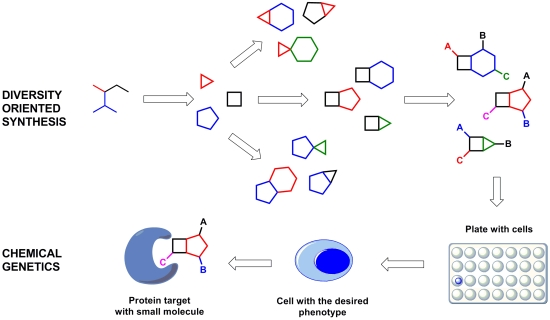
Diversity-Oriented Synthesis as a Tool for Chemical Genetics



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

1.1. Plasmodium Falciparum: An Example of the Application of Both Forward and Reverse Chemical Genetics Studies

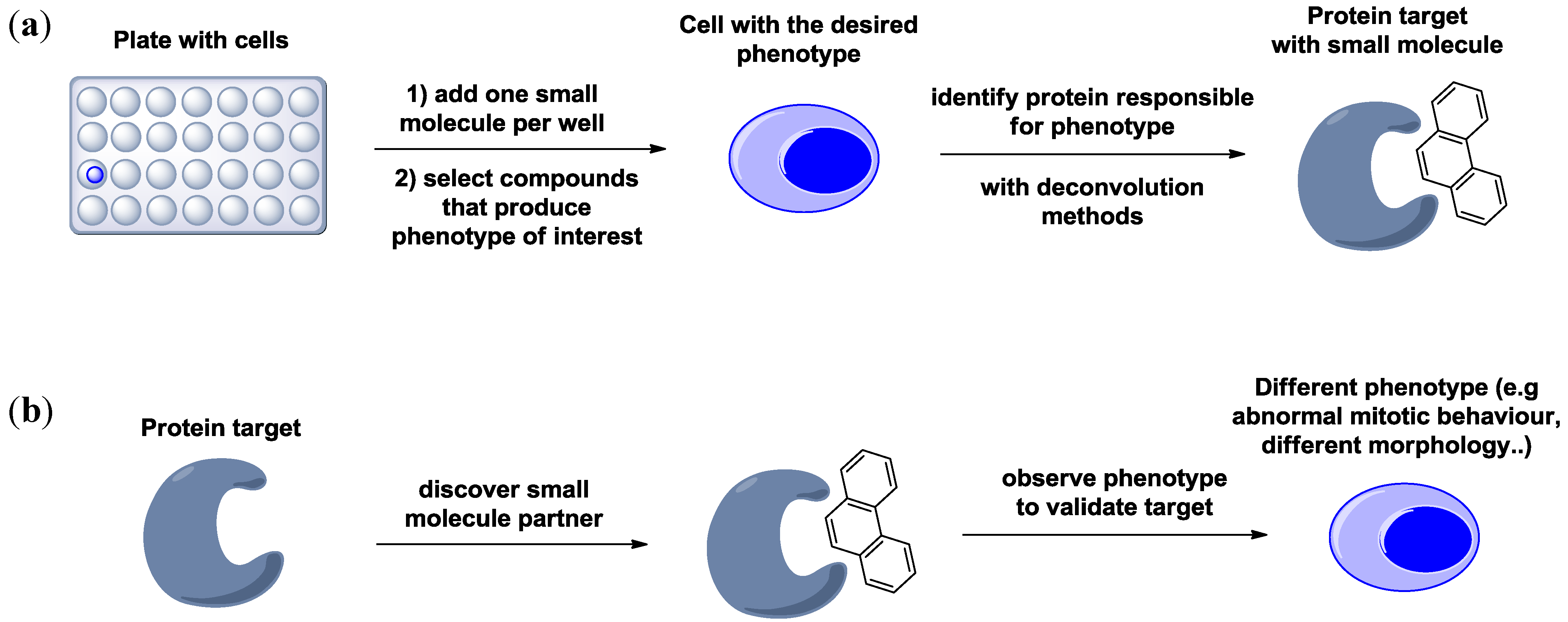
1.2. Generating Libraries for Chemical Genetics Studies. The Importance of Diversity-Oriented Synthesis

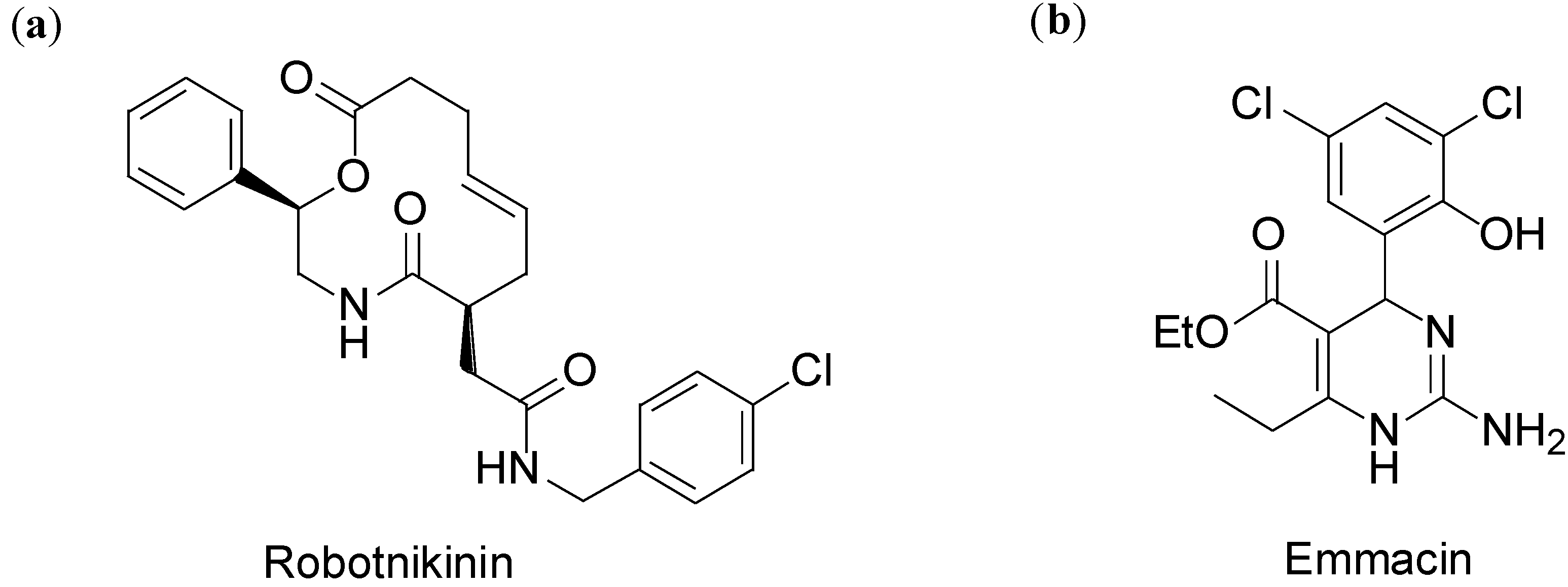
2. Case Study 1: Pioneering Work of Stuart L. Schreiber


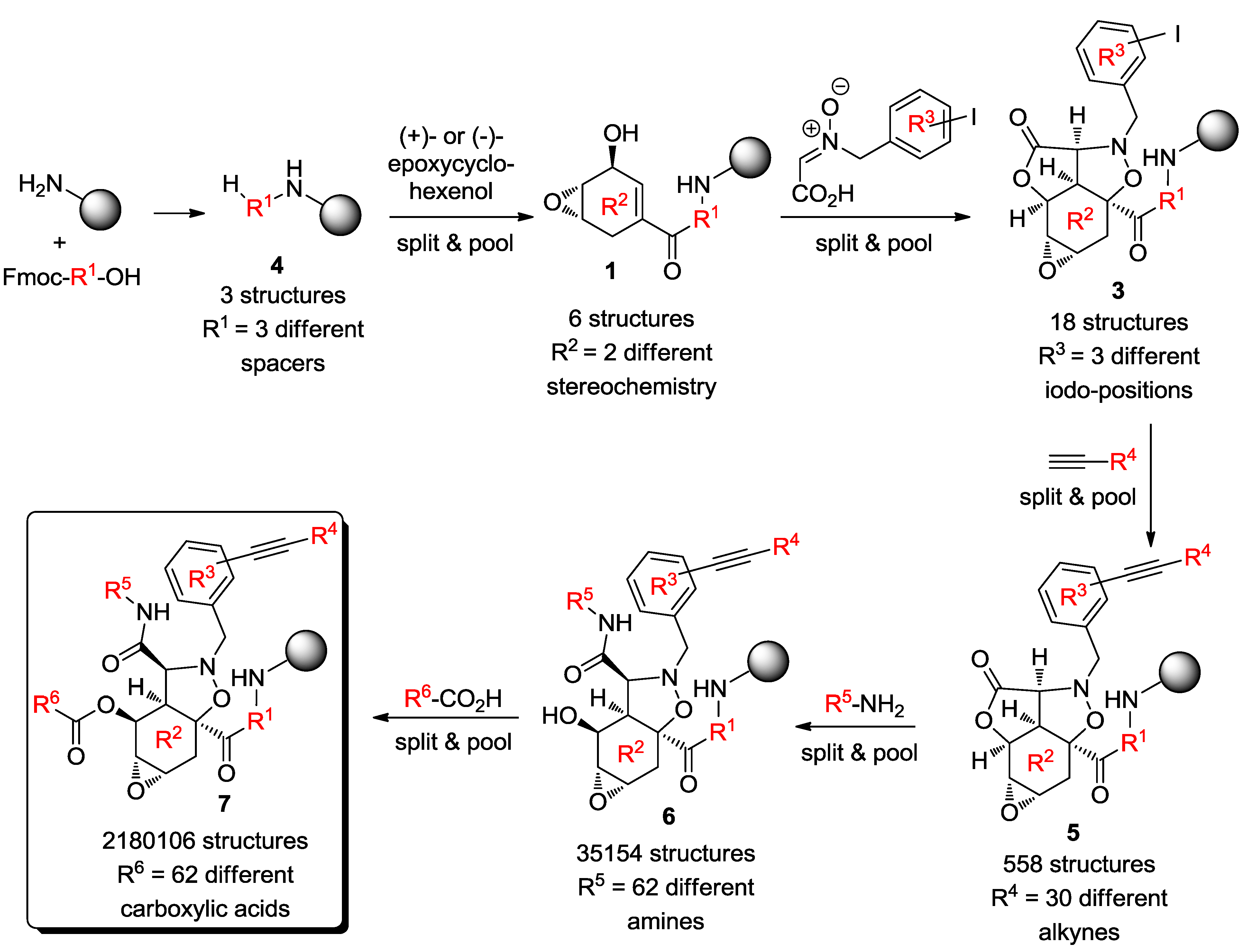

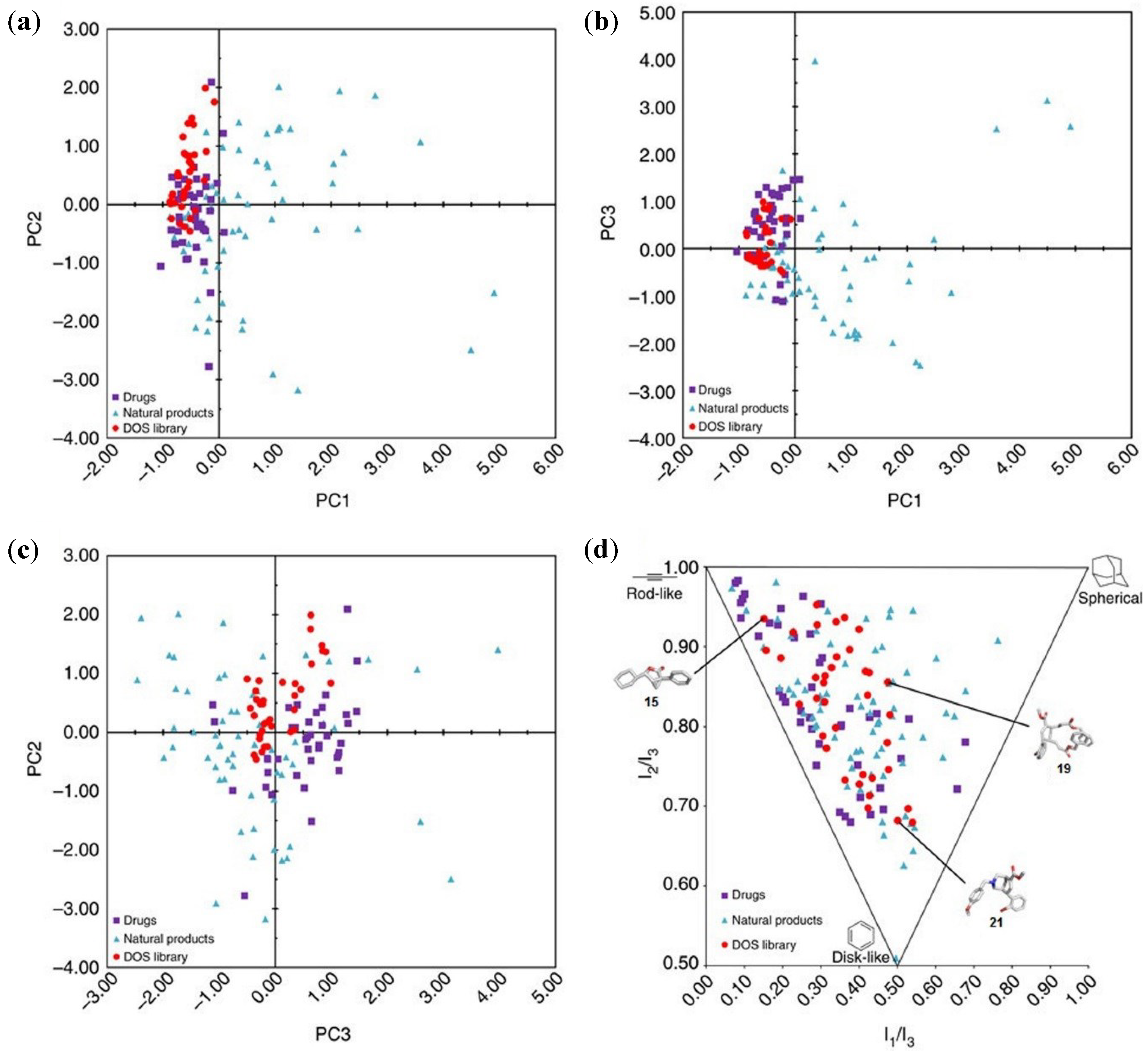
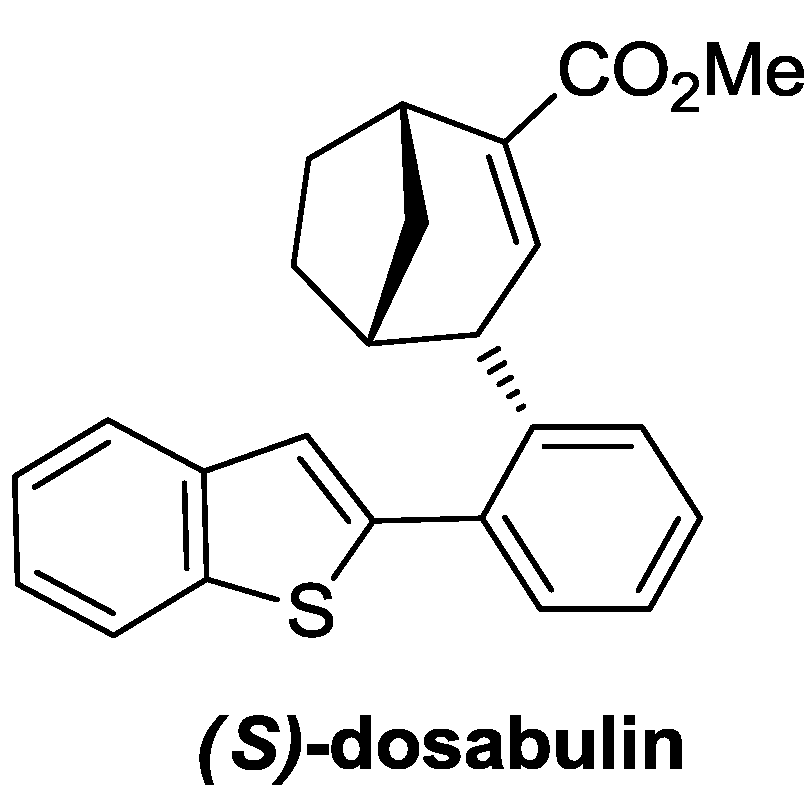
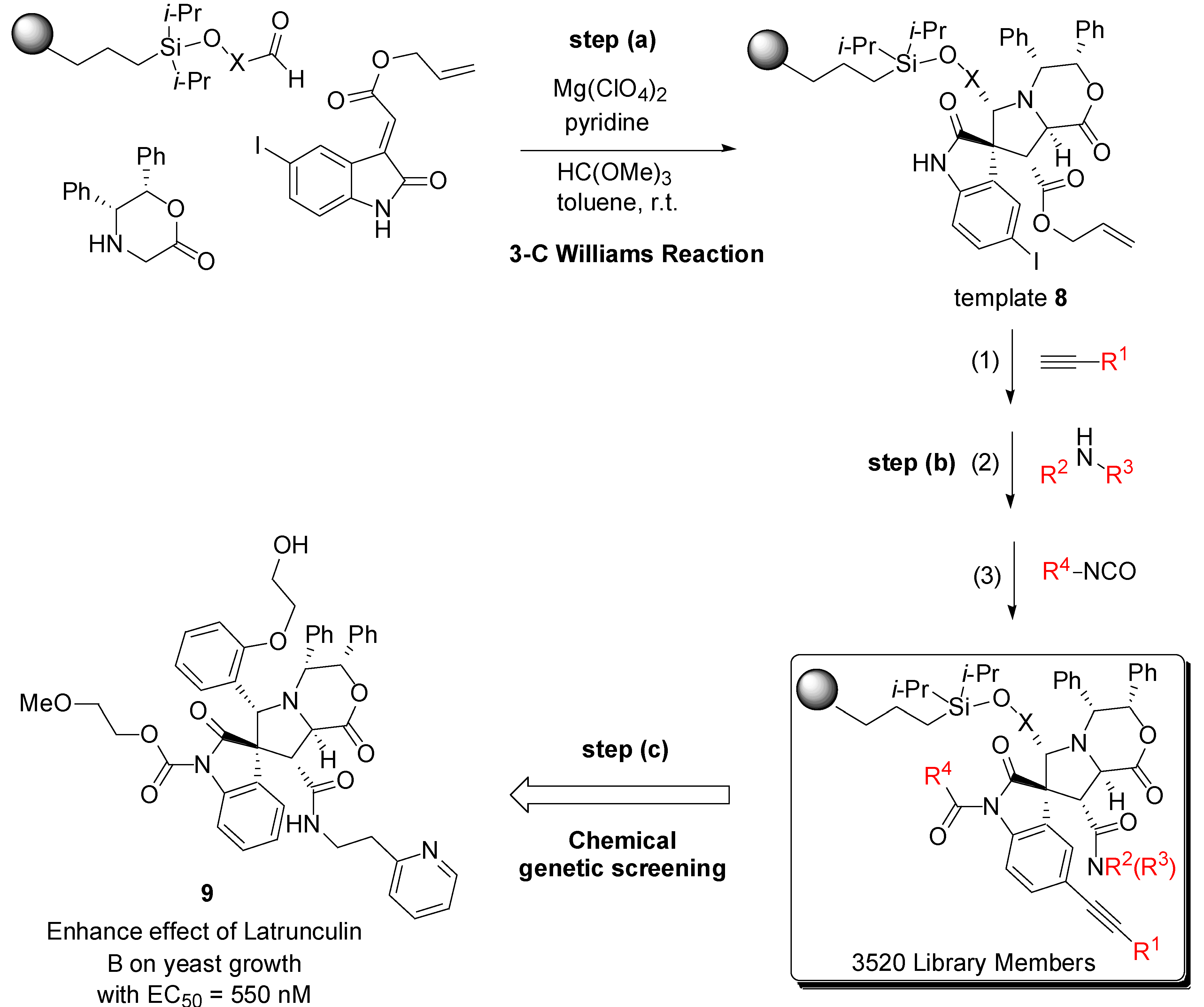
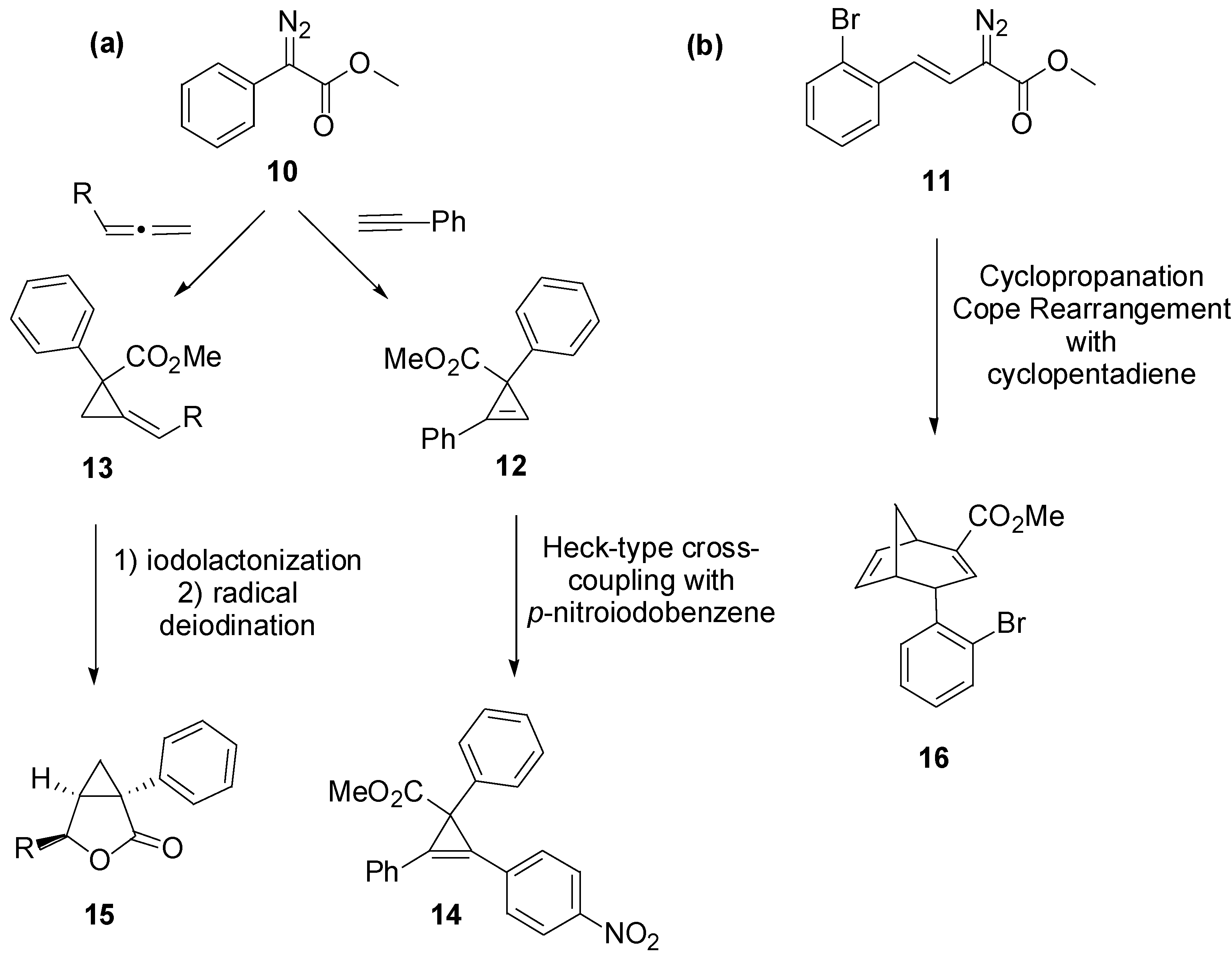
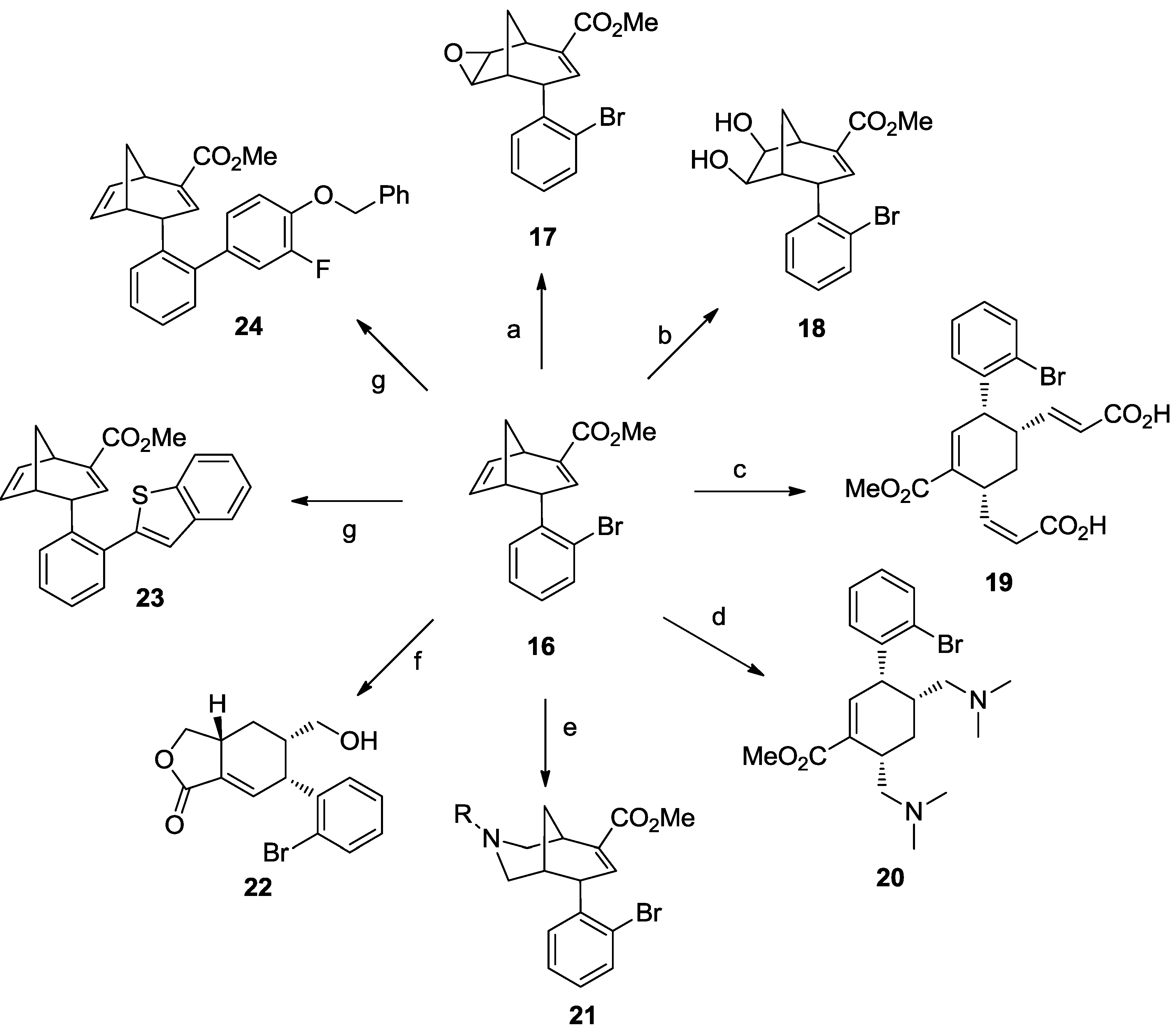
3. Case Study 2: Contribution of David R. Spring to the Discovery of Antimitotic Agents through Chemical Genetics Screening of DOS Libraries




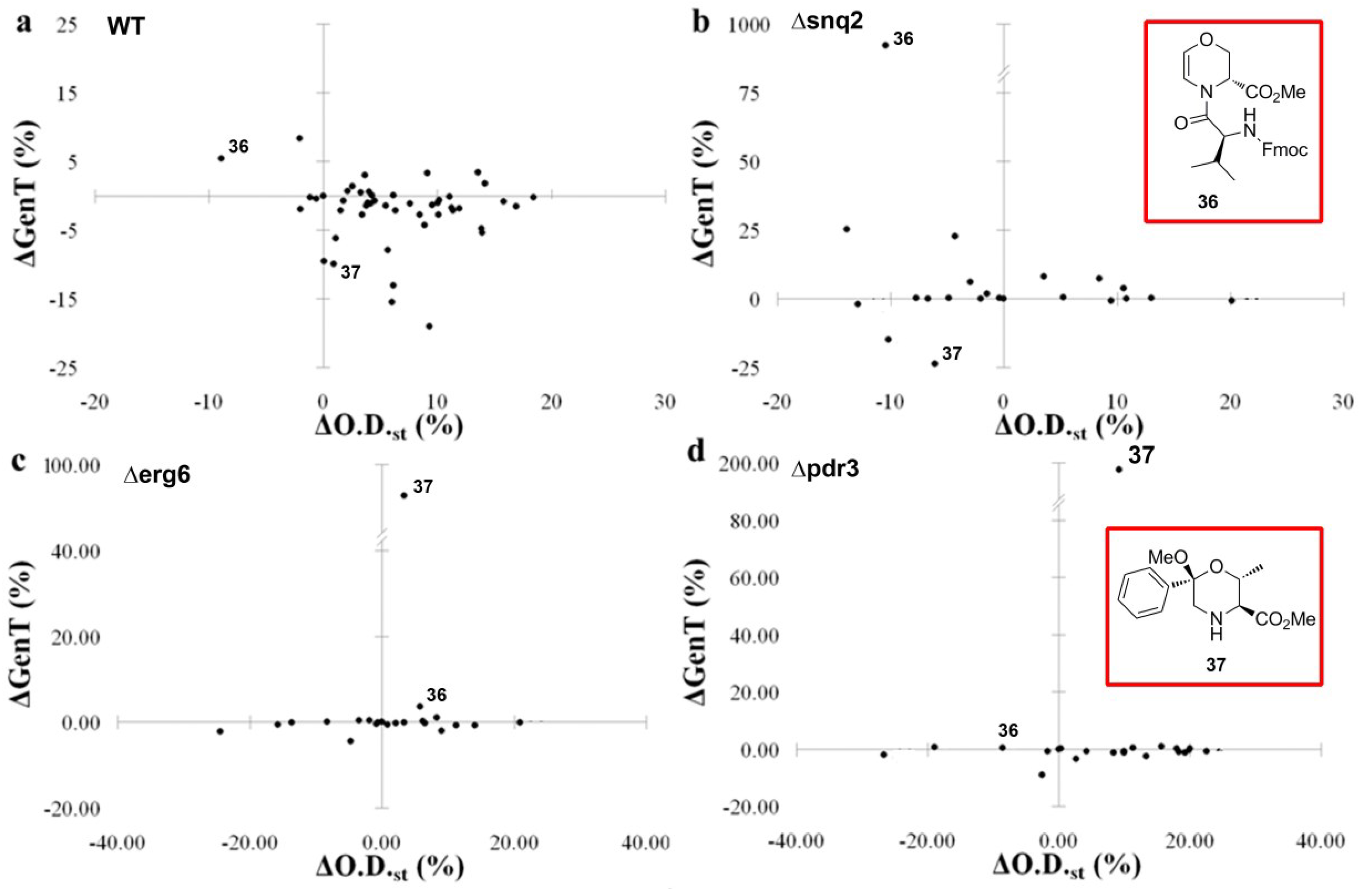
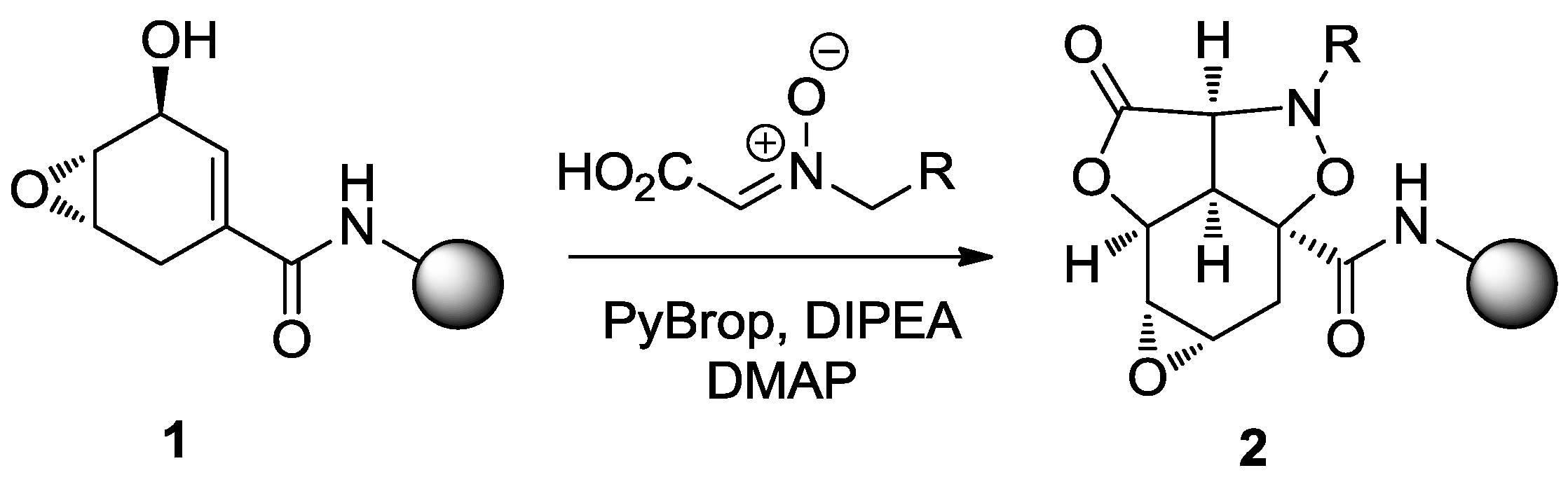
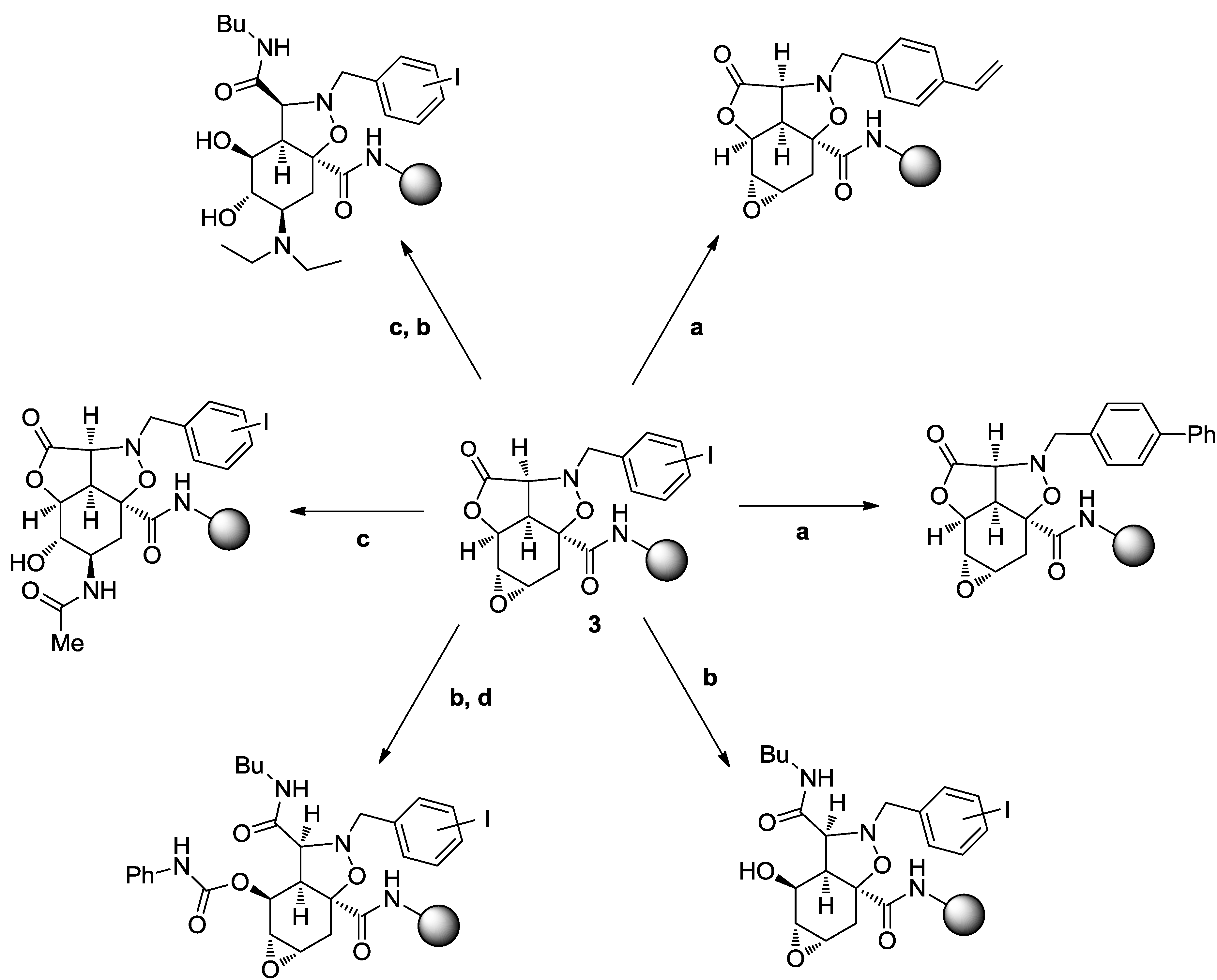
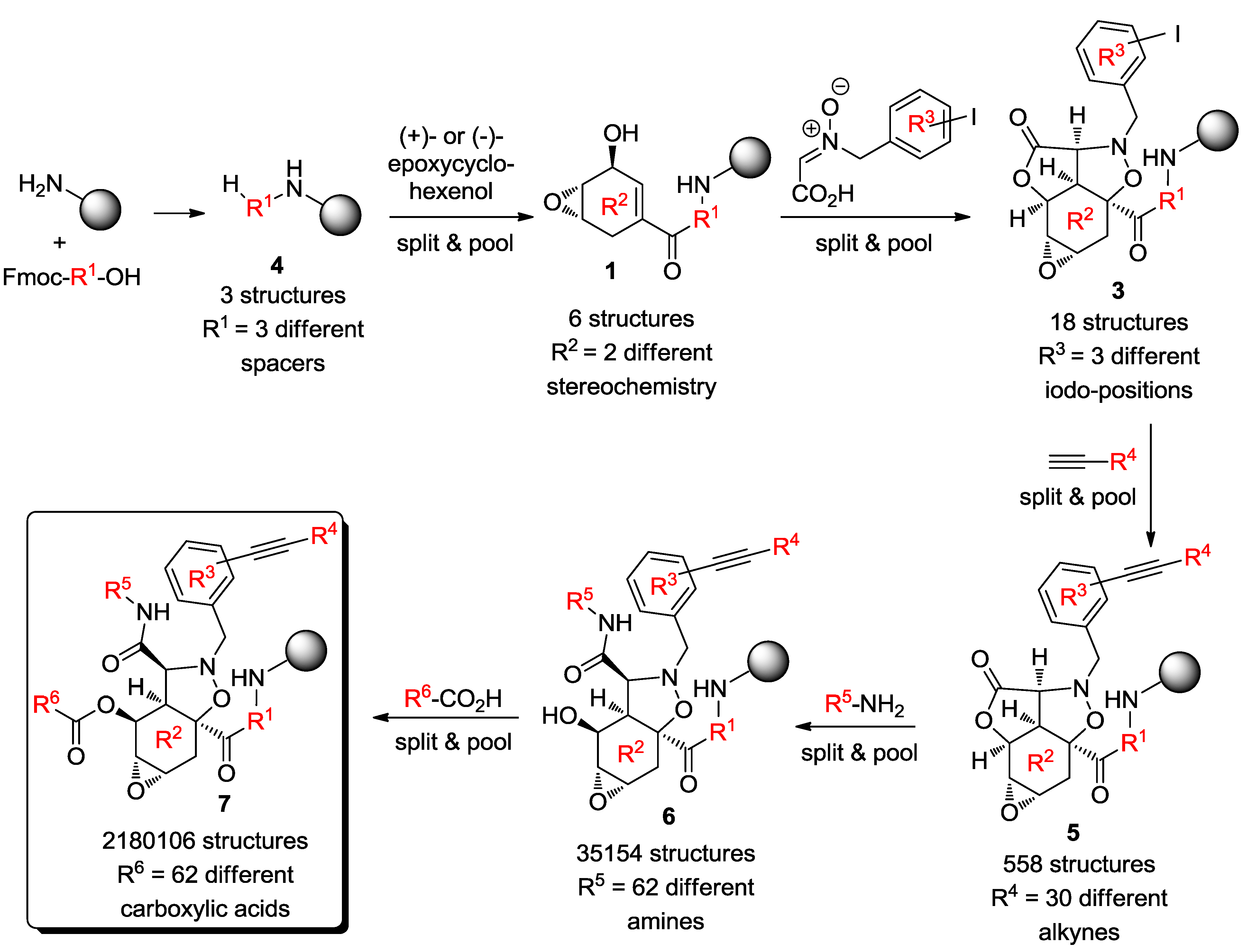
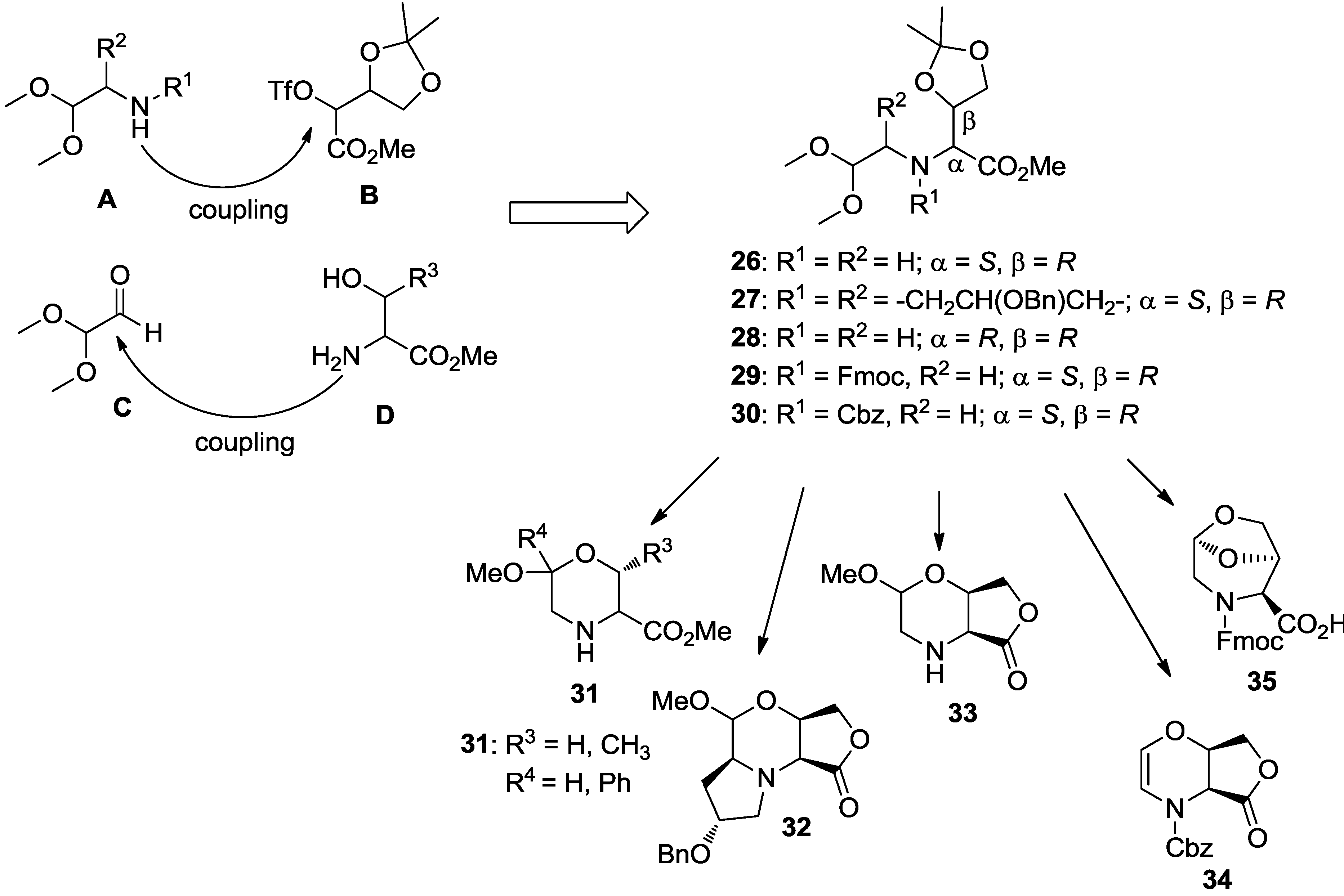
4. Case Study 3: Our Contribution to the Chemical Genetics Screening of Peptidomimetic DOS Libraries towards Yeast Deletant Strains
