Synthetic Advances in Macrosphelides: Natural Anticancer Agents

College of Pharmacy and Research Institute of Pharmaceutical Sciences, Gyeongsang National University, Jinju daero, Jinju, Gyeongnam 660-701, Korea
Molecules 2014, 19(10), 15982-16000; https://doi.org/10.3390/molecules191015982
Submission received: 11 August 2014
/
Revised: 23 September 2014
/
Accepted: 26 September 2014
/
Published: 8 October 2014
(This article belongs to the Collection Bioactive Compounds)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Total synthesis of macrosphelides is summarized. Synthetic approaches contain the preparation of key fragments and the final ring-closure reaction for unique 16- or 15-membered macrolactone skeletons.
1. Introduction
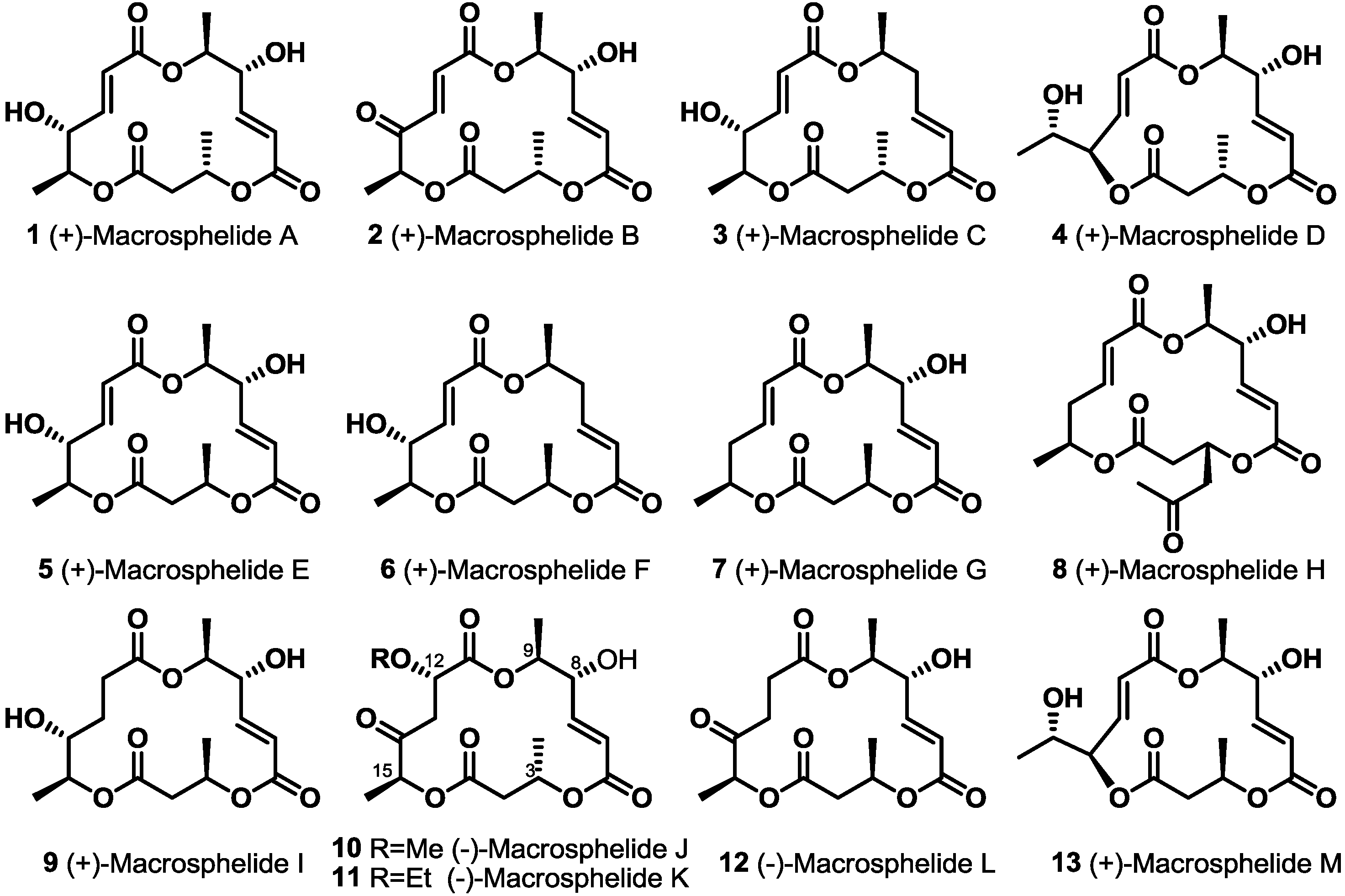
Macrosphelides (MS), natural macrolide polyketides, were first reported as new anticancer metastasis agents in 1995 [1]. Because this new macrolide showed promising biological activities, such as inhibition of the adhesion of HL60 cells to human umbilical-vein endothelial cells (HUVEC) [2,3,4,5,6,7], no acute toxicity by i.p. injection of MSA into BDF1 mice at 200 mg/kg for five days [8], potent immunosuppressant activity in vivo [9], anticancer activity against lung metastasis of B16/BL6 melanoma in mice [10] and antimicrobial activity [11], significant endeavors to synthesize this natural product family and isolate-related MS isotypes have been carried out [12,13]. So far, 13 natural isomers have been reported from Microsphaeropsis sp. FO-5050, Periconia byssoides, Coniothyrium minitans and the fungus, Tritirachium sp. HKI 0317 [14]. In addition, about 20 synthetic approaches have been developed for more efficient preparation. Herewith, synthetic advances in natural MS are summarized (Figure 1).
Figure 1.
Natural macrosphelides (MSs).

2. Results and Discussion
2.1. Synthetic Features of MS
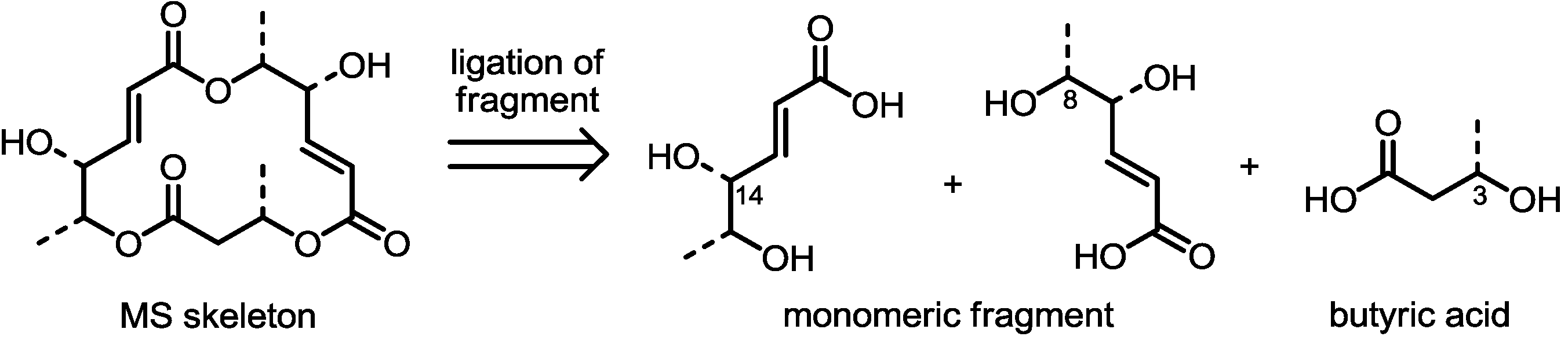
As shown in Figure 2, MS usually possesses a 16-membered macrolactone core with three ester linkages. In the retrosynthetic view, this skeleton can be divided into three hydroxy acids. It is important to note that two of them feature an almost identical carbon framework and oxidation state. This means that just one efficient synthesis of the fragment may complete full synthesis of MS itself after appropriate protection group modifications and ligation of the fragments. Actually, most methods of MS synthesis involve a unique preparation method of the monomeric fragment. For the final macrocyclization reaction, the Yamaguchi macrolactonization reaction [15] was chosen as the first option. Recently, however, other useful C-C bond formations have been developed to create special 16-membered macrolactones efficiently. These variations in the synthetic procedure make it possible to prepare a variety of MS-related compounds and improve their activities.
Figure 2.
Synthetic features of MS preparation.

2.2. The First Total Synthesis of MSA and MSB
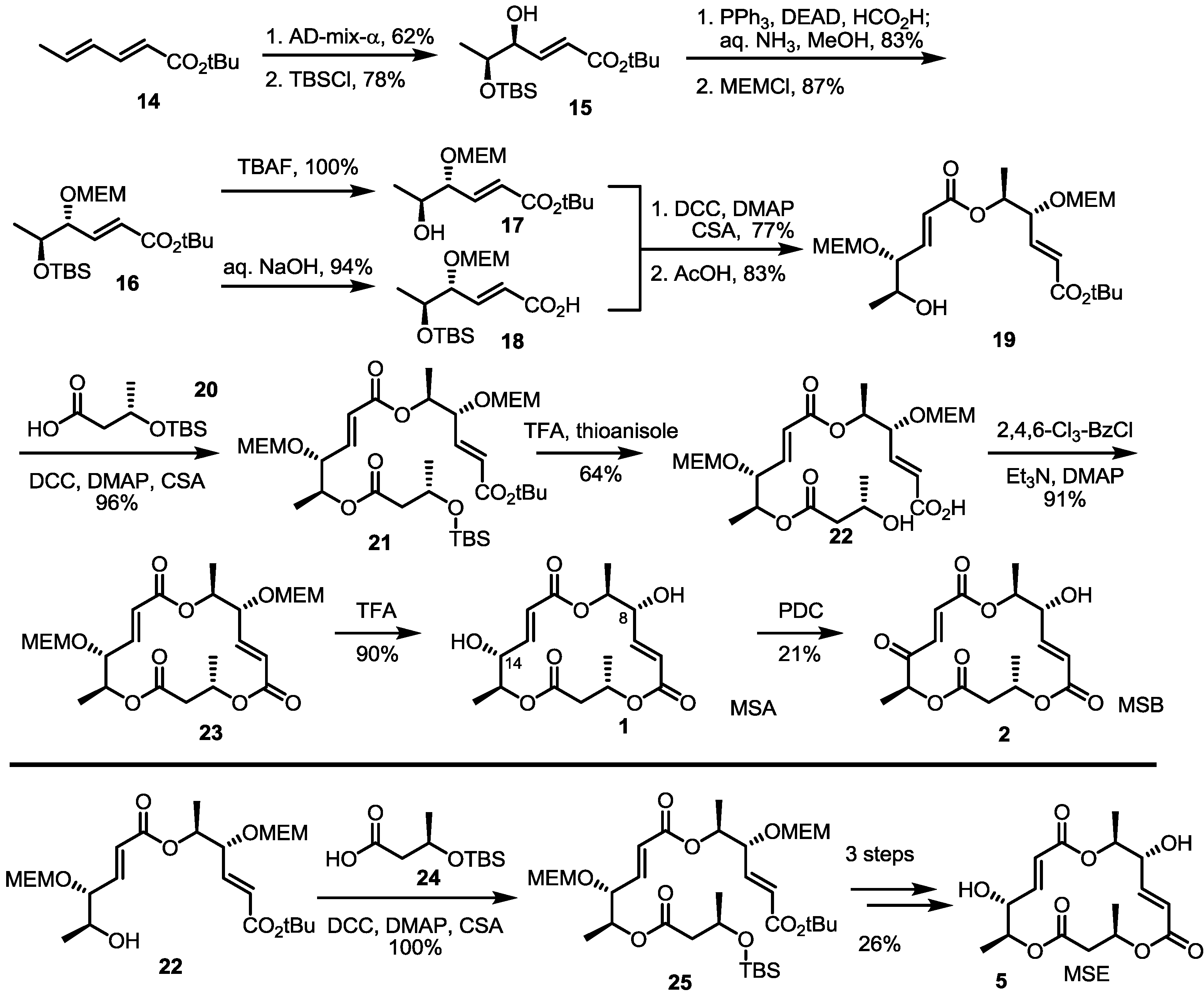
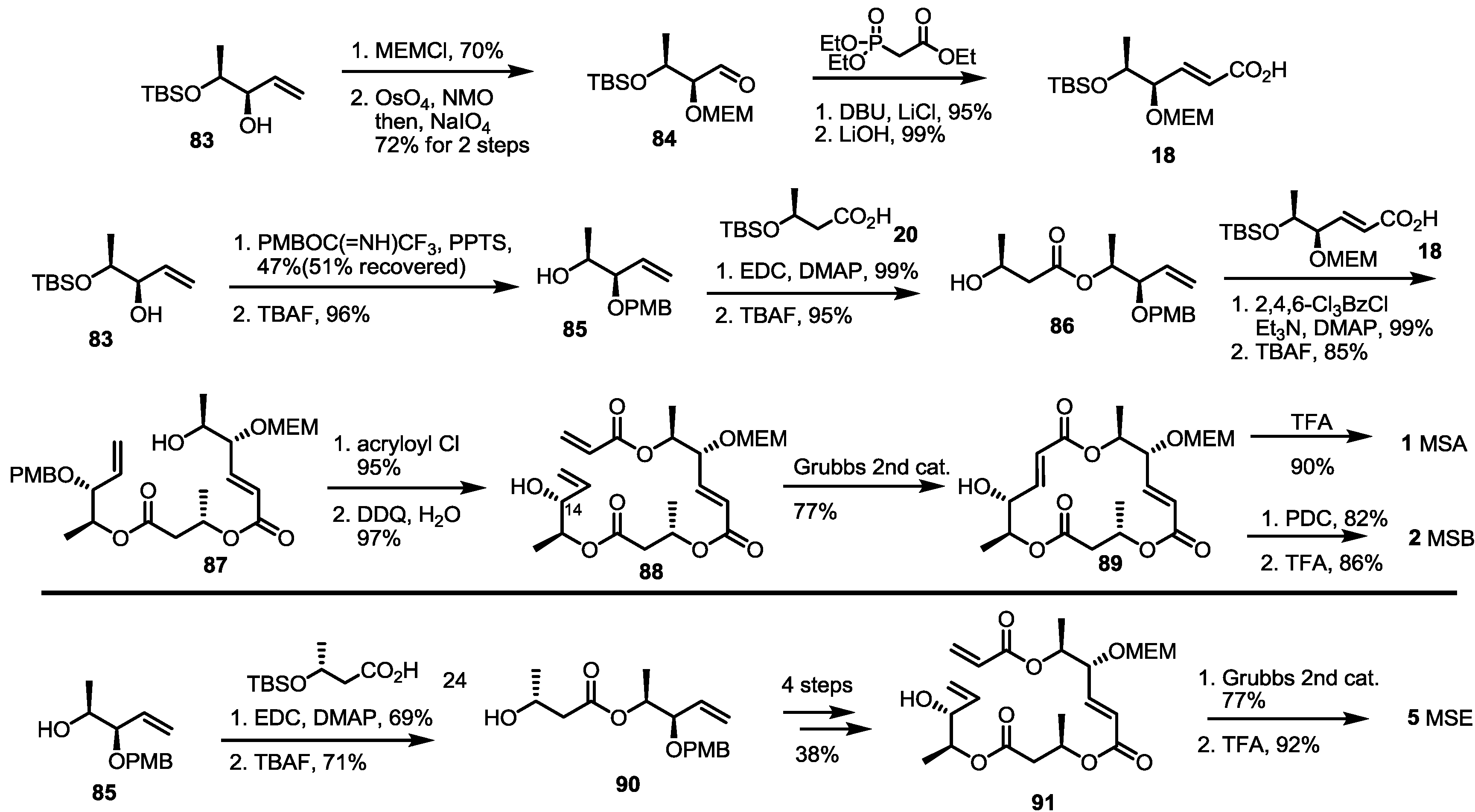
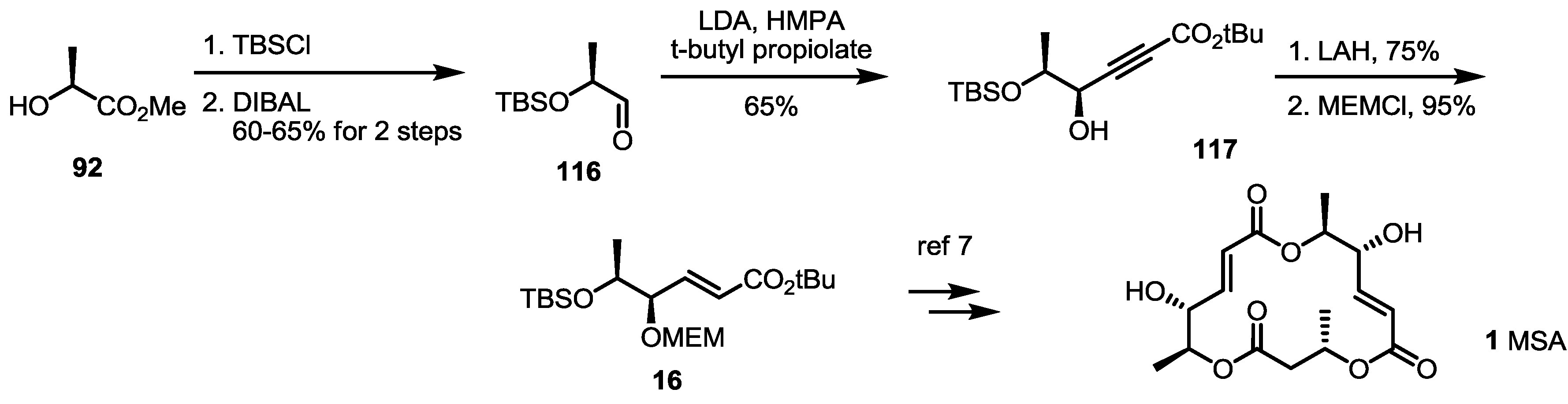
As the main isomers of the MS family, the first synthetic approach was developed through a collaboration of the Smith and Omura groups, as shown in Scheme 1 [8]. This first synthesis contains not only the synthetic route to MSA/B, but also a determination of the absolute structures of these natural products.
Scheme 1.
The first total synthesis of MSA, B and E.

