar] [CrossRef]
- Su, Y.; Bouma, M.J.; Alcaraz, L.; Stocks, M.; Furber, M.; Masson, G.; Zhu, J. Organocatalytic enantioselective One-pot four-component Ugi-type multicomponent reaction for the synthesis of epoxy-tetrahydropyrrolo [3,4-β]pyridine-5-ones. Chem. Eur. J. 2012, 18, 12624–12627. [Google Scholar] [CrossRef]
- Hashimoto, T.; Kimura, H.; Kayamata, Y.; Marouka, K. A catalytic assymetric Ugi-type reaction with acyclic azomethine imines. Angew. Chem. Int. Ed. 2012, 51, 7279–7281. [Google Scholar] [CrossRef]
- Banfi, L.; Riva, R. The Passerini reaction. Org. React. 2005, 65, 1–140. [Google Scholar]
- Sehlinger, A.; Kreye, O.; Meier, M.A.R. Tunable polymers obtained from Passerini multicomponent reaction derived acrylate monomers. Macromolecules 2013, 46, 6031–6037. [Google Scholar] [CrossRef]
- Kaicharla, T.; Yetra, S.R.; Roy, T.; Biju, A.T. Engaging insatins in solvent-free, sterically congested Passerini reaction. Green Chem. 2013, 15, 1608–1614. [Google Scholar] [CrossRef]
- Alcaide, B.; Alemendros, P.; Arrangoncillo, C.; Callejo, R.; Ruiz, M.P.; Torres, M.P. Regio- and diastereoselective synthesis of β-lactam-triazole hybrids via Passerini/CuAAC sequence. J. Org. Chem. 2012, 77, 6917–6928. [Google Scholar] [CrossRef]
- Jee, J.-A.; Spagnoulo, L.A.; Rudick, J.G. Convergent synthesis of dendrimers via the Passerini Three-component reaction. Org. Lett. 2012, 14, 3292–3295. [Google Scholar] [CrossRef]
- Rostovstev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I) catalyzed regioselective ligation of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornoe, C.; Christensen, C.; Medal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click chemistry: 1,2,3-Triazoles as pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef]
- Liu, J.-F; Sang, C.-Y.; Xu, X.-H; Zhang, L.-L.; Yang, X.; Hui, L.; Zhang, J.-B.; Chen, S.-W. Synthesis and cytotoxic activity of human cancer cells of carbamate derivatives of 4-b-(1,2,3-triazol-1-yl) podophylloxotin. Eur. J. Med. Chem. 2013, 64, 621–628. [Google Scholar] [CrossRef]
- Li, J.; Kaoud, T.S.; LeVieux, J.; Gilbreath, B.; Swapna, M.; Dalby, K.N.; Kerwin, S.M. A Fluorescence-Based assay for p38α recruitment site binders: Identification of rooperol as novel p38α kinase inhibitor. ChemBioChem 2013, 14, 66–71. [Google Scholar] [CrossRef]
- Binder, W.H.; Kluger, C. Azide/Alkyne-click reactions: Applications in material science and organic synthesis. Curr. Org. Chem. 2006, 10, 1791–1815. [Google Scholar] [CrossRef]
- Dickschat, A.T.; Behrends, F.; Bühner, M.; Ren, J.; Weiß, M.; Eckert, H.; Studer, A. Preparation of bifunctional mesoporous silica nanoparticles by orthogonal click reactions and their application in cooperative catalysis. Chem. Eur. J. 2012, 18, 16689–16697. [Google Scholar] [CrossRef]
- Abdulkin, P.; Moglie, Y.; Knappet, B.R.; Jefferson, D.A.; Yus, M.; Alonso, F.; Wheatley, A.E.H. New routes to Cu(I)/Cu nanocatalysts for the multicomponent synthesis of 1,2,3-triazoles. Nanoscale 2013, 5, 342–350. [Google Scholar]
- Moorhouse, A.D.; Santos, A.M.; Gunaratman, M.; Moore, M.; Neidle, S.; Moses, J.E. Stabilization of G-quadruplex DNA by highly selective ligands via click chemistry. J. Am. Chem. Soc. 2006, 128, 15972–15973. [Google Scholar] [CrossRef]
- Lee, L.V.; Mitchell, M.L.; Huang, S.-J.; Fokin, V.V.; Sharpless, K.B. A potent and highly selective inhibitor of human α-1,3-Fucosyltransferase via click chemistry. J. Am. Chem. Soc. 2003, 125, 9588–9589. [Google Scholar] [CrossRef]
- Wu, P.; Feldman, A.K.; Nugent, A.K.; Hawker, C.J.; Scheel, A.; Voit, B.; Pyun, J.; Frechet, J.M.J.; Sharpless, K.B.; Fokin, V.V. Efficiency and fidelity in a click chemistry route to triazole dendrimers by the copper(I)-catalyzed ligation of azides and alkynes. Angew. Chem. Int. Ed. 2004, 43, 3928–3932. [Google Scholar] [CrossRef]
- Wu, P.; Malkoch, M.; Hunt, J.N.; Vestberg, R.; Kaltgrad, E.; Finn, M.G.; Fokin, V.V.; Sharpless, K.B.; Hawker, C.J. Multivalent, bifunctional dendrimers prepared by click chemistry. Chem. Commun. 2005, 5775–5777. [Google Scholar] [CrossRef]
- Rozkiewickz, D.I.; Janczewski, D.; Verboom, W.; Ravoo, B.J.; Reinhoudt, D.N. Click chemistry by microcontact printing. Angew. Chem. Int. Ed. 2006, 45, 5292–5296. [Google Scholar] [CrossRef]