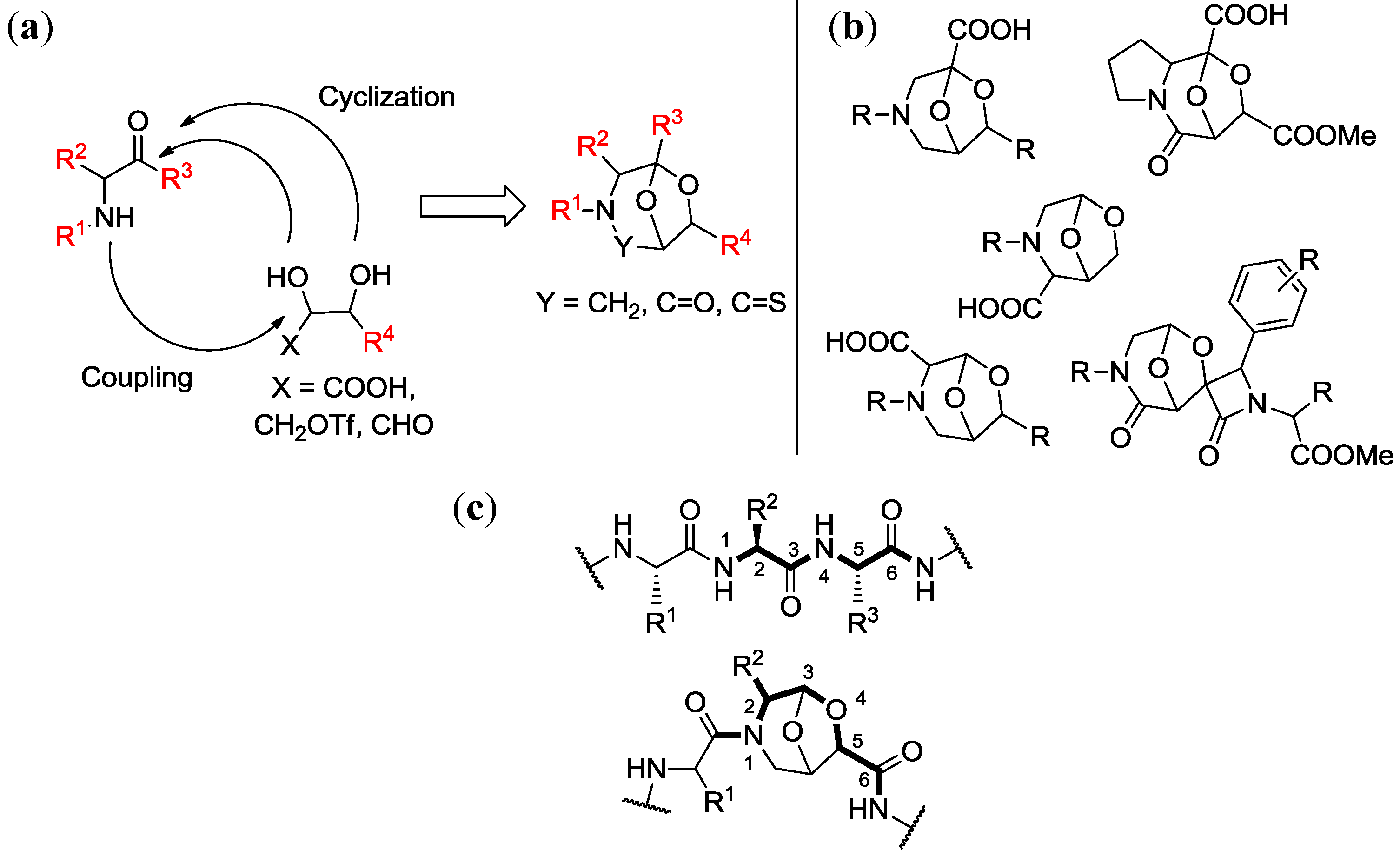
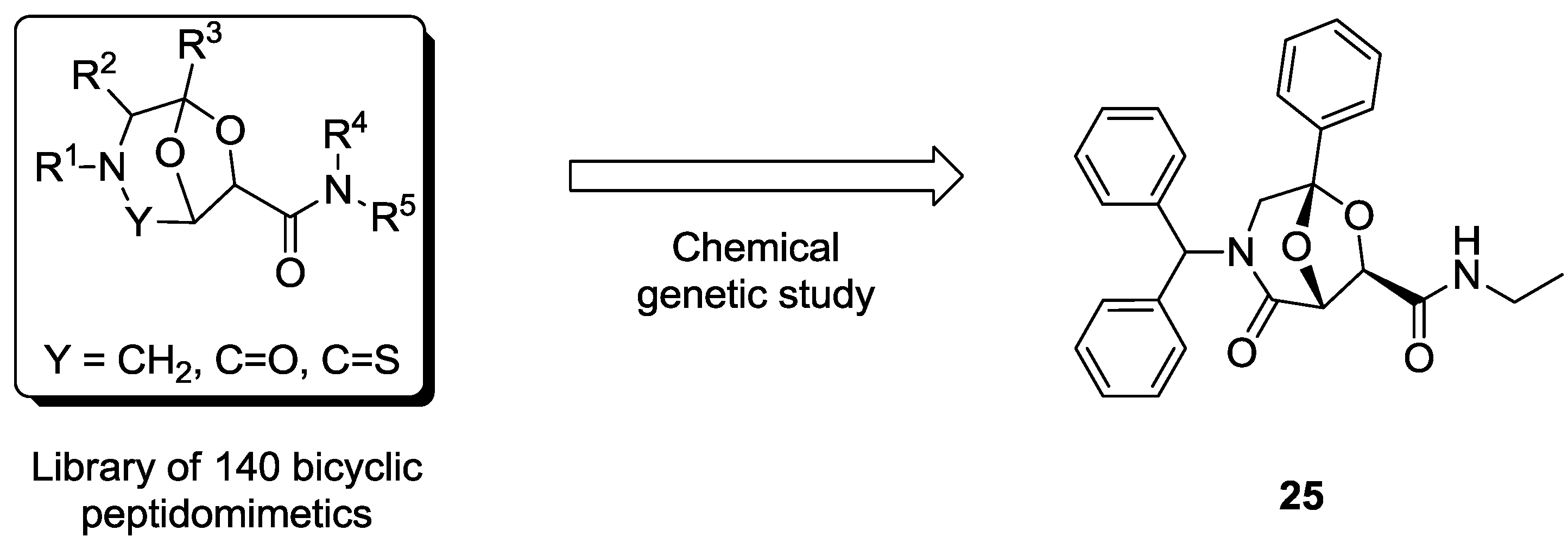
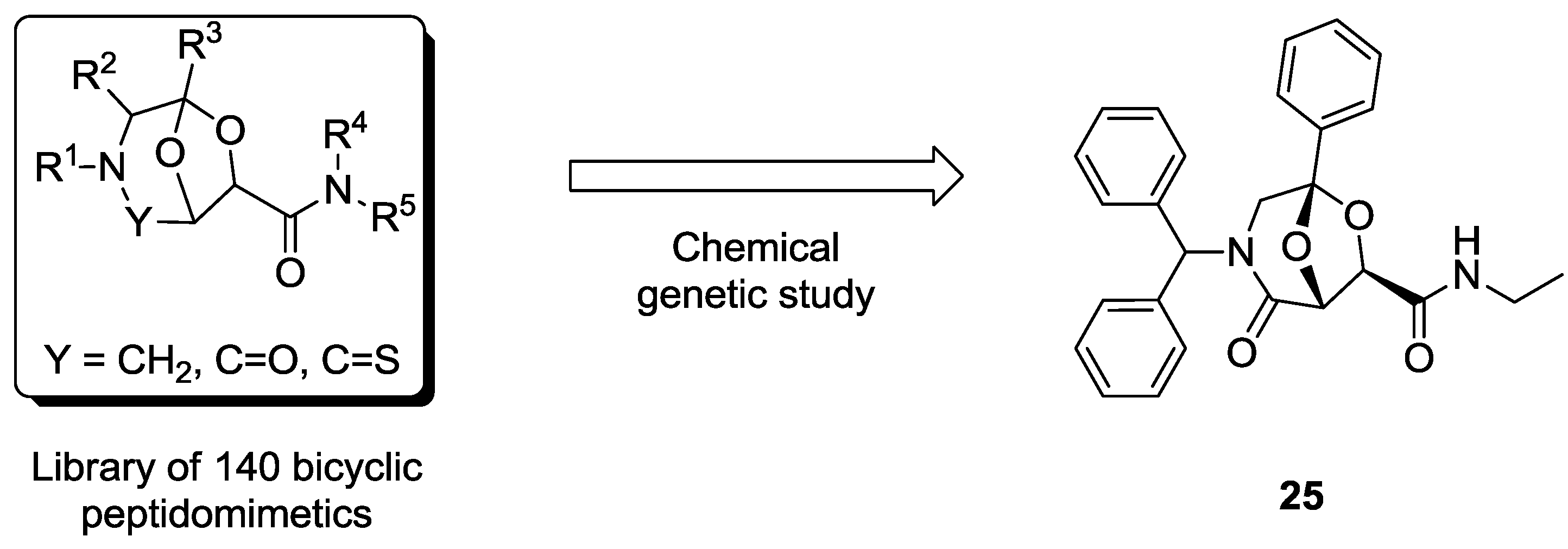
4.1. Chemical Genetics of a Bicyclic Peptidomimetic DOS Library
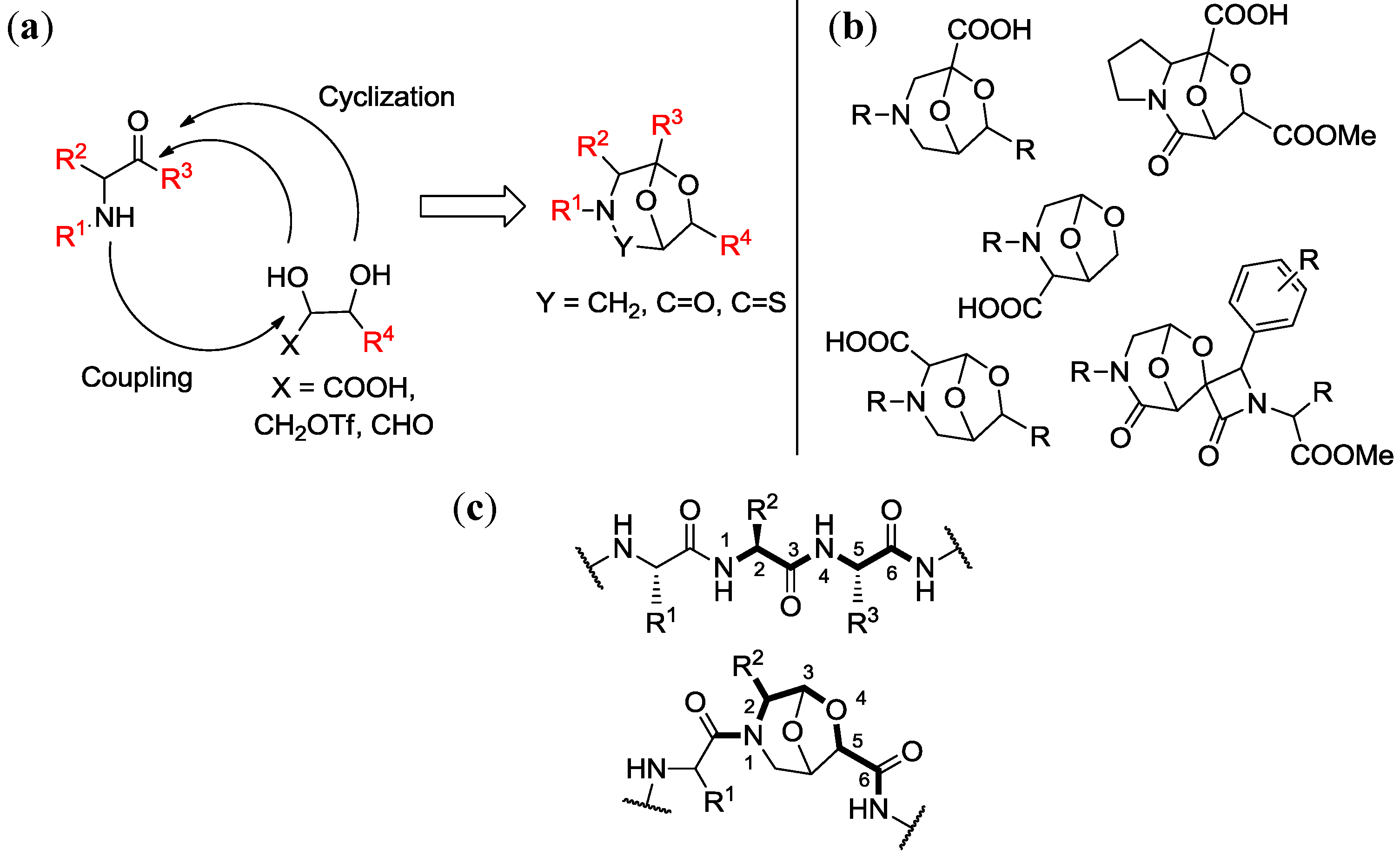
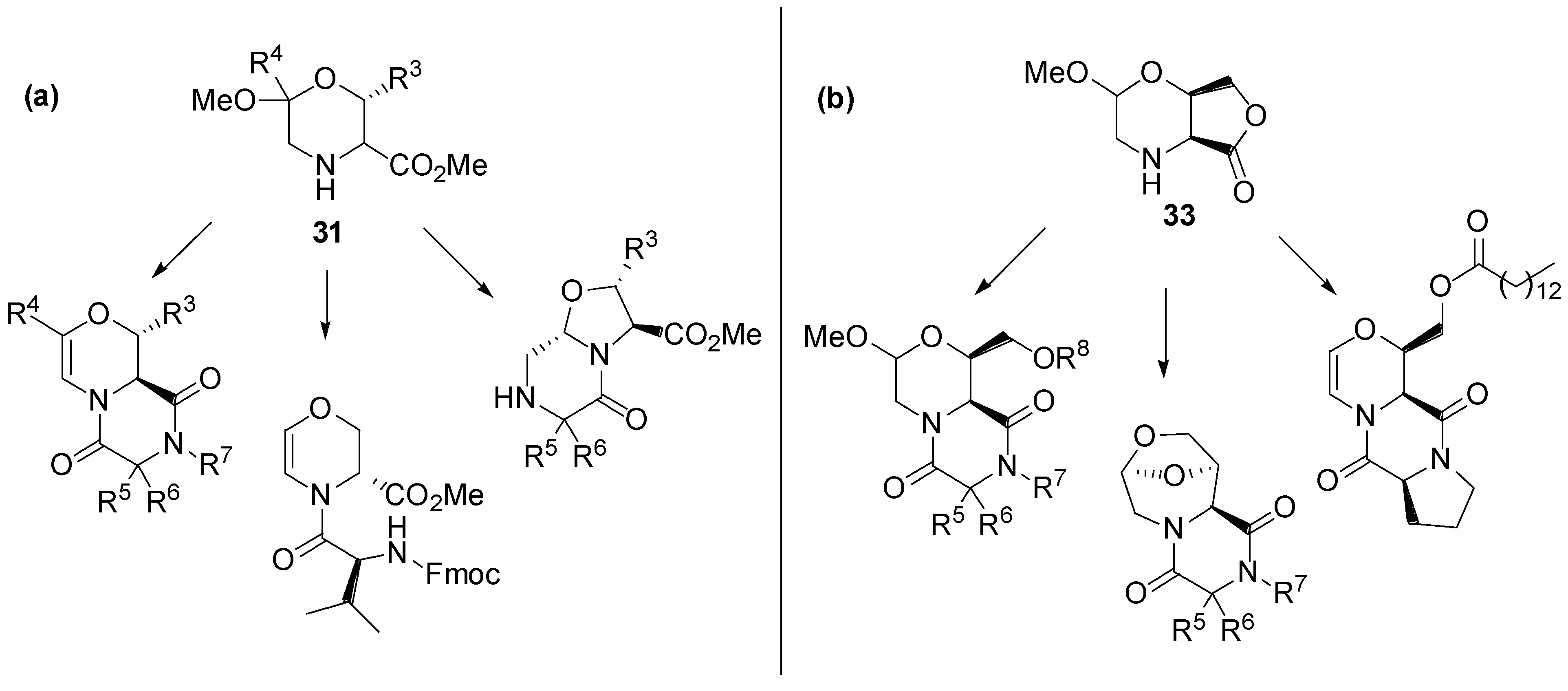
4.2. Chemical Genetics of Morpholine-Based DOS Library



5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 6, 203–214. [Google Scholar]
- Ryan, D.P.; Matthews, J.M. Protein-protein interactions in human disease. Curr. Opin. Struct. Biol. 2005, 15, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.H.; Buchner, J.; Kessler, H.; Diederich, F.; Krautler, B.; Lippard, S.; Liskamp, R.; Muller, K.; Nolan, E.M.; Samorì, B.; et al. The state of the art of chemical biology. ChemBioChem 2009, 10, 16–29. [Google Scholar] [CrossRef]
- Stockwell, B.R. Chemical genetics: Ligand-based discovery of gene function. Nat. Rev. Genet. 2000, 1, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Exploring biology with small organic molecules. Nature 2004, 432, 847–870. [Google Scholar] [CrossRef]
- Walsh, D.P.; Chang, Y.-T. Chemical genetics. Chem. Rev. 2006, 106, 2476–2530. [Google Scholar] [CrossRef]