MEM: 2-Methoxyethoxymethyl, TBS: tert-Butyldimethylsilyl, DCC: Dicyclohexylcarbodiimide, TBAF: Tetrabutylammonium Fluoride, TFA: Trifluoro aceticacid, DMAP: 4-(Dimethylamino)pyridine, CSA: Camphor-10-sulfonic acid, DEAD: Diethyl azodicarboxylate, PDC: Pyridinium dichromate.
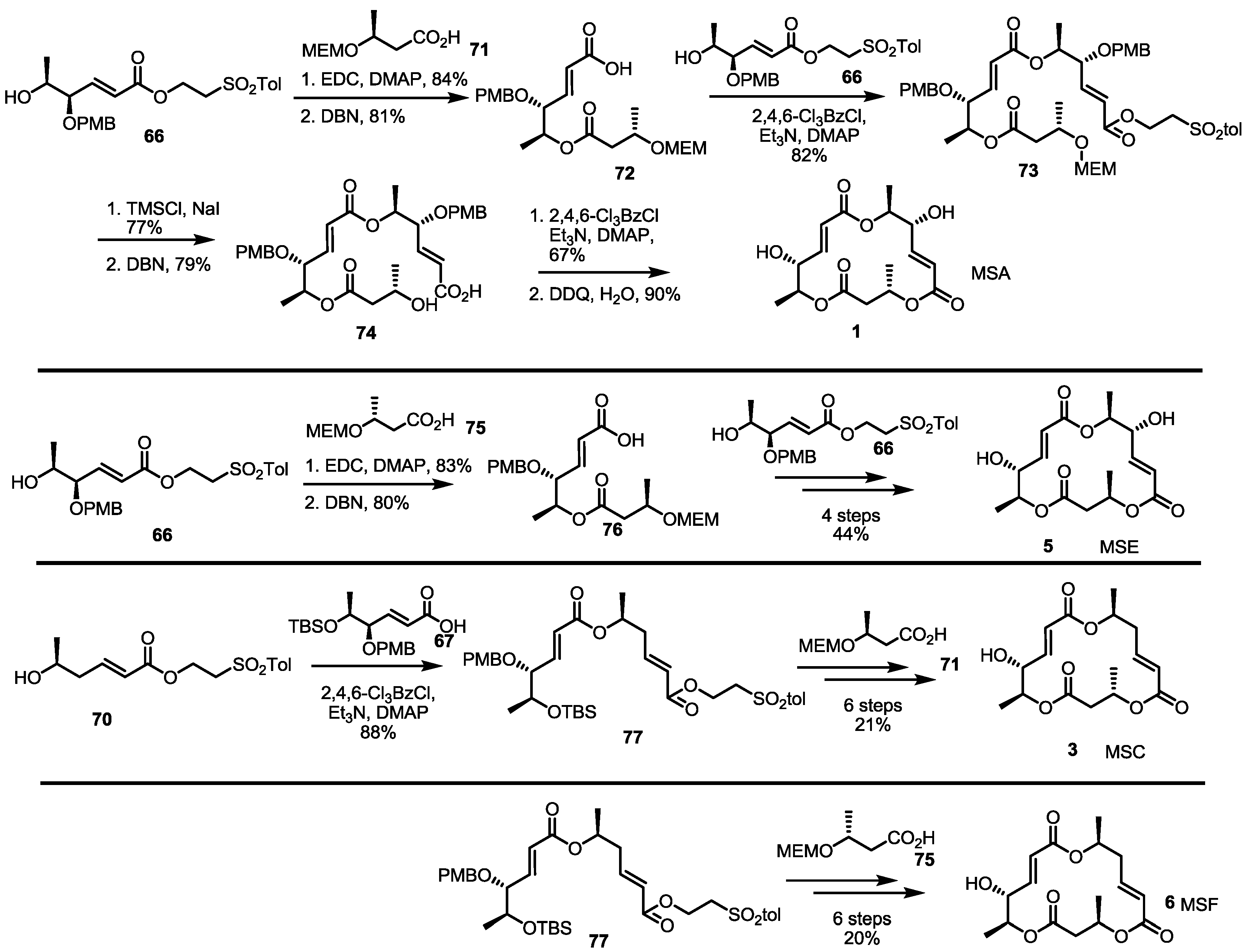
Asymmetric dihydroxylation of t-butyl sorbate and chemoselective silyl protection produced allylic alcohol 15 in a good yield. Mitsunobu inversion of the allylic alcohol configuration and MEM protection produced bis-protected diol 16, which could be used as a common intermediate for further transformation. Silyl deprotected alcohol 17 and hydrolyzed carboxylic acid 18 were coupled to produce a dimerized ester that was converted into secondary alcohol 19 using AcOH in 83% yield. Esterification of 19 with butyric acid 20 under Keck conditions [16], followed by acidic silyl deprotection and concomitant t-butyl hydrolysis, gave the crucial seco acid in moderation. Finally, Yamaguchi macrolactonization and MEM deprotection produced MSA 1 in an excellent yield. MSA could be oxidized to MSB 2 using PDC (Pyridinium dichromate) reagent. Although this final oxidation produced a C-8 oxidized product in high yield, it is important that this conversion first confirmed the absolute structure of MSB.
Eight years later, total synthesis of MSE was also reported by employing a similar reaction sequence [17]. Instead of 20, 24 was coupled with the secondary hydroxyl ether 25, which was transformed into MSE 5 via the same conversion protocol as MSA.
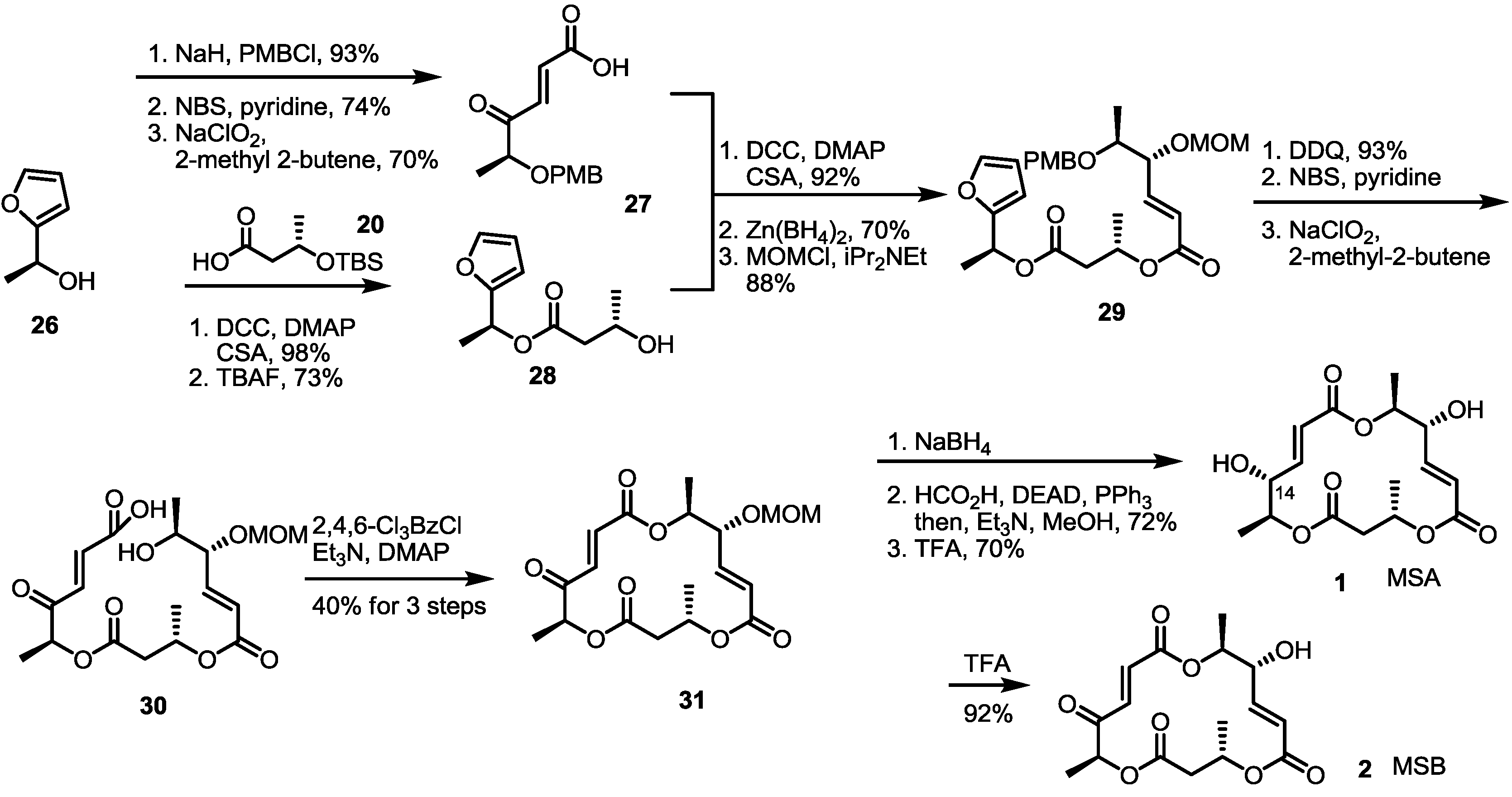
2.3. Synthesis of MSA and MSB by Furan Oxidation
The second total synthesis of MS was accomplished using the furan oxidation strategy [18,19]. The Kobayashi group reported conversion of furan to γ-keto-α,β-unsaturated carboxylic acid using NBS, and then Pinnick oxidation [20] could be used for efficient preparation of the MS skeleton framework (Scheme 2). Actually, versatile conversion of the hydroxyfuran 26 produced not only carboxylic acid 27 through the PMB (p-Methoxybenzyl) protection/furan oxidation sequence, but also secondary alcohol 28 through the esterification/silyl deprotection sequence, respectively. They were connected together under Keck conditions to give a dimeric ester, which was reduced, and the resulting hydroxyl group was protected with the MOM (Methoxymethyl) group to form 29. It was possible to create new stereogenic centers in high selectivity (>15:1) using Zn(BH4)2, −90 °C and the reverse addition strategy. To produce seco acid 30, PMB deprotection/furan oxidation of dimer 29 was carried out successfully. The 16-memberd macro-ring closure reaction under Yamaguchi conditions produced MOM-protected MSB 31. Unlike MSB, which was synthesized by simple acidic deprotection (92%), however, MSA was difficult to produce, because the reduction of the ketone moiety gave the undesired C14 epimer exclusively. Finally, Mitsunobu inversion of the C14 hydroxy group and deprotection were executed to create MSA from the MOM-protected MSB 31.
Scheme 2.
Total synthesis of MSA and MSB through furan oxidation.