- Speers, A.E.; Adam, G.C.; Cravatt, B.F. Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 4686–4687. [Google Scholar] [CrossRef]
- Speers, A.E.; Cravatt, B.F. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004, 11, 535–546. [Google Scholar]
- Burley, G.A.; Gierlich, J.; Mofid, M.R.; Nir, H.; Tal, S.; Eichen, Y.; Carell, T. Directed DNA metallization. J. Am. Chem. Soc. 2006, 128, 1398–1399. [Google Scholar] [CrossRef]
- Wang, Q.; Chan, T.R.; Hilgraf, R.; Fokin, V.V.; Sharpless, K.B.; Finn, M.G. Biconjugation by Copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [Google Scholar] [CrossRef]
- Megia-Fernandez, A.; Ortega-Munos, M.; Hernadez-Mateo, F.; Santoyo-Gonzalez, F. One-pot three-component click reaction of cyclic sulphates and cyclic sulphamides. Adv. Synth. Catal. 2012, 354, 1797–1803. [Google Scholar]
- Wang, W.; Wu, J.; Xia, C.; Li, F. Reusable ammonium salt-tagged-NHC-Cu(I) complexes: Preparation and application in the three component reaction. Green Chem. 2011, 13, 3440–3445. [Google Scholar] [CrossRef]
- Alonso, F.; Moglie, Y.; Radivoy, G.; Yus, M. Multicomponent click synthesis of potentially biologically active triazoles catalyzed by copper nanoparticles on activated carbon in water. Heterocycles 2012, 84, 1033–1044. [Google Scholar] [CrossRef]
- Shargi, H.; Beyzavi, M.S.; Safavi, A.; Doroodman, M.; Mohammad, M.; Khalifeh, R. Immobilization of porphyniratocopper nanoparticles onto activated multi-walled carbon nanotubes and a study of its catalytic activity as an efficient heterogeneous catalyst for a click approach to the three-component synthesis of triazoles in water. Adv. Synth. Catal. 2009, 351, 2391–2410. [Google Scholar] [CrossRef]
- Kumar, D.; Reddy, V.B.; Varma, R.J. A facile and regioselective synthesis of 1,4-disubstituted 1,2,3-triazoles using click chemistry. Tetrahedron Lett. 2009, 50, 2065–2068. [Google Scholar] [CrossRef]
- Quan, Z.-J.; Quion, X.; Zhang, Z.; Da, Y.-X.; Wang, X.-C. Copper-catalyzed click synthesis of functionalized 1,2,3-triazoles with 3,4-dihydropyrimidinone or amide group via a one-pot four-component reaction. Tetrahedron 2013, 69, 881–887. [Google Scholar] [CrossRef]
- Bahulayan, D.; Arun, S. An easy two step synthesis of macrocyclic peptidotriazoles via a four-component reaction and catalyzed intramolecular alkyne-azide [3+2] click cycloaddition. Tetrahedron Lett. 2012, 53, 2850–2855. [Google Scholar] [CrossRef]
- Dabiri, M.; Salehi, P.; Bahramnejad, M.; Sherafat, F. Synthesis of diheterocyclic compounds based on triazolyl methoxy Phenylquinazolines via a one-pot four-component-click reaction. J. Comb. Chem. 2010, 12, 638–642. [Google Scholar] [CrossRef]
- Yadav, J.S.; Subba Reddy, B.V.; Madhusudhan Reddy, G.; Rehana Anjum, S. Cu(OTf)2/Cu-catalyzed four-component reaction: A facile synthesis of α-alkoxytriazoles via click chemistry. Tetrahedron Lett. 2009, 50, 6029–6031. [Google Scholar] [CrossRef]
- Cantet, A.-C.; Carreyre, H.; Gesson, J.P.; Jouannetaud, M.-P.; Renoux, B. gem-difluorination of aminoalkynes via highly reactive dicationic species in super acid HF-SbF5: Application to the efficient synthesis of difluorinated chinchona alkaloid derivatives. J. Org. Chem. 2008, 73, 2875–2878. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mendoza-Espinosa, D.; Negron-Silva, G.E.; Lomas-Romero, L.; Gutierrez-Carrillo, A.; Santillán, R. Pseudo-Four Component Synthesis of Mono- and Di-Benzylated-1,2,3-Triazoles Derived from Aniline. Molecules 2014, 19, 55-66. https://doi.org/10.3390/molecules19010055
Mendoza-Espinosa D, Negron-Silva GE, Lomas-Romero L, Gutierrez-Carrillo A, Santillán R. Pseudo-Four Component Synthesis of Mono- and Di-Benzylated-1,2,3-Triazoles Derived from Aniline. Molecules. 2014; 19(1):55-66. https://doi.org/10.3390/molecules19010055
Chicago/Turabian StyleMendoza-Espinosa, Daniel, Guillermo E. Negron-Silva, Leticia Lomas-Romero, Atilano Gutierrez-Carrillo, and Rosa Santillán. 2014. "Pseudo-Four Component Synthesis of Mono- and Di-Benzylated-1,2,3-Triazoles Derived from Aniline" Molecules 19, no. 1: 55-66. https://doi.org/10.3390/molecules19010055
APA StyleMendoza-Espinosa, D., Negron-Silva, G. E., Lomas-Romero, L., Gutierrez-Carrillo, A., & Santillán, R. (2014). Pseudo-Four Component Synthesis of Mono- and Di-Benzylated-1,2,3-Triazoles Derived from Aniline. Molecules, 19(1), 55-66. https://doi.org/10.3390/molecules19010055