- O’Connor, C.J.; Laraia, L.; Spring, D.R. Chemical genetics. Chem. Soc. Rev. 2011, 40, 4332–4345. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L. The small-molecule approach to biology. Chem. Eng. News 2003, 81, 51–61. [Google Scholar]
- Spring, D.R. Chemical genetics to chemical genomics: Small molecules offer big insights. Chem. Soc. Rev. 2005, 34, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, S.; O’Donnell, R.A.; Koussis, K.; Dluzewski, A.R.; Ansel, K.H.; Osborne, S.A.; Hackett, F.; Withers-Martinez, C.; Mitchell, G.H.; Bannister, L.H.; et al. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell 2007, 131, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Arastu-Kapur, S.; Ponder, E.L.; Fonović, U.P.; Yeoh, S; Yuan, F.; Fonović, M.; Grainger, M.; Phillips, C.; Powers, J.C.; Bogyo, M. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat. Chem. Biol. 2008, 4, 203–213. [Google Scholar] [CrossRef]
- Gamo, F.J.; Sanz, L.M.; Vidal, J.; de Cozar, C.; Alvarez, E.; Lavandera, J.L.; Vanderwall, D.E.; Green, D.V.; Kumar, V.; Hasan, S.; et al. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Rottmann, M.; McNamara, C.; Yeung, B.K.; Lee, M.C.