PMB: p-Methoxybenzyl, NBS: N-Bromosuccinimide, DDQ: 2,3-Dichloro-5,6-dicyanobenzoquinone, MOM: Methoxymethyl.
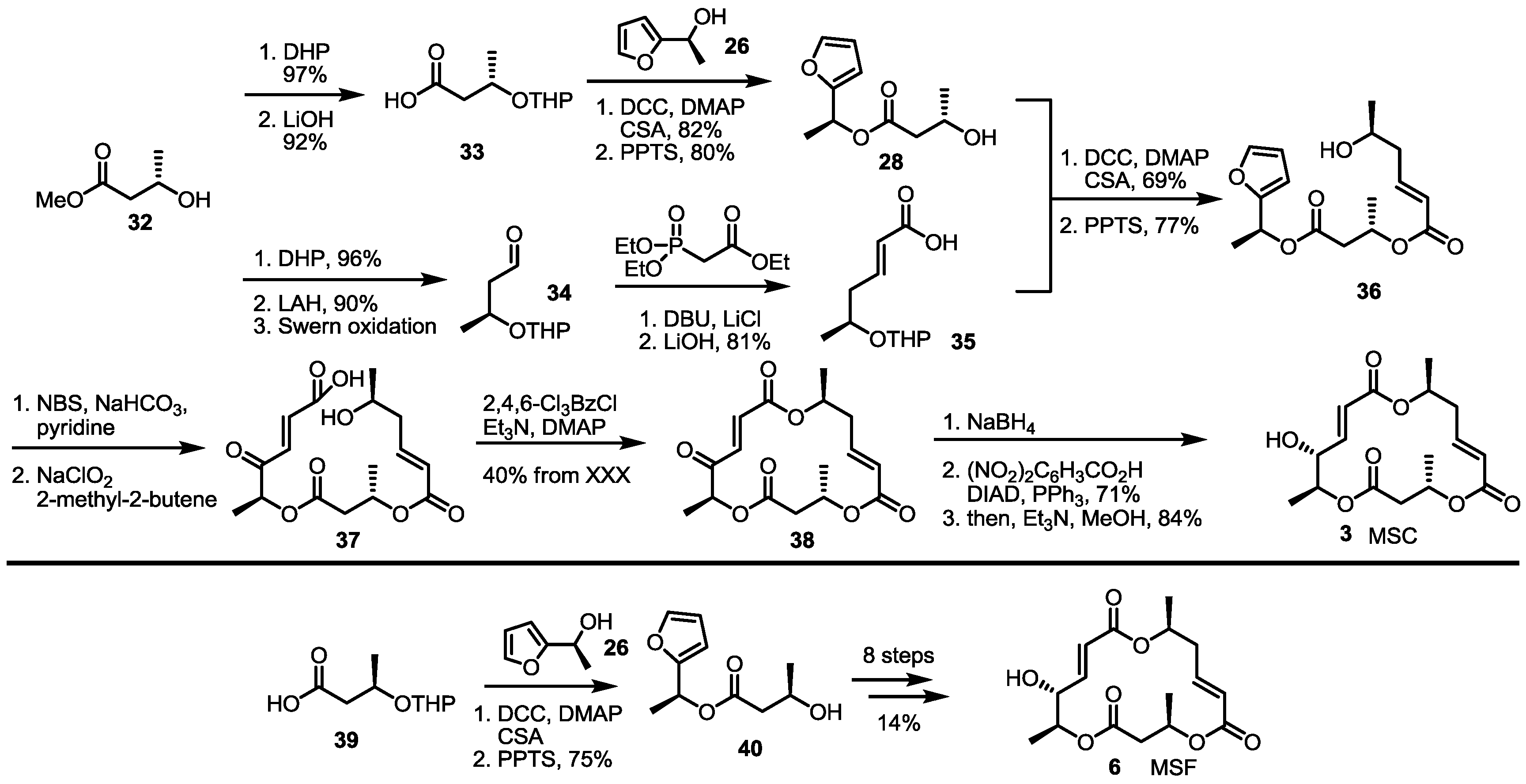
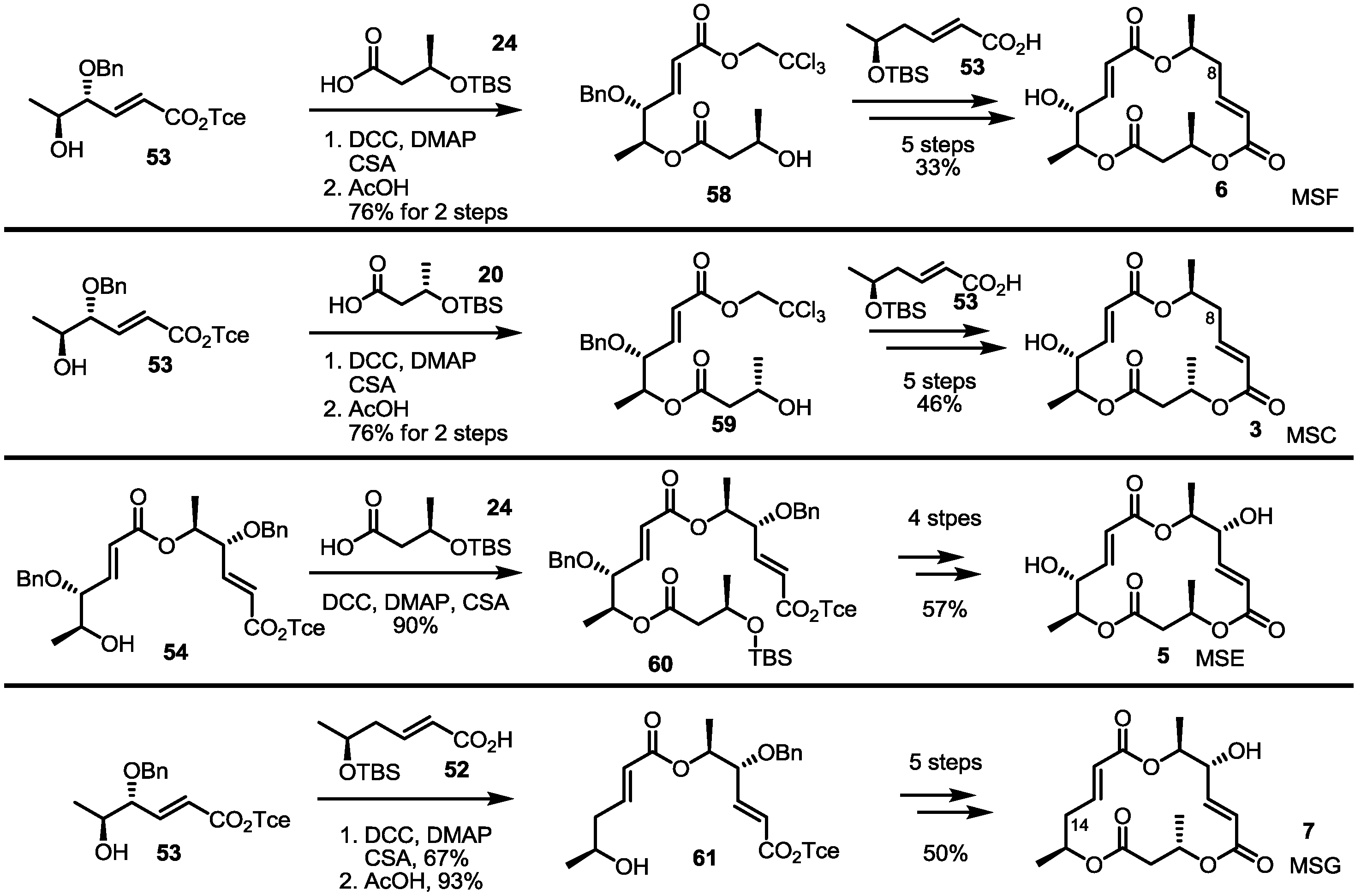
Employing the late-stage furan oxidation protocol, 8-dehydroxy MS was prepared [21] (Scheme 3). The hydroxy butyrate 32 was protected and hydrolyzed to produce butyric acid 33 to be converted into hydroxyfuran 28 through esterification and THP deprotection. This alcohol 28 was transformed into dimeric alcohol 36 via esterification/deprotection with carboxylic acid 35, which were prepared by Horner-Emmons olefination and hydrolysis of the known aldehyde 34 [22]. The furan moiety of alcohol 36 was oxidized with NBS and the Pinnick oxidation sequence to produce seco acid 37. The well-established Yamaguchi lactonization, reduction and the Mitsunobu reaction [23] produced MSC in a good yield. Instead of butyric acid 33, enantiomeric acid 39 was utilized to give MSF possessing a C-13 epimeric chiral center of MSC. The established procedure made it possible to produce MSF in 10 steps.
Scheme 3.
Total synthesis of MSC and MSF through furan oxidation.

DHP: Dihydropyran, LAH: Lithium aluminium hydride, PPTS: Pyridinium p-toluenesulfonate, DBU: 1,8-Diazabicyclo[5.4.0]undec-7-ene, DIAD: Diisopropyl azodicarboxylate.
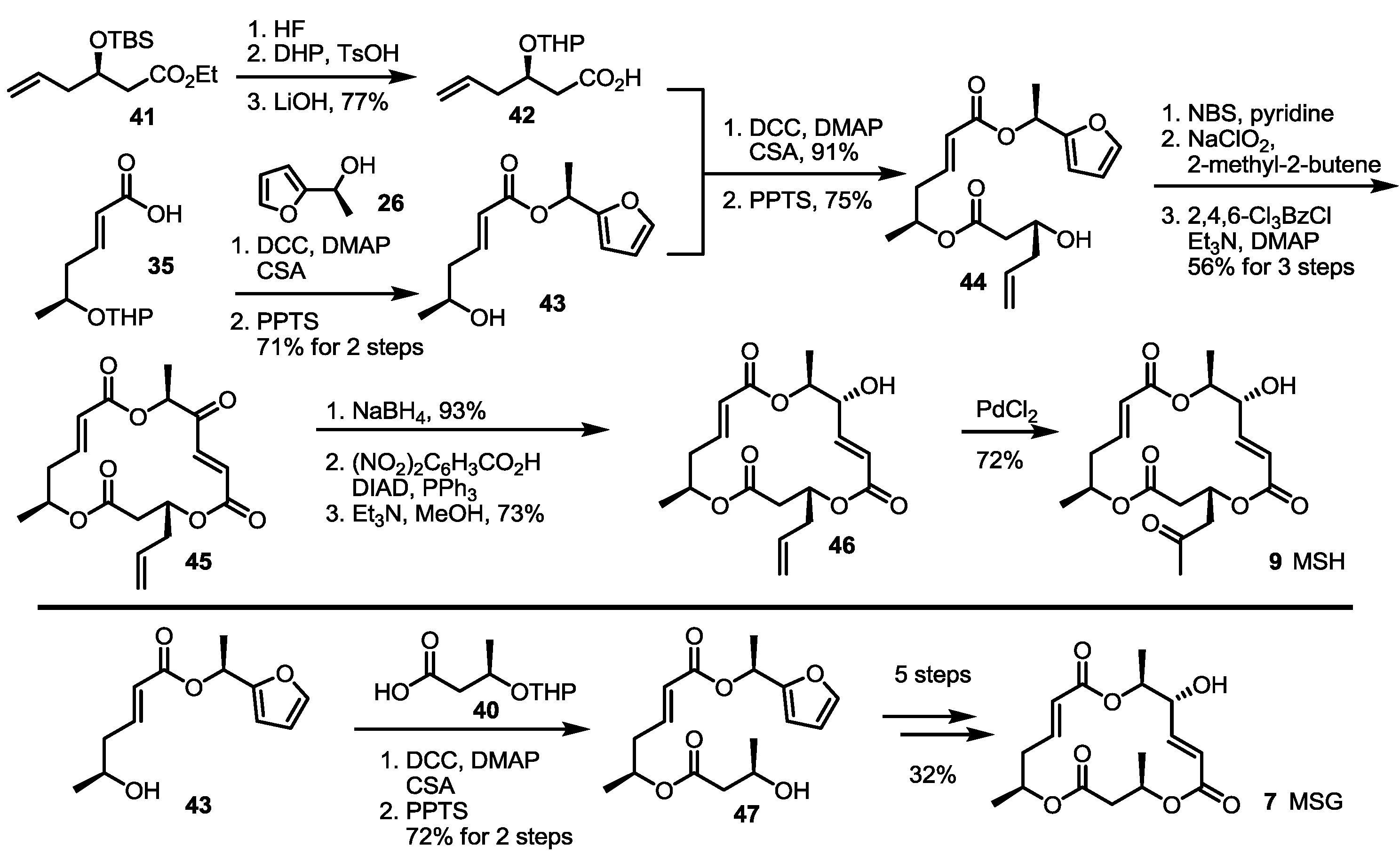
The Kobayashi group also reported total synthesis of MSH/G in 2002 [24]. The known hexenoate 41 [25] was converted into carboxylic acid 42 through protection group exchange and hydrolysis. This carboxylic acid was esterified with the furan 43, prepared from the previously synthesized 35 via esterification, followed by THP deprotection, to produce the alcohol 44, after treatment with PPTS, in a 75% yield. Crucial NBS-mediated furan oxidation/Pinnick oxidation/Yamaguchi lactonization gave trimeric ester 45 in 56% yield. After introduction of the C-14 hydroxy group, the terminal alkene was oxidized to a ketone through Wacker oxidation to form MSH 9. Instead of 42 as its esterification partner, simple carboxylic acid 39 was chosen to produce MSG 7, employing the well-established seven steps shown in Scheme 4.
Scheme 4.
Total synthesis of MSH and MSG through furan oxidation.

THP: Tetrahydropyran, HF: Hydrogen fluoride.
2.4. Synthesis of MS by Enzymatic Resolution
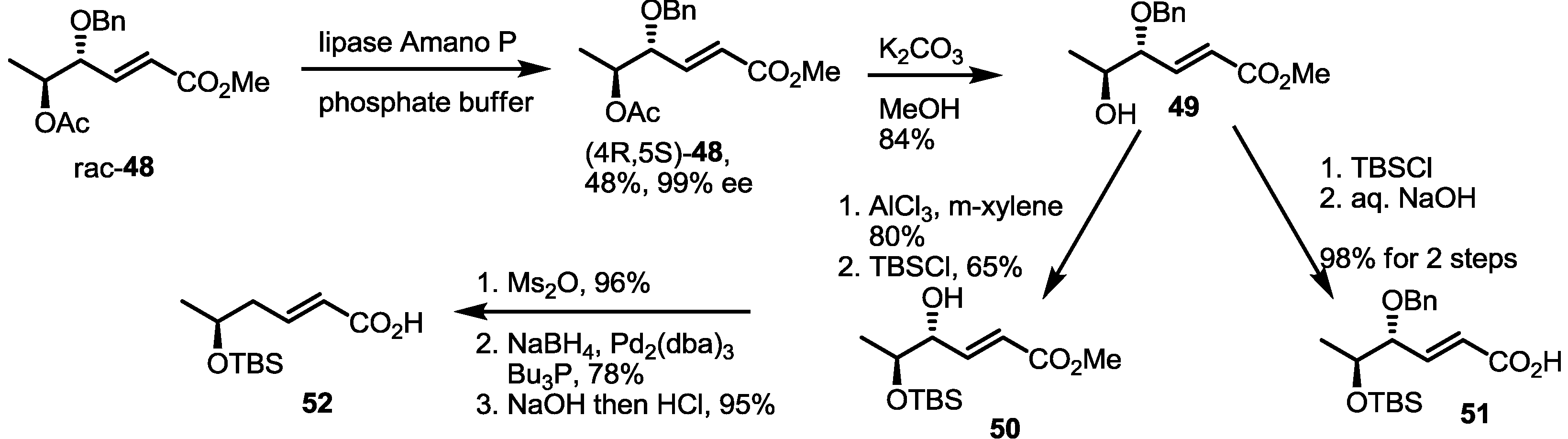
Enzymatic resolution of racemic 48 was also developed for MS synthesis [26,27] (Scheme 5). Acetylated anti-diol 48 was differentiated under catalytic hydrolysis of lipase Amano P to produce optically active (4R,5S)-48 in an excellent selectivity. Deacetylation of (4R,5S)-48 produced versatile synthon 49. Actually, the TBS protection/hydrolysis protocol gave the key monomeric acid 51, used for general synthesis of MS. At the same time, protection group exchange of 49 was carried out to prepare 50. Then, deoxygenation was executed via mesylation followed by Pd-mediated hydride insertion [28]. After hydrolysis of the methyl ester, monohydroxy carboxylic acid 52 was obtained in a good yield and utilized for the synthesis of deoxygenated MSs.
Scheme 5.
Chiral resolution of racemic 48 and preparation of building blocks.

Ac: Acetyl, Bn: Benzyl.
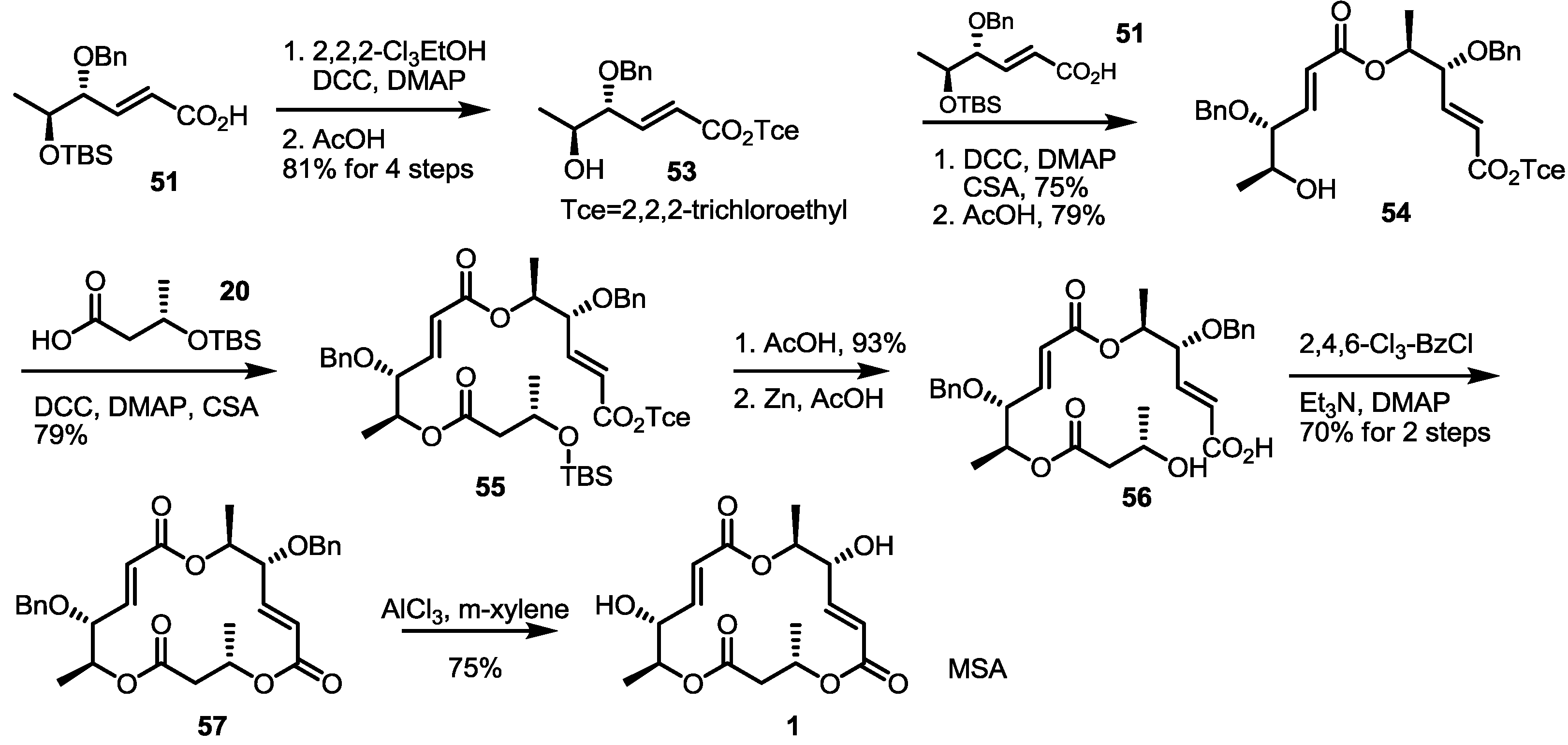
Employing these building blocks, MS synthesis was carried out (Scheme 6). Carboxylic acid 51 was transformed into 53 via trichloroethyl esterification and acidic deprotection. Iterative esterification of 53 with acid 51, deprotection of the TBS group, esterification with acid 20, deprotection of the TBS group and treatment with zinc produced seco acid 56 in a good yield. Finally, macrolactonization and benzyl deprotection gave MSA in a good yield.
Scheme 6.
Total synthesis of MSA and MSE through an enzymatic resolution route.

Similarly, MSE was also prepared by esterification with 20 and butyric acid 24 and additional transformations (four steps, 57% yield) (Scheme 7). Deoxygenated MSs was also prepared using this synthetic protocol and building blocks. Esterification of 53 with deoxygenated carboxylic acid 52 and deprotection gave dimer 61 in a good yield. The established 5-step procedure produced MSG. Coupling of butyric acid 20/24 with alcohol 53 produced 59 and 58, depending on the chiral center of butyric acid [29]. They were successively converted into MSF/C through normal transformations.
Scheme 7.
Total synthesis of MSE, G, F and C.

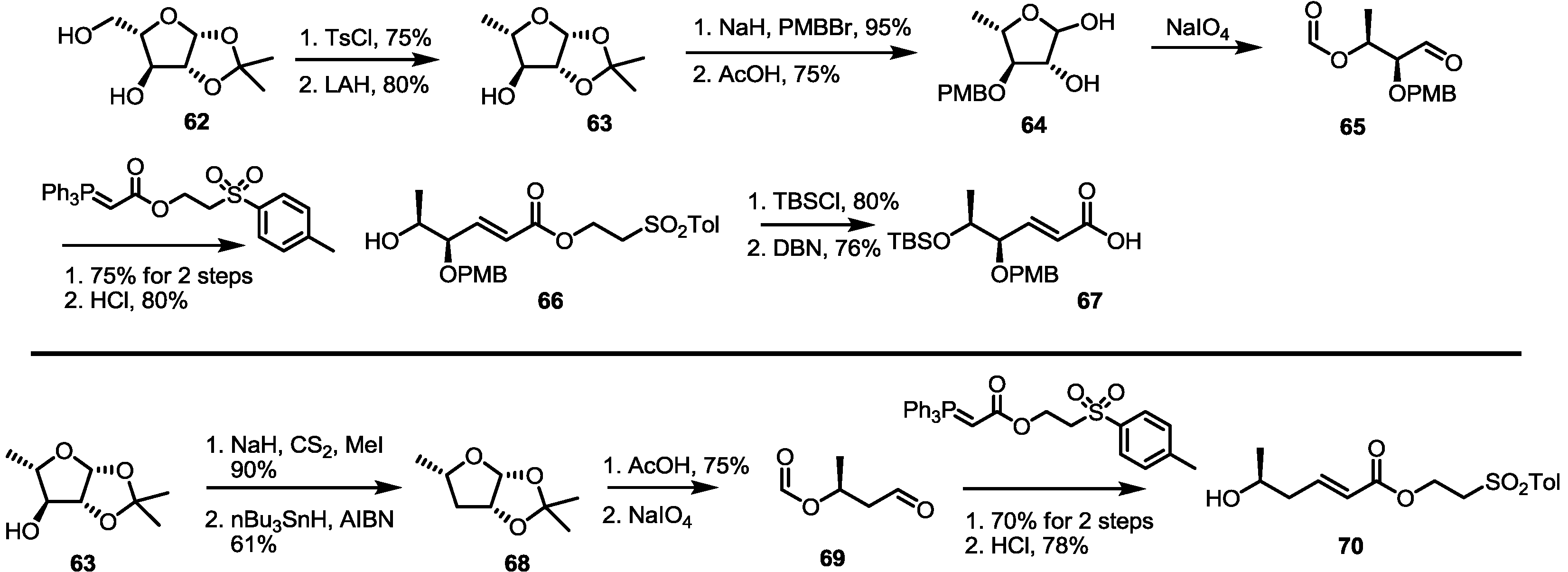
Another approach that used sugar building blocks was also developed for MS synthesis by the Sharma group [30,31]. To prepare the 1,2-antihydroxy carboxylic acid moiety, pentose 62 was tosylated and reduced, as shown in Scheme 8. PMB protection and deacetalization, followed by the diol cleavage reaction, gave unstable aldehyde 65. Immediate Wittig olefination and deformylation produced the desired bis-hydroxy ester 66 easily. TBS protection and de-esterification under basic conditions also gave another building block, 67. Deoxygenated sugar 63 was also utilized to prepare mono-hydroxy ester 70. Xanthate formation and radical-mediated deoxygenation produced deoxy sugar 68 in a good yield. After application of the previously established four-step procedure, mono-hydroxy ester 70 was quickly prepared.
Scheme 8.
Preparation of building blocks from sugar species.

AIBN: Azobisisobutyronitrile, DBN: 1,5-Diazabicyclo[4.3.0]non-5-ene.
MS synthesis using sugar synthon is summarized in Scheme 9. Hydroxy ester 66 was coupled with protected butyric ester and treated with DBN to give dimeric acid 73. Again, esterification with hydroxyl ester 66 produced the fully substituted ester 73 in an 82% yield. Macrolactonization, deprotection and de-esterification were executed to produce seco acid 74, which was converted into MSA 1 after Yamaguchi lactonization and the PMB deprotection sequence. After establishing the synthetic protocol, MS isomers E, C and F were also synthesized. Instead of 71, enantiomeric acid 75 was utilized in the synthesis of MSE 5. Actually, the normal deprotection/esterification protocol produced MSE 5. For deoxygenated isomer MSC/F, deoxygenated fragment 70 was used. Esterification of 70 with carboxylic acid 67 produced a versatile intermediate 77 in an excellent yield. Employing butyric acid 71 or 75, MSC 3 or MSF 6 was synthesized in six steps, depending on the chirality of the coupling partner.
Scheme 9.
Total synthesis of MSA, E, C and F using building blocks from sugar species.

EDC: N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride.
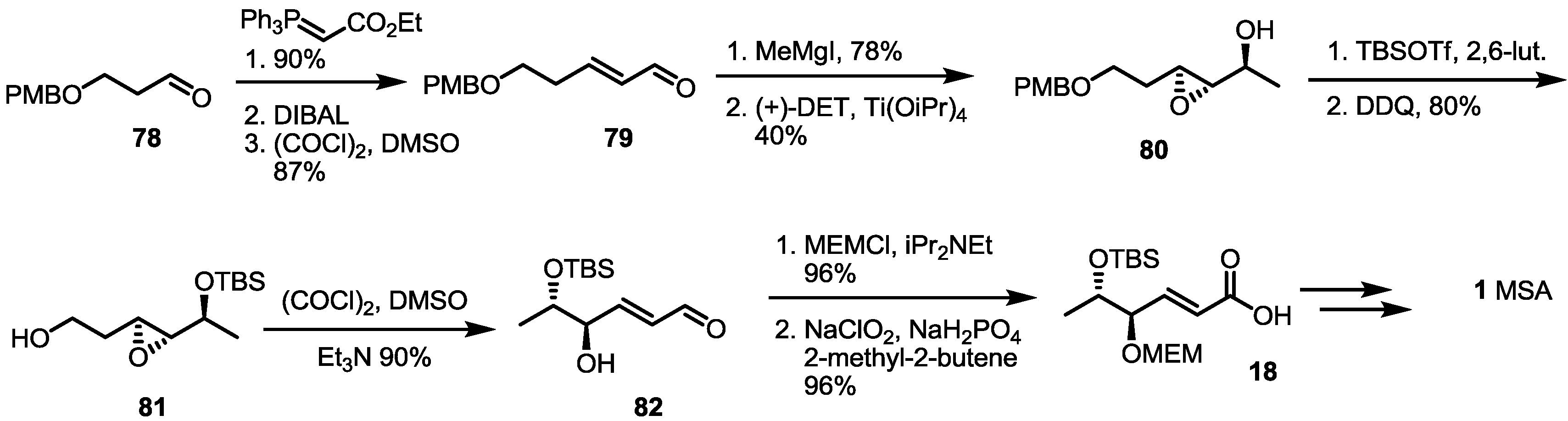
Another synthetic approach using chiral resolution was reported in 2003 [32] (Scheme 10). A simply protected butanal 78 was treated with Wittig reagent, followed by DIBAL (Diisobutyl aluminium hydride) reduction/Swern oxidation to produce hexenal 79 in an excellent yield. After Grignard reaction with MeMgI, the resulting allylic alcohol was exposed to Sharpless asymmetric epoxidation for chiral resolution (94% enantiomeric excess). An optically active alcohol 80 was then protected with TBS, and primary alcohol was liberated with the help of DDQ to give epoxy alcohol 81. Free alcohol 81 was then oxidized under Swern oxidation conditions, and concomitant aldehyde was spontaneously transformed into the desired hexenal 82 via beta-elimination. This aldehyde was protected and oxidized to produce the known carboxylic acid 18 in an excellent yield [8].
Scheme 10.
Formal synthesis of MSA using an oxidation/elimination protocol.