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D.M.; Dharia, N.V.; Tan, J.; et al. Spiroindolones, a potent compound class for the treatment of malaria. Science 2010, 329, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Guiguemde, W.A.; Shelat, A.A.; Bouck, D.; Duffy, S.; Crowther, G.J.; Davis, P.H.; Smithson, D.C.; Connelly, M.; Clark, J.; Zhu, F.; et al. Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Gujjar, R.; Marwaha, A.; el Mazouni, F.; White, J.; White, K.L.; Creason, S.; Shackleford, D.M.; Baldwin, J.; Charman, W.N.; Buckner, F.S.; et al. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J. Med. Chem. 2009, 52, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Sijwali, P.S.; Rosenthal, P.J. Gene disruption confirms a critical role for the cysteine protease falcipain-2 in hemoglobin hydrolysis by Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2004, 101, 4384–4389. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, J.M.; Coppens, I.; Tripathi, A.K.; Scholl, P.F.; Shuman, J.; Bajad, S.; Shulaev, V.; Sullivan, D.J. The role of neutral lipid nanospheres in Plasmodium falciparum haem crystallization. Biochem J. 2007, 402, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Weissbuch, I.; Leiserowitz, L. Interplay between malaria, crystalline hemozoin formation, and antimalarial drug action and design. Chem. Rev. 2008, 108, 4899–4914. [Google Scholar] [CrossRef] [PubMed]
- Kugawa, F.; Watanabe, M.; Tamanoi, F. Chemical biology/chemical genetics/chemical genomics: Importance of chemical libraries. Chem-Bio Inform. J. 2007, 3, 49–68. [Google Scholar] [CrossRef]
- Bunin, B.A.; Ellman, J.A. A general and expedient method for the solid-phase synthesis of 1,4-benzodiazepine derivatives. J. Am. Chem. Soc. 1992, 114, 10997–10998. [Google Scholar] [CrossRef]
- De Witt, S.H.; Kiely, J.S.; Stankovic, C.J.; Schroeder, M.C.; Reynolds Cody, D.M.; Pavia, M.R. “Diversomers”: An approach to nonpeptide, nonoligomeric chemical diversity. Proc. Natl. Acad. Sci. USA 1993, 90, 6909–6913. [Google Scholar] [CrossRef] [PubMed]
- Balkenholh, F.; von dem Bussche-Hunnefeld, C.; Lansky, A.; Zechel, C. Combinatorial synthesis of small organic molecules. Angew. Chem. Int. Ed. 1996, 35, 2288–2337. [Google Scholar] [CrossRef]
- Plumkett, M.J.; Ellman, J.A. Solid-phase synthesis of structurally diverse 1,4-benzodiazepine derivatives using the Stille coupling reaction. J. Am. Chem. Soc. 1995, 117, 3306–3307. [Google Scholar] [CrossRef]