DET: Diethyl tartrate, DIBAL: Diisobutyl aluminium hydride.
Instead of the Yamaguchi lactonization reaction as macrocyclization, another strategy has been studied since 2003. For this purpose, ring-closing metathesis (RCM) was applied to synthesize the MS core structure successfully, as shown in Scheme 11 [33]. The Nemoto group reported that the known allyl alcohol 83 could be converted into MS using this strategy. Protection of the allyl alcohol 83 with the MEM group and oxidative cleavage produced bis-protected aldehyde 84, which can be homologated using Horner–Emmons reagent to produce hexenoic acid 18 after basic hydrolysis. Allyl alcohol 83 was also protected with the PMB group, and TBS was eliminated to prepare homoallylic alcohol 85. Esterification with butyric acid 20 and iterative TBS deprotection gave hydroxyl ester 86 in an excellent yield. Again, esterification with acid 18 and deprotection gave the secondary alcohol 87, which was converted into pivotal acrylate 88 after acryloylation and PMB deprotection. After an extensive substrate and reaction conditions survey, the RCM reaction was carried out sufficiently to give the macrolactone skeleton 89 using Grubbs second catalyst and refluxing CH2Cl2. It is important to note that the RCM did not work well in the presence of the PMB group, due to the C-14 protection group. This discrepancy means that insertion of the substrate into the ruthenium core depends on the steric factor of the substrate. Except for Yamaguchi cyclization, the RCM strategy was the first protocol to create an MS macrolactone core. In addition, the RCM strategy was applied to the synthesis of other MS and MS artifacts, because of its efficiency and easy preparation of the corresponding substrates, as will be shown later (Scheme 16). Finally, MSA 1 was prepared from the macrolactone 89 after acidic deprotection, while MSB 2 was prepared using oxidation/deprotection reactions. In addition, MSE 5 was synthesized from homoallylic alcohol 85 using butyric acid 24 instead of 20 after a similar reaction protocol [34].
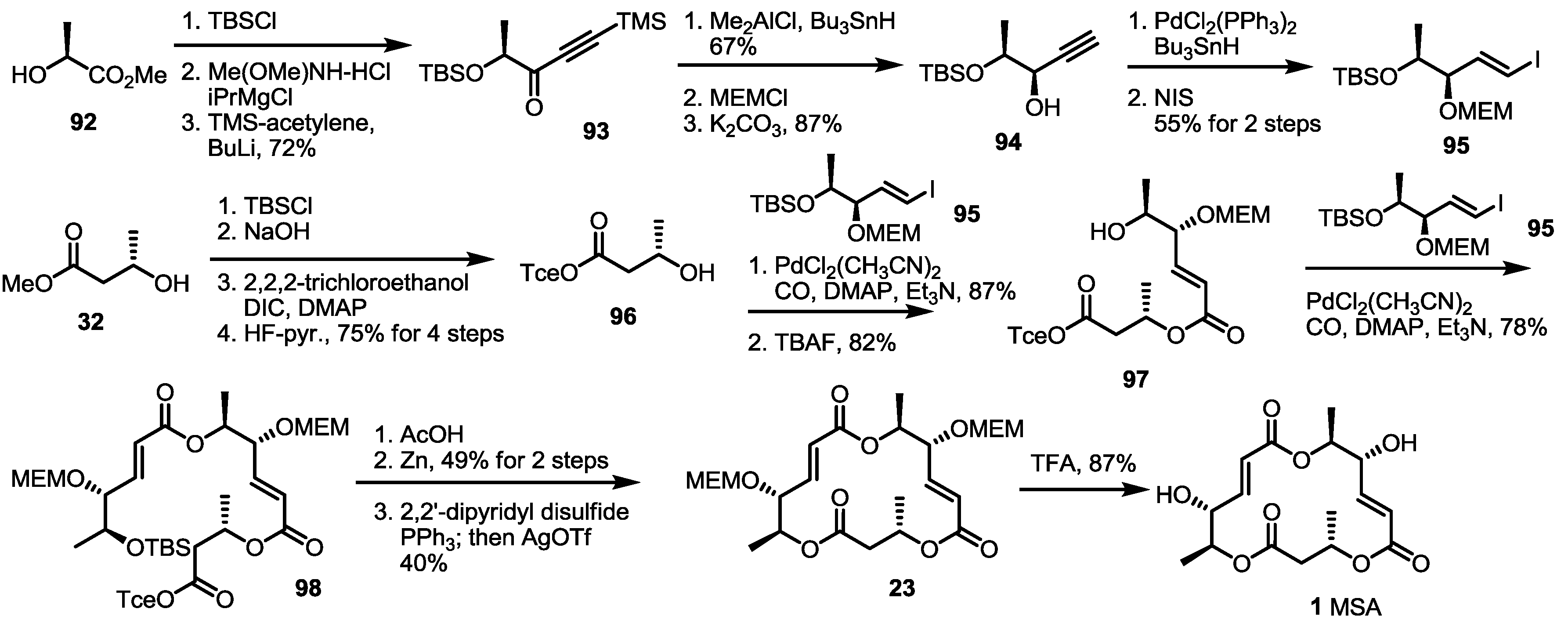
Pd-mediated CO insertion was also developed for MS synthesis by the Takahashi group [35] (Scheme 12). Methyl lactate 92 was transformed into alkynone 93 by TBS protection, Weinreb amide formation and alkynylation. After selective reduction of the carbonyl group to a hydroxyl group, free alcohol was protected using MEM to give propargylic alcohol 94 after TMS deprotection. This alkyne was then converted into vinyl iodide 95 via a Pd-catalyzed reaction and NIS (N-Iodosuccinimide) treatment. The vinyl iodide 95 was coupled with hydroxyl butyrate 96, prepared from methyl ester 32 and the ester exchange reaction, to produce dimer 97 after TBS deprotection. This C-C and C-O bond formation reaction with CO insertion into the vinyl iodide produced the secondary alcohol in an excellent yield. The same reaction was utilized again to give trimeric ester 98 in 78% yield. After TBS/trichloroethyl deprotection, the corresponding seco acid was cyclized to the bis-protected macrolactone 23 [8] under a modified Mukaiyama–Corey lactonization condition in a moderate yield. It is noteworthy that the addition of AgOTf was crucial to perform cyclization reaction. Finally, acidic deprotection produced MSA 1 in a good yield. This Pd(0)-mediated carbonylative insertion strategy also utilized the combinatorial chemistry of MS to prepare the MS-related chemical library successfully [36].
Scheme 11.
Total synthesis of MSA, B and E by the ring-closing metathesis (RCM) strategy.

Scheme 12.
Total synthesis of MSA using the CO insertion strategy by the Takahashi group.

NIS: N-Iodosuccinimide, DIC: N,N′-Diisopropylcarbodiimide.
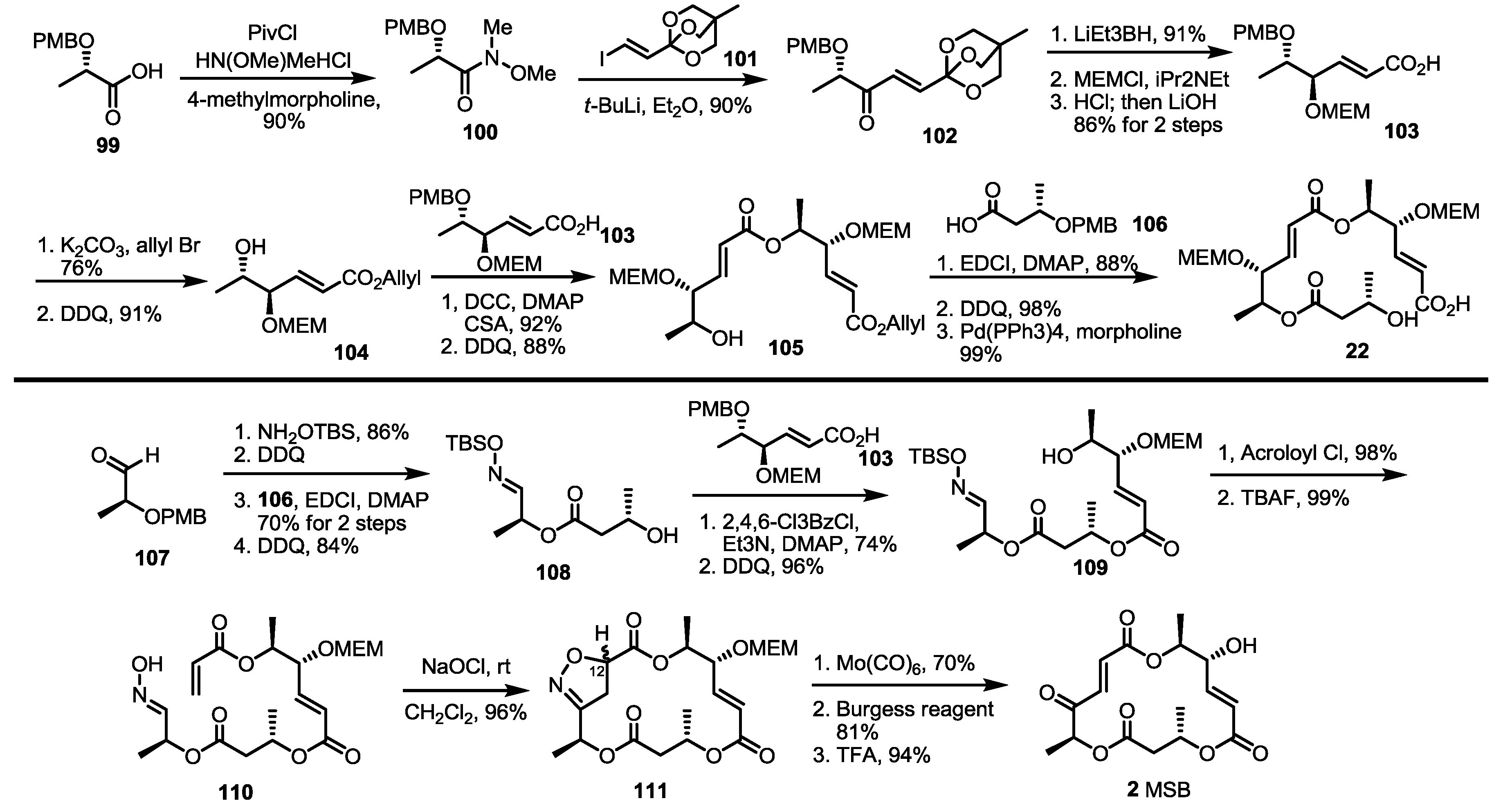
A vinylogous ester anion was found to be a useful synthon for the MS skeleton in 2005 [37]. Protected lactic acid 99 was converted into Weinreb amide 100 (90%), which was treated with the lithiated vinylogous orthoester anion to give unsaturated ketone 102 in a 90% yield [38,39]. After chelation-controlled reduction, MEM protection and hydrolysis, monomeric acid 103 was obtained quickly. This building block was esterified and deprotected to homoallyl ester 104, which was transformed into dimeric ester 105 via iterative esterification/deprotection reactions. Finally, esterification, PMB deprotection and allyl deprotection produced the known seco acid 22 in an excellent yield [8].
A more important advance was the finding that nitrile oxide cycloaddition could be used to construct the MS skeleton, generating an unstable ketone moiety at the same time (Scheme 13). The known aldehyde 107 was converted into alcohol 108 using oxime formation, esterification and deprotection [40]. This alcohol was also coupled with carboxylic acid 103 to give dimeric alcohol 109 after PMB deprotection. Acryloylation and TBS deprotection produced the desired oxime 110 quickly. Pivotal intramolecular nitrile oxide cycloaddition was carried out at this stage to prepare isoxazoline 111, which possesses latent unsaturated ketone functionality. Fortunately, isoxazoline 111 was obtained in an excellent regioselectivity, while stereoselectivity varied depending on the reaction conditions, such as the solvent and additives [41,42]. Although C12 stereogenic center would be eliminated for MSB 2 synthesis, it was important for MSJ/K synthesis (Scheme 14). Reductive cleavage of the N-O bond, elimination of the secondary alcohol and acidic deprotection produced MSB 2 directly.
Scheme 13.
Synthesis of MSA and MSB using orthoester addition and cycloaddition.

Scheme 14.
Structural determination of MSJ and MSK through total synthesis.

Tf: Trifluoromethanesulfonate, CAN: Ceric ammoniumnitrate.
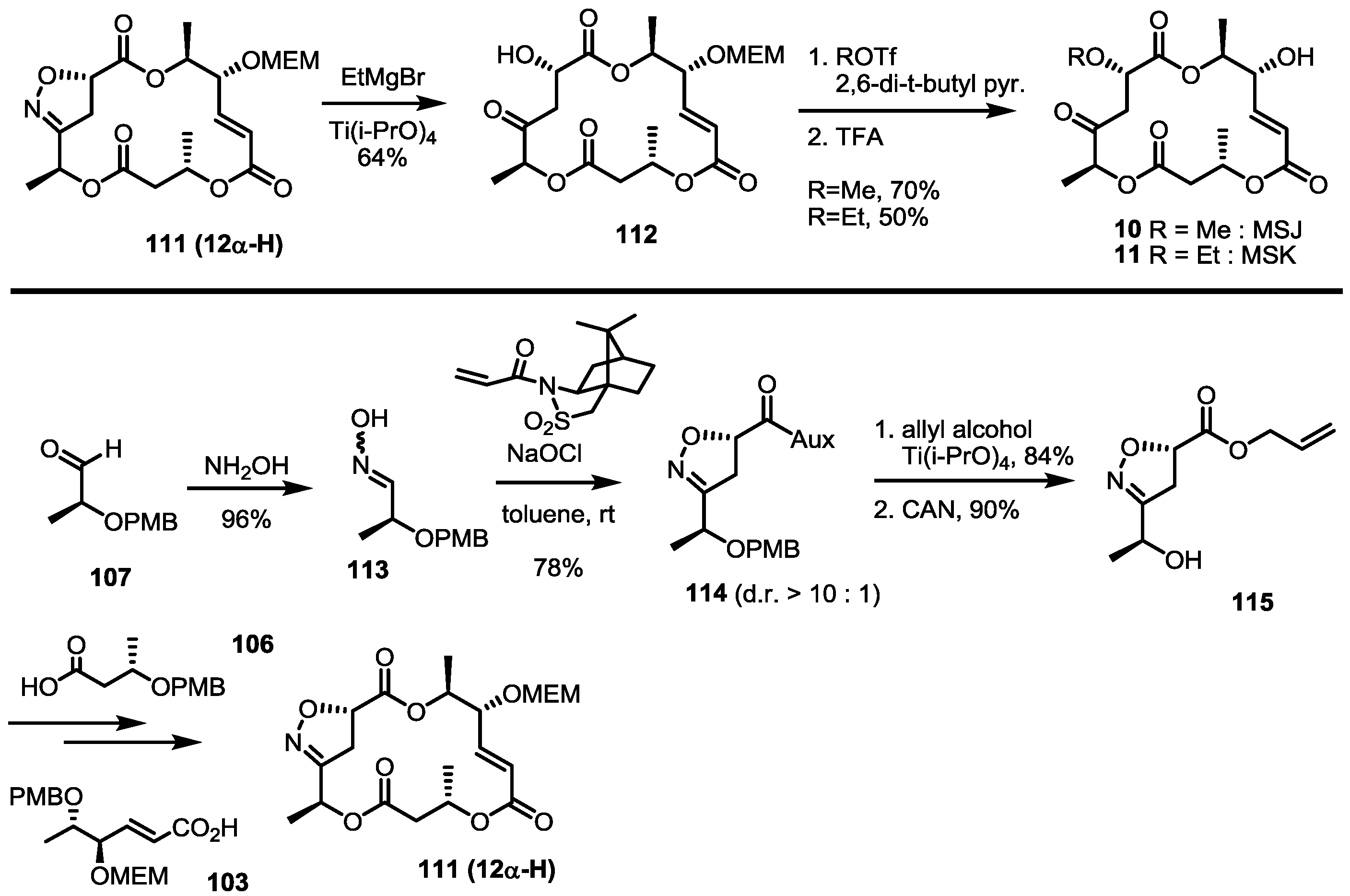
After total synthesis of MSA and B was completed, the synthetic intermediate 111 was utilized to elucidate the structure of MSJ 10 and K 11 [43]. As expected, reductive cleavage of isoxazoline, O-alkylation and the acidic deprotection sequence produced the desired product, which possesses all spectral data matching natural MSJ and K, allowing confirmation of the exact structure of MSJ and K. Because the 12-α isoxazoline 111 was usually a minor product of the corresponding cycloaddition, auxiliary-assisted cycloaddition was also designed (Scheme 14). The free oxime 113 and acrylated sultam were ligated to give isoxazoline 114 in an excellent diastereoselectivity via intermolecular cycloaddition. Ester exchange and PMB deprotection produced allyl ester 115, which was converted into isoxazoline 111 using the established esterification/deprotection sequence.
As another formal synthesis method for MSA, an alkyne reduction strategy was also reported in 2005 [44] (Scheme 15). TBS-protected aldehyde 116 was prepared from methyl lactate 92 and TBS protection/semi-reduction. Alkyne addition under basic conditions was carried out to get propargylic alcohol 117, which was reduced and protected to allylic ether 16 [8]. This known ether 16 could be used to synthesize MSA 1, as reported previously.
Scheme 15.
Formal synthesis of MSA using an alkyne reduction strategy.

LDA: Lithium diisopropylamide, HMPA: Hexamethylphosphoramide.
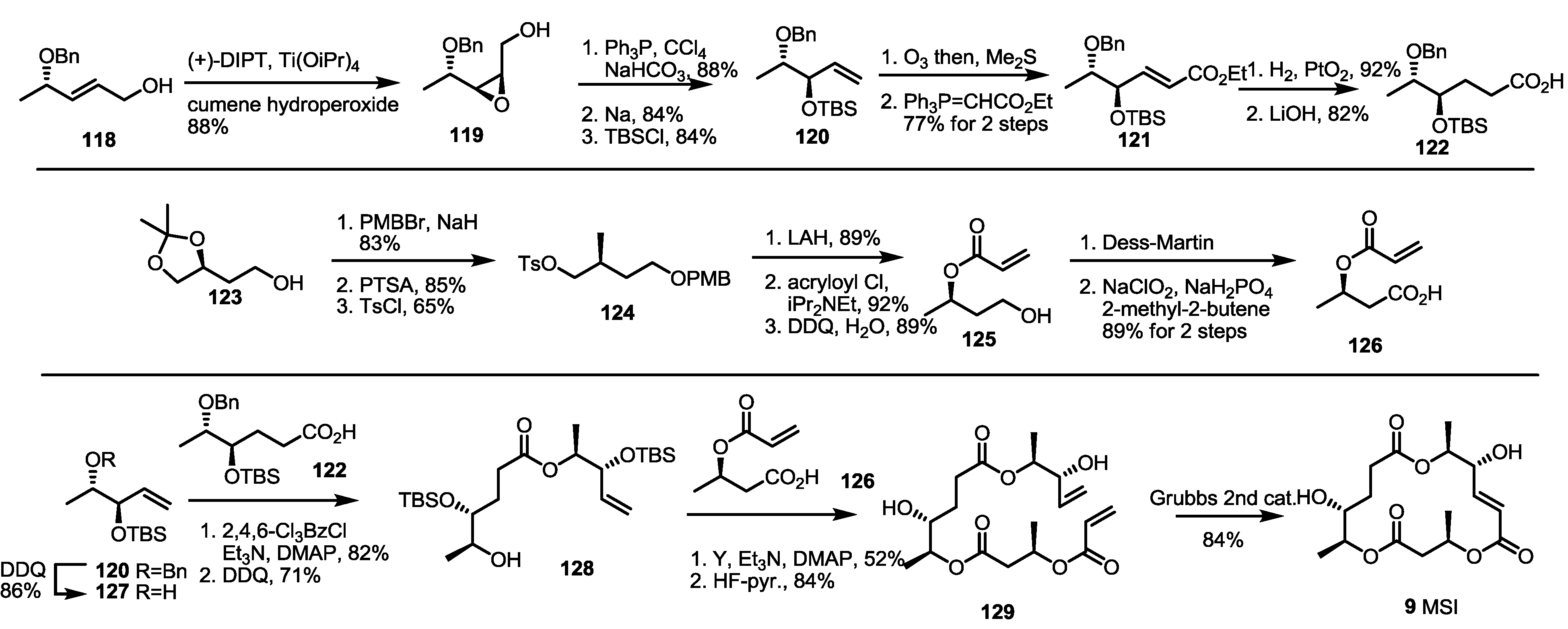
A dihydro-MS core structure was synthesized in 2007 by the Sharma group [45] (Scheme 16). They used Sharpless asymmetric epoxidation of the known alcohol 118 to obtain epoxy alcohol 119. The terminal hydroxyl group was chlorinated and eliminated with the assistance of Na to produce terminal alkene 120 after TBS protection. Ozonolysis and Wittig homologation produced hexenoate 121, which was transformed to hexanoic acid 122 by hydrogenation and hydrolysis. At the same time, the known primary alcohol 123 was protected with the PMB group (83%) [46]. After deacetalization and tosylation of another primary alcohol, tosylate 124 was obtained in a good overall yield and converted into acrylate 125 through reduction/acryloylation/PMB deprotection. Two-step oxidation of the primary alcohol into carboxylic acid also produced another building block, 126. Ligation of the three fragments commenced with debenzylation of ether 121 with DDQ. The corresponding alcohol 127 was esterified with carboxylic acid 128 to give dimeric alcohol after PMB deprotection. The second esterification with acrylic acid 126 was performed under the same conditions to give trimeric ester 129 after TBS deprotection again. Finally, RCM of the terminal diene 129 delivered MSI 9 in an excellent yield, as expected.
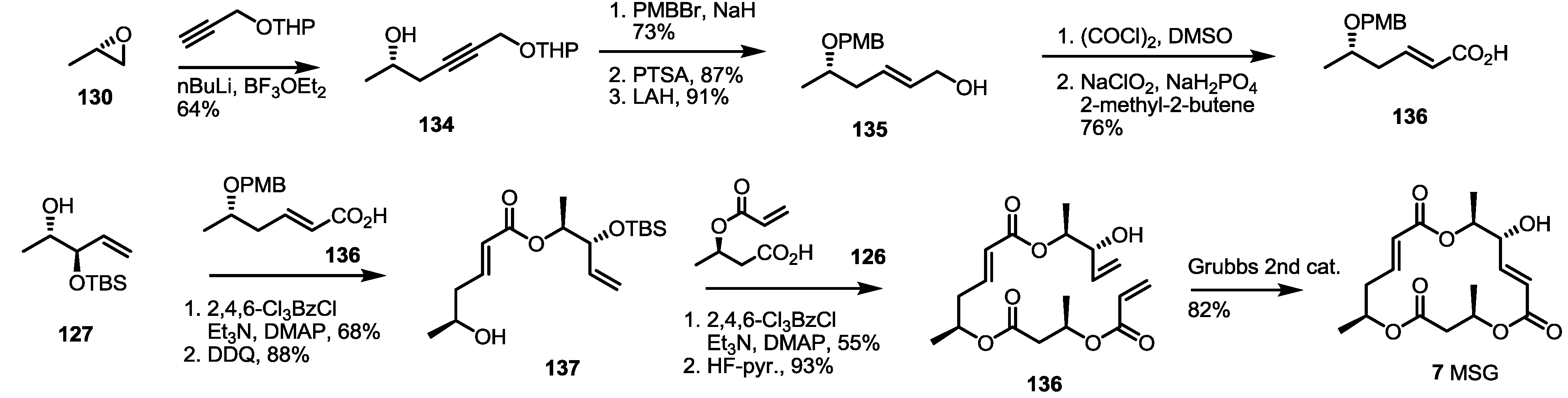
The building blocks were also utilized to synthesize MSG, as shown below (Scheme 17). Chiral epoxide 130 was treated with acetylide anion to give homopropargylic alcohol 134. After protection group exchange, the liberated primary alcohol 135 was oxidized to produce conjugated acid 136 after Swern/Pinnick oxidation. This carboxylic acid 136 was esterified with the previous building block 127 using Yamaguchi reagents. After PMB deprotection, free alcohol 137 was coupled with another previous building block 126 under the same conditions as for MSI (Scheme 16). Finally, RCM was carried out to synthesize the deoxygenated MS product, MSG 7, in an 82% yield.
Scheme 16.
Total synthesis of MSI and dihydro-MSE.

PTSA: p-Toluenesulfonic acid.
Scheme 17.
Total synthesis of MSG.

During the medicinal chemistry approach of MS, MSI and L were synthesized through the RCM route [47] (Scheme 18). The Nemoto group reported previously that the synthesized allylic alcohol 83 could be converted into 141 using protection group exchange and esterification with carboxylic acid 140. After iterative esterification/deprotection, RCM was carried out to produce a dihydro-MS skeleton 143 in an excellent yield. Acidic hydrolysis of the MEM protection group produced MSI, while protection group exchange, oxidation and the final deprotection sequence produced MSL in a moderate yield.
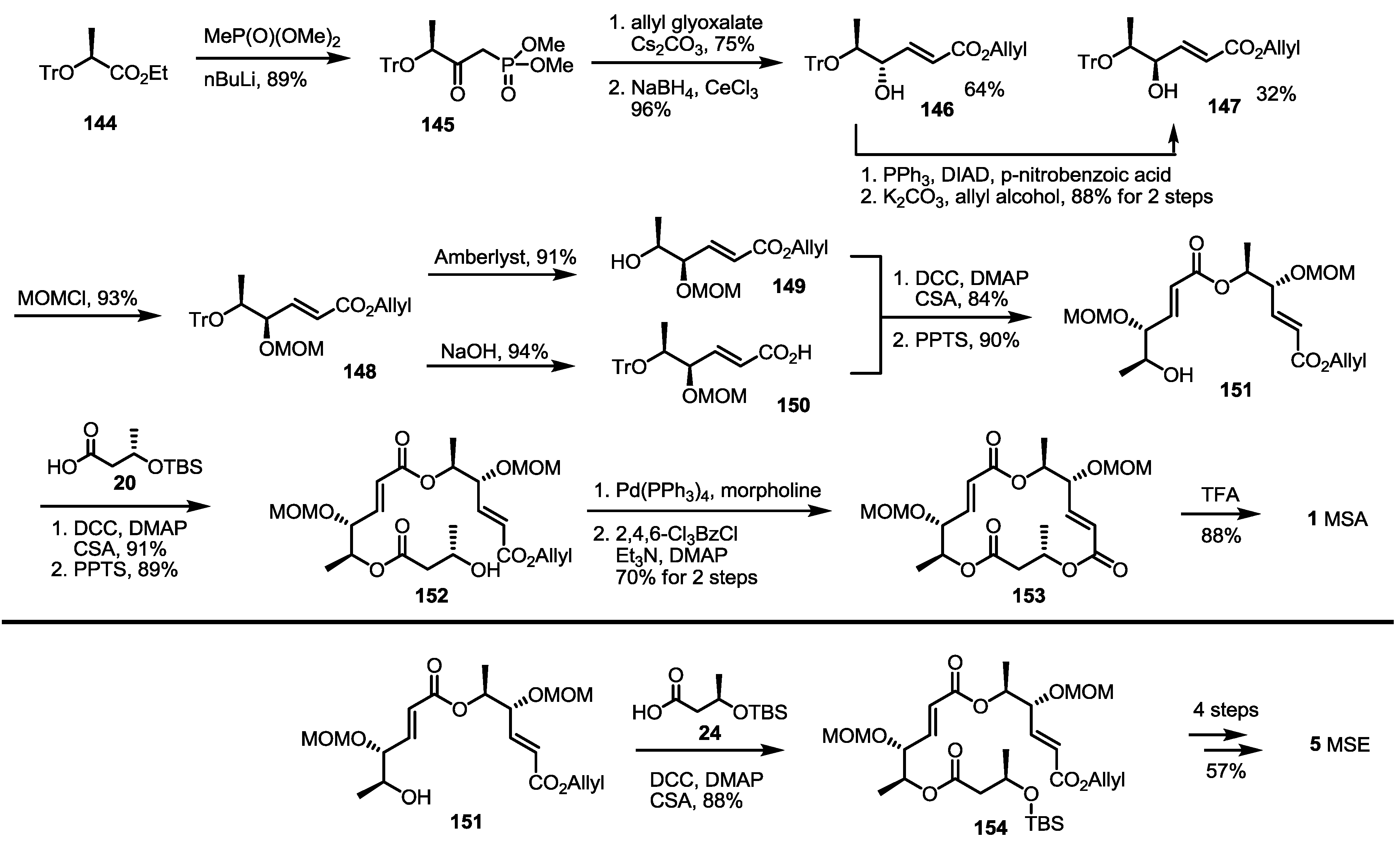
Traditional Wittig/Horner–Emmons olefination was also reported to be applicable for the synthesis of the MS skeleton, as shown in Scheme 19 [48]. A phosphonate synthon 147 was prepared from ethyl ester 144 [49] and the addition of methyl phosphonate in 89% yield. Horner–Emmons olefination of the phosphonate 145 with allyl glyoxalate, followed by NaBH4 reduction, produced allylic alcohol 146 and 147 together. Because the undesired isomer 146 was a major product, Mitsunobu inversion of the chiral center was carried out in 88% for two steps. The desired allylic alcohol 147 was then protected with the MOM group to produce bis-protected allyl ester 148 as the common substrate for further synthesis. Acidic deprotection of the trityl group produced free alcohol, 149, which was then esterified with carboxylic acid 150, prepared from 148 and hydrolysis, to give dimeric ester 151 after further PPTS-mediated trityl (triphenylmethyl) deprotection. The iterative esterification/deprotection sequence produced allyl ester 152 in an excellent yield. Finally, routine allyl deprotection/macrolactonization/MOM deprotection reactions produced MSA 1 in a good yield. As a coupling partner, butyric acid 24 was used to give MSE 5 after the conventional four-step sequence.
Scheme 18.
Total synthesis of MSI and L.