- Haggarty, S.J. The principle of complementarity: Chemical versus biological space. Curr. Opin. Chem. Biol. 2005, 9, 296–303. [Google Scholar] [CrossRef]
- Trabocchi, A. (Ed.) Diversity-Oriented Synthesis: Basics and Applications in Organic Synthesis, Drug Discovery and Chemical Biology, 1st ed.; John Wiley and Sons: Hoboken, NJ, USA, 2013; p. 664.
- Galloway, W.R.J.D.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Spring, D.R. Diversity-oriented synthesis; a challenge for synthetic chemists. Org. Biomol. Chem. 2003, 1, 3867–3870. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1968. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.D.; Schreiber, S.L. A planning strategy for diversity-oriented synthesis. Angew. Chem. Int. Ed. 2004, 43, 46–58. [Google Scholar] [CrossRef]
- Spandl, R.J.; Bender, A.; Spring, D.R. Diversity-oriented synthesis: A spectrum of approach and results. Org. Biomol. Chem. 2007, 6, 1149–1158. [Google Scholar] [CrossRef]
- Nielsen, T.E.; Schreiber, S.L. Towards the optimal screening collection: A synthesis strategy. Angew. Chem. Int. Ed. 2008, 47, 48–56. [Google Scholar] [CrossRef]