TBDPS: tert-Butyldiphenylsilyl.
Scheme 19.
Horner–Emmons approach to MSA and E.

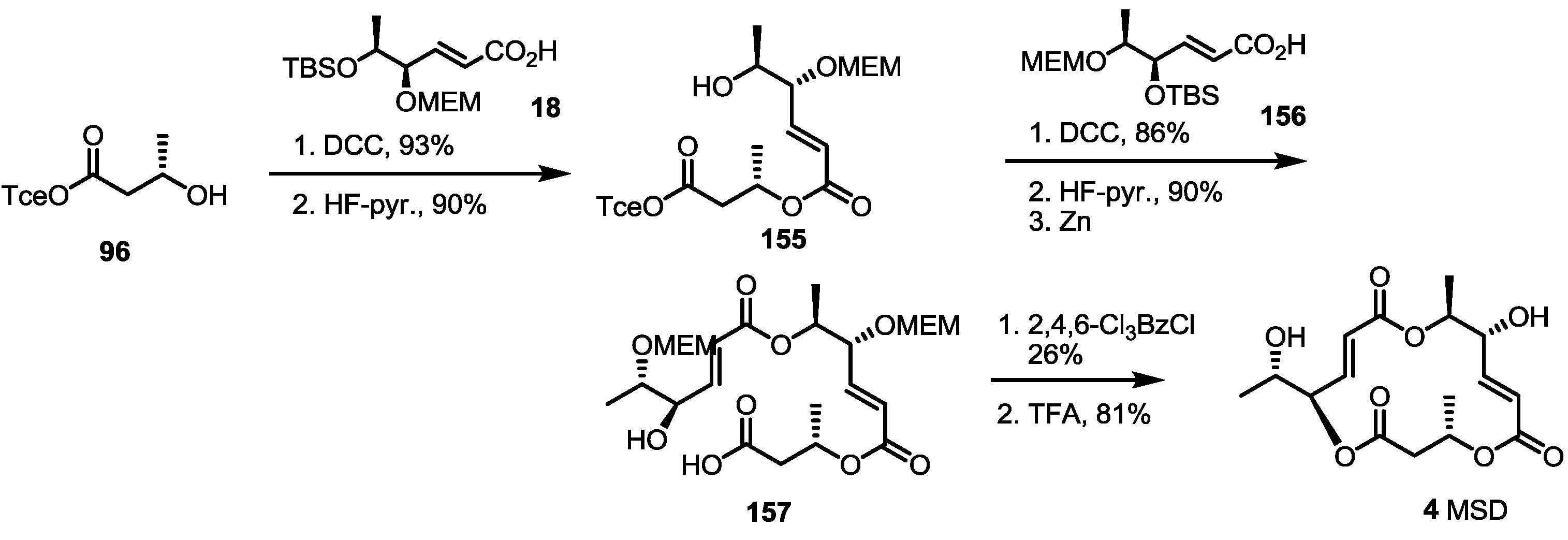
Recently, the Curran group in Pittsburgh also reported the first total synthesis and structural elucidation of MSD [50] (Scheme 20). Conventional esterification of the hydroxyl butyrate 96 with the known carboxylic acid 18 gave dimer 155 in an excellent yield after TBS deprotection. Another carboxylic acid 156 was also attached using dimer 155 under similar conditions, and the resulting product was deprotected with HF-pyridine/Zn sequentially. Finally, Yamaguchi lactonization and global deprotection produced MSD 4 in a moderate yield. It is noteworthy that the research was conducted after the fluorous mixture synthesis of MS. They synthesized all of the 16-stereoiosmer library of MS skeletons using their own fluorous tagging technique. As all of the synthetic isomers did not match with natural MSD, however, they hypothesized that MSD is a regioisomeric 15-membered lactone MS framework. After extensive NMR study, they were able to synthesize MSD by conventional methods and to revise the originally proposed structure.
Scheme 20.
First synthesis and confirmation of the structure of MSD.

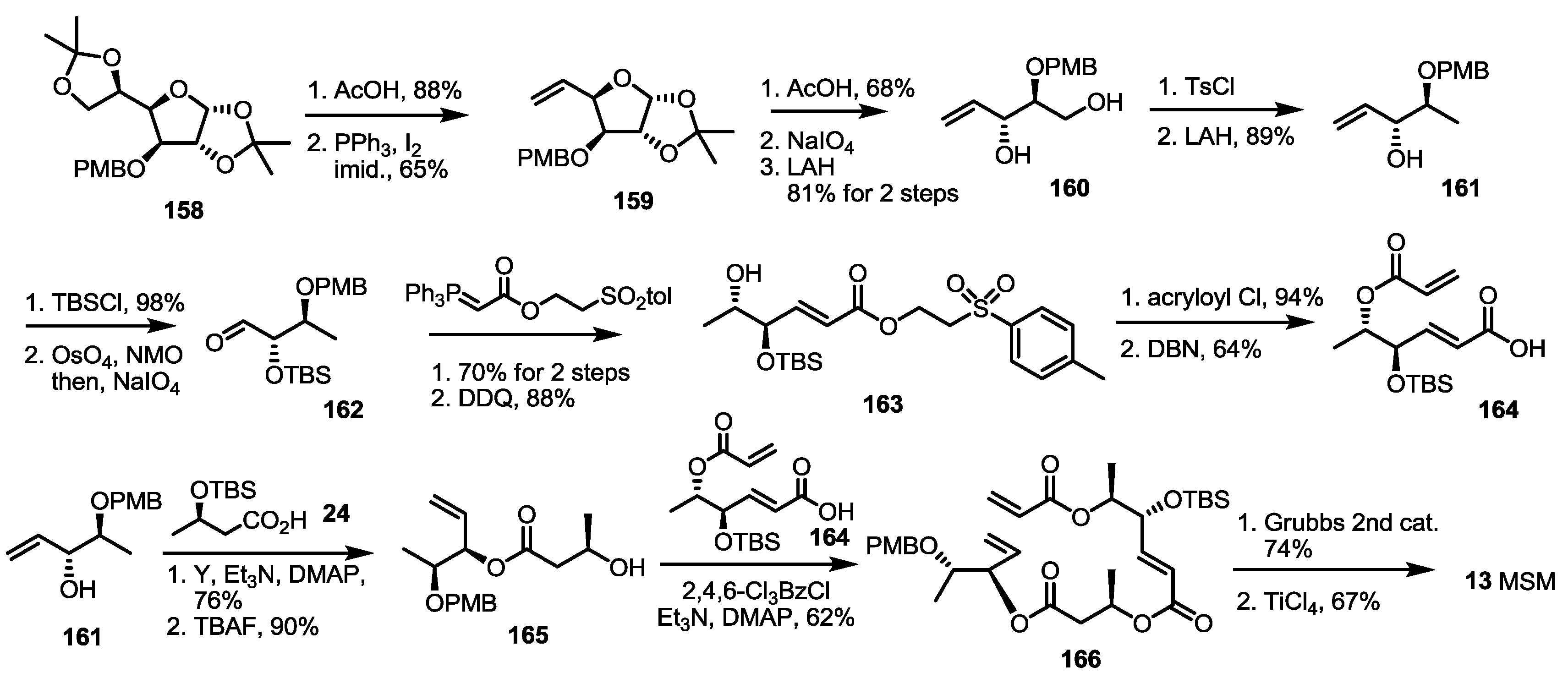
The last synthesis so far was reported by the Sharma group [51] (Scheme 21). They published a regioisomeric 15-membered lactone MSM from the sugar skeleton again. The known furanose 158 was transformed into terminal alkene 159 via hydrolysis of acetal and the elimination reaction [52]. As for the previous synthesis of MSA by the same group (Scheme 8), acidic hydrolysis/oxidative cleavage and the reduction sequence produced diol 160 in a good yield. Selective tosylation and LAH reduction was used to obtain the key building block, 161. This allylic alcohol 161 was protected and oxidized to aldehyde 162, which was converted into 163 via Wittig olefination/PMB deprotection sequence. Acryloylation of the secondary alcohol and basic elimination produced acryloyl acid 164 in a good yield. The allylic alcohol 161 was also used to prepare another secondary alcohol 165 via esterification with butyric acid 24 and TBS deprotection. The resulting alcohol 165 was coupled with the acryloyl acid 164 under Yamaguchi conditions to give pivotal terminal diene 166 in a moderate yield. Finally, RCM and acidic deprotection produced the 15-membered macrocycle MSM 13.
Scheme 21.
Total synthesis of MSM.

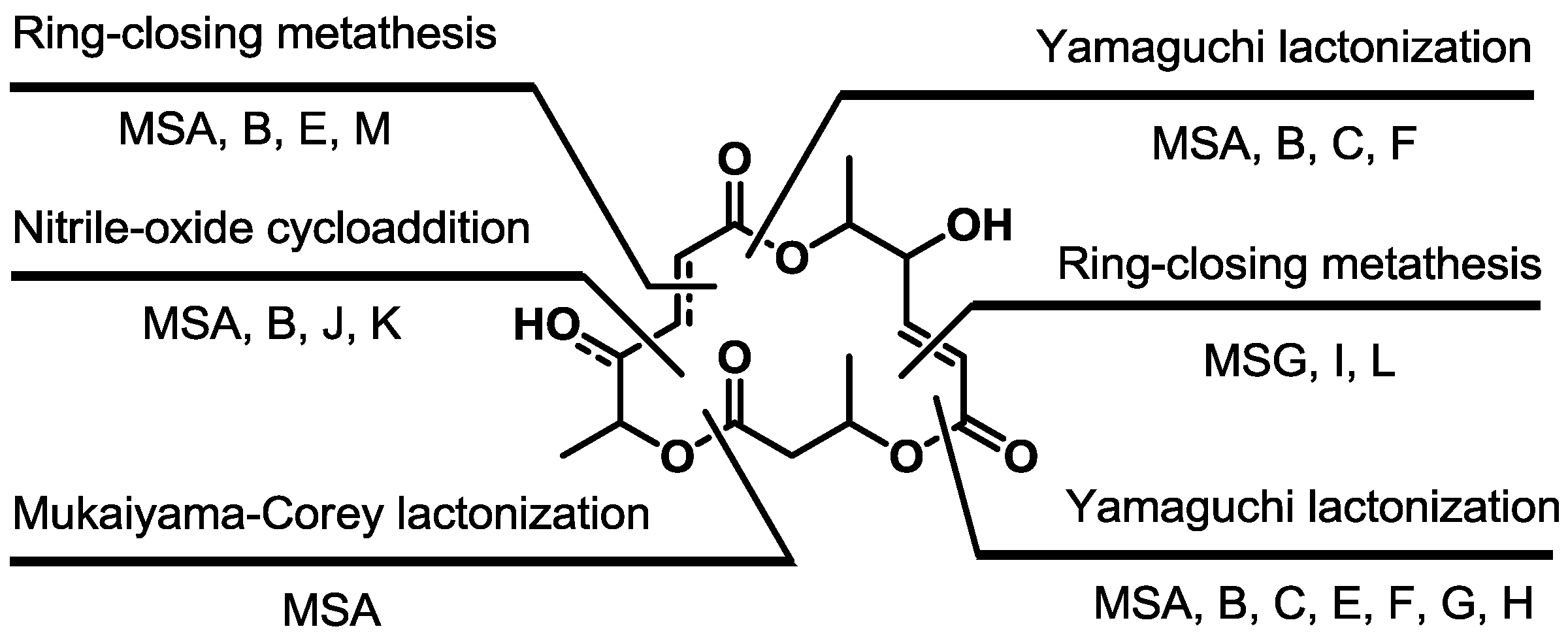
The macrocyclization strategy is summarized in Scheme 22. This shows that various approaches have been employed to construct a unique skeleton of the macrosphelide family.
Scheme 22.
Macrocyclization of the MS skeleton.

3. Conclusions
In conclusion, the total synthesis of MS was summarized. Efficient synthesis of monomeric fragments and macrolactonization was usually employed. For the preparation of monomers, unique synthetic methods, such as asymmetric dihydroxylation, furan oxidation, enzymatic chiral resolution, usage of sugar chirality, vinylogous orthoester addition, reduction of propargylic alcohol and Wittig olefination, were developed. For final macrolactonization, Yamaguchi-type esterification, RCM and nitrile-oxide cycloaddition have been developed so far. This synthetic study made it possible to prepare not only natural MS in a large scale, but also artificial MS with advanced pharmacological activities. Based on the accumulated results, more improved agents from MS are anticipated for the development of related molecules. The medicinal synthetic approach and more advanced synthetic research will be carried out in due course.
Acknowledgments
This work was supported by a grant from the Next-Generation BioGreen 21 Program (SSAC, No. PJ0090882014).
Conflicts of Interest
The author declares no conflict of interest.
References
- Hayashi, M.; Kim, Y.-P.; Hiraoka, H.; Natori, M.; Takamatsu, S.; Kawakubo, T.; Masuma, R.; Komiyama, K.; Omura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 1995, 48, 1435–1439. [Google Scholar] [CrossRef]
- Takamatsu, S.; Kim, Y.-P.; Hayashi, M.; Hiraoka, H.; Natori, M.; Komiyama, K.; Omura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule II. Physicochemical properties and structural elucidation. J. Antibiot. 1996, 49, 95–98. [Google Scholar]
- Takamatsu, S.; Hiraoka, H.; Kim, Y.-P.; Hayashi, M.; Natori, M.; Komiyama, K.; Omura, S. Macrosphelides C and D, novel inhibitors of cell adhesion. J. Antibiot. 1997, 50, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Numata, A.; Iritani, M.; Yamada, T.; Minoura, K.; Matsumura, E.; Yamori, T.; Tsuruo, T. Novel antitumour metabolites produced by a fungal strain from a sea hare. Tetrahedron Lett. 1997, 38, 8215–8218. [Google Scholar] [CrossRef]
- Fukami, A.; Taniguchi, Y.; Nakamura, T.; Rho, M.-C.; Kawaguchi, K.; Hayashi, M.; Komiyama, K.; Omura, S. New members of the macrosphelides from microsphaeropsis sp. FO-5050 IV. J. Antibiot. 1999, 52, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Iritani, M.; Doi, M.; Minoura, K.; Ito, T.; Numata, A. Absolute stereostructures of cell-adhesion inhibitors, macrosphelides C, E–G and I, produced by a Periconia species separated from an Aplysia sea hare. J. Chem. Soc. Perkin Trans. 1 2001, 3046–3053. [Google Scholar]
- Yamada, T.; Iritani, M.; Minoura, K.; Numata, A.; Kobayashi, Y.; Wang, Y.-G. Absolute stereostructures of cell adhesion inhibitors, macrosphelides H and L, from Periconia byssoides OUPS-N133. J. Antibiot. 2002, 55, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Sunazuka, T.; Hirose., T.; Harigaya, Y.; Takamatsu, S.; Hayashi, M.; Komiyama, K.; Omura, S.; Sprengeler, P.A.; Smith, A.B. Relative and absolute stereochemistries and total synthesis of (+)-macrosphelides A and B, potent, orally bioavailable inhibitors of cell-cell adhesion. J. Am. Chem. Soc. 1997, 119, 10247–10248. [Google Scholar] [CrossRef]
- Omura, S.; Komiyama, K. Immunosuppressant Macrosphelide B. WO 0147516 A1, 5 July 2001. [Google Scholar]
- Fukami, A.; Iijima, K.; Hayashi, M.; Komiyama, K.; Omura, S. Macrosphelide B suppressed metastasis through inhibition of adhesion of sLex/E-Selectin molecules. Biochem. Biophy. Res. Commun. 2002, 291, 1065–1070. [Google Scholar]
- Tomprefa, N.; McQuilken, M.; Hill, R.; Whipps, J. Antimicrobial activity of Coniothyrium minitans and itsmacrolide antibiotic macrosphelide A. J. Appl. Microbiol. 2009, 106, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Akita, H.; Nakamura, H.; Ono, M. Total synthesis of (+)-macrosphelides A, C, E, F, and G based on enzymatic function. Chirality 2003, 15, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Matsuya, Y.; Nemoto, H. Recent advances in macrosphelide synthesis. Heterocycles 2005, 65, 1741–1749. [Google Scholar]
- Ivanova, V.; Kolarova, M.; Aleksieva, K.; Graefe, U.; Schlegel, B. Diphenylether and macrotriolides occurring in a fungal isolate from the antarctic lichen Neuropogon. Prep. Biochem. Biotechnol. 2007, 37, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A rapid esterification by mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar]
- Boden, E.P.; Keck, G.E. Proton-transfer steps in Steglich esterification: A very practical new method for macrolactonization. J. Org. Chem. 1985, 50, 2394–2395. [Google Scholar] [CrossRef]
- Sunazuka, T.; Hirose, T.; Chikaraishi, N.; Harigaya, Y.; Hayashi, M.; Komiyama, K.; Sprengeler, P.A.; Smith, A.B., III; Ōmura, S. Absolute stereochemistries and total synthesis of (+)/(−)-macrosphelides, potent, orally bioavailable inhibitors of cell-cell adhesion. Tetrahedron 2005, 61, 3789–3803. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kumar, B.G.; Kurachi, T. Total synthesis of macrosphelides B and A. Tetrahedron Lett. 2000, 41, 1559–1563. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kumar, G.B.; Kurachi, T.; Acharya, H.P.; Yamazaki, T.; Kitazume, T. Furan ring oxidation strategy for the synthesis of macrosphelides A and B. J. Org. Chem. 2001, 66, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Bal, B.S.; Childers, W.E., Jr.; Pinnick, H.W. Oxidation of α, β-unsaturated aldehydes. Tetrahedron 1981, 37, 2091–2096. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Acharya, H.P. First total synthesis of macrosphelides C and F. Tetrahedron Lett. 2001, 42, 2817–2820. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Matsuumi, M. First total synthesis of grahamimycin A. J. Org. Chem. 2000, 65, 7221–7224. [Google Scholar] [CrossRef] [PubMed]
- Mitsunobu, O.; Yamada, M. Preparation of esters of carboxylic and phosphoric acid via quaternary phosphonium salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Wang, Y.-G. Synthesis of macrosphelides H and G. Tetrahedron Lett. 2002, 43, 4381–4384. [Google Scholar] [CrossRef]
- Hareau, G.P.; Koiwa, M.; Hikichi, S.; Sato, F. Synthesis of optically active 5-(tert-butyldimethylsiloxy)-2-cyclohexenone and its 6-substituted derivatives as useful chiral building blocks for the synthesis of cyclohexane rings. Syntheses of carvone, penienone, and penihydrone. J. Am. Chem. Soc. 1999, 121, 3640–3650. [Google Scholar] [CrossRef]
- Ono, M.; Nakamura, H.; Konno, F.; Akita, H. Total syntheses of macrosphelides (+)-A, (−)-A and (+)-E. Tetrahedron: Asymmetry 2000, 11, 2753–2764. [Google Scholar] [CrossRef]
- Ono, M.; Nakamura, H.; Arakawa, S.; Honda, N.; Akita, H. Formal total synthesis of (+)-macrosphelide A based on regioselective hydrolysis using lipase. Chem. Pharm. Bull. 2002, 50, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Ono, M.; Shida, Y.; Akita, H. New total syntheses of (+)-macrosphelides C, F and G. Tetrahedron: Asymmetry 2002, 13, 705–713. [Google Scholar] [CrossRef]
- Nakamura, H.; Ono, M.; Makino, M.; Akita, H. Formal total synthesis of macrosphelide (+)-A, effect on macrolactonization depended upon the lactone formation position. Heterocycles 2002, 57, 327–336. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Mouli, C.C. The total synthesis of macrosphelides A and E from carbohydrate precursors. Tetrahedron Lett. 2002, 43, 9159–9161. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Mouli, C.C. A total synthesis of macrosphelides C and F from l-(+)-arabinose. Tetrahedron Lett. 2003, 44, 8161–8163. [Google Scholar] [CrossRef]
- Chakraborty, T.K.; Purkait, S.; Das, S. Synthesis of chiral 4-hydroxy-2,3-unsaturated carbonyl compounds from 3,4-epoxy alcohols by oxidation: application in the formal synthesis of macrosphelide A. Tetrahedron 2003, 59, 9127–9135. [Google Scholar] [CrossRef]
- Matsuya, Y.; Kawaguchi, T.; Nemoto, H. New strategy for the total synthesis of macrosphelides A and B based on ring-closing metathesis. Org. Lett. 2003, 5, 2939–2941. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Funamori, N.; Matsuya, Y.; Nemoto, H. Total synthesis of macrosphelides A, B, and E: First application of ring-closing metathesis for macrosphelide synthesis. J. Org. Chem. 2004, 69, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Kusaka, S.-I.; Dohi, S.; Doi, T.; Takahashi, T. Total synthesis of macrosphelide A by way of palladium-catalyzed carbonylative esterification. Tetrahedron Lett. 2003, 44, 8857–8859. [Google Scholar] [CrossRef]
- Takahashi, T.; Kusaka, S.-I.; Doi, T.; Takayuki, T.; Sunazuka, T.; Omura, S. A combinatorial synthesis of a macrosphelide library utilizing a palladium-catalyzed carbonylation on a polymer support. Angew. Chem. Int. Ed. Engl. 2003, 42, 5230–5234. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.-M.; Seo, S.-Y.; Kim, S.-H.; Jung, J.-W.; Lee, Y.-S.; Jung, J.-K.; Suh, Y.-G. Concise syntheses of (+)-macrosphelides A and B. Org. Lett. 2005, 7, 3159–3162. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Rajapakse, H.A.; Chiu, A.; Stenkamp, D. Asymmetric syntheses of pectenotoxins-4 and -8, Part II: Synthesis of the C20-C30 and C31-C40 subunits and fragment assembly. Angew. Chem. Int. Ed. Engl. 2002, 114, 4755–4758. [Google Scholar] [CrossRef]
- Suh, Y.-G.; Jung, J.-K.; Seo, S.-Y.; Min, K.-H.; Shin, D.-Y.; Lee, Y.-S.; Kim, S.-H.; Park, H.-J. Total synthesis of (+)-brefeldin A. J. Org. Chem. 2002, 67, 4127–4137. [Google Scholar] [CrossRef]
- Maezaki, N.; Hirose, Y.; Tanaka, T. Stereoselective synthesis of 1,4-bifunctional compounds by regioselective Pd-catalyzed allylic substitution reaction. Org. Lett. 2004, 6, 2177–2180. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.-M.; Yun, H.; Kim, N.-J.; Jung, J.-W.; Chang, D.-J.; Lee, S.; Yoo, J.; Park, H.-J.; Suh, Y.-G. Concise syntheses of (+)-macrosphelides A and B: Studies on the macro-ring closure strategy. J. Org. Chem. 2009, 74, 554–561. [Google Scholar] [CrossRef]
- Paek, S.-M.; Suh, Y.-G. Synthetic studies on bioactive natural polyketides: Intramolecular nitrile oxide-olefin cycloaddition approach for construction of a macrolactone skeleton of macrosphelide B. Molecules 2011, 16, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.; Paek, S.-M.; Jung, J.-W.; Kim, N.-J.; Kim, S.-H.; Suh, Y.-G. First total syntheses of (−)-macrosphelides J and K and elucidation of their absolute configuration. Chem. Commun. 2009, 2463–2465. [Google Scholar]
- Rao, K.S.; Mukkanti, K.; Reddy, D.S.; Pal, M.; Iqbal, J. A simple procedure for the synthesis of γ-hydroxy-α, β-(E)-alkenoic esters: Formal synthesis of (+)-macrosphelides A and B. Tetrahedron Lett. 2005, 46, 2287–2290. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Babu, K.V. RCM mediated synthesis of macrosphelides I and G. Tetrahedron: Asymmetry 2007, 18, 2175–2184. [Google Scholar] [CrossRef]
- Hanessian, S.; Ugolini, A.; Dubé, D.; Glamyan, A. Facile access to (S)-1, 2, 4-butanetriol and its derivatives. Can. J. Chem. 1984, 62, 2146–2147. [Google Scholar] [CrossRef]
- Matsuya, Y.; Kobayashi, Y.; Kawaguchi, T.; Hori, A.; Watanabe, Y.; Ishihara, K.; Ahmed, K.; Wei, Z.-L.; Yu, D.-Y.; Zhao, Q.-L.; et al. Design, synthesis, and biological evaluation of artificial macrosphelides in the search for new apoptosis-inducing agents. Chem.-Eur. J. 2009, 15, 5799–5819. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.R.; Gutala, P. Enantioselective total synthesis of macrosphelides A and E. Tetrahedron 2011, 67, 4514–4520. [Google Scholar] [CrossRef]
- Colin-Messager, S.; Girard, J.-P.; Rossi, J.-C. Convenient method for the preparation of trityl ethers from secondary alcohols. Tetrahedron Lett. 1992, 33, 2689–2692. [Google Scholar] [CrossRef]
- Curran, D.P.; Sinha, M.K.; Zhang, K.; Sabatini, J.J.; Cho, D.-H. Binary fluorous tagging enables the synthesis and separation of a 16-stereoisomer library of macrosphelides. Nat. Chem. 2012, 4, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.V.M.; Reddy, P.S. Total synthesis of macrosphelide M from diacetone glucose. Eur. J. Org. Chem. 2012, 2012, 2414–2421. [Google Scholar] [CrossRef]
- Garegg, P.J.; Samuelsson, B. Conversion of vicinal diols into olefins using triphenylphosphine and triiodoimidazole. Synthesis 1979, 1979, 813–814. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Paek, S.-M. Synthetic Advances in Macrosphelides: Natural Anticancer Agents. Molecules 2014, 19, 15982-16000. https://doi.org/10.3390/molecules191015982
AMA Style
Paek S-M. Synthetic Advances in Macrosphelides: Natural Anticancer Agents. Molecules. 2014; 19(10):15982-16000. https://doi.org/10.3390/molecules191015982
Chicago/Turabian StylePaek, Seung-Mann. 2014. "Synthetic Advances in Macrosphelides: Natural Anticancer Agents" Molecules 19, no. 10: 15982-16000. https://doi.org/10.3390/molecules191015982