- O’Connor, C.J.; Beckmann, H.S.G.; Spring, D.R. Diversity-oriented synthesis: Producing chemical tools for dissecting biology. Chem. Soc. Rev. 2012, 41, 4444–4456. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.Z.; Peng, L.F.; Maloof, N.; Nakai, K.; Wang, X.; Duffner, J.L.; Taveras, K.M.; Hyman, J.M.; Lee, S.W.; Koehler, A.N.; et al. A small molecule that binds Hedgehog and block its signaling in human cells. Nat. Chem. Biol. 2009, 5, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, E.E.; Fergus, S.; Galloway, W.R.J.D.; Bender, A.; Fox, D.J.; Plowright, A.T.; Jessiman, A.S.; Welch, M.; Spring, D.R. Skeletal diversity construction via a branching synthetic strategy. Chem. Commun. 2006, 31, 3296–3298. [Google Scholar] [CrossRef]
- Wyatt, E.E.; Galloway, W.R.J.D.; Thomas, G.L.; Welch, M.; Loiseleur, O.; Plowright, A.T.; Spring, D.R. Identification of an anti-MRSA dihydrofolate reductase inhibitor from a diversity-oriented synthesis. Chem. Commun. 2008, 40, 4962–4964. [Google Scholar] [CrossRef]
- Tan, D.S.; Foley, M.A.; Shair, M.D.; Schreiber, S.L. Stereoselective synthesis of over two million compounds having structural features both reminiscent of natural products and compatible with miniaturized cell-based assays. J. Am. Chem. Soc. 1998, 120, 8565–8566. [Google Scholar] [CrossRef]
- Tan, D.S.; Foley, M.A.; Stockwell, B.R.; Shair, M.D.; Schreiber, S.L. Synthesis and preliminary evaluation of a library of polycyclic small molecules for use in chemical genetics assays. J. Am. Chem. Soc. 1999, 121, 9073–9087. [Google Scholar]
- Tamura, O.; Okabe, T.; Yamaguchi, T.; Gotanda, K.; Noe, K.; Sakamoto, M. Studies on tandem transesterification and intramolecular cycloaddition of nitrones. 1. Sequential bicyclization of α-methoxycarbonylnitrones with allyl alcohols. Tetrahedron 1995, 51, 107–118. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Stille, J.K. The palladium-catalyzed cross-coupling reactions of organotin reagents with organic electrophiles. Angew. Chem. Int. Ed. Engl. 1986, 25, 508–524. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 50, 4467–4470. [Google Scholar] [CrossRef]
- Stephens, R.D.; Castro, C.E. The substitution of aryl iodides with cuprous acetylides. A synthesis of tolanes and heterocyclics. J. Org. Chem. 1963, 28, 3313–3315. [Google Scholar] [CrossRef]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierski, W.M.; Knapp, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Furka, A.; Sebestyen, F.; Asgedom, M.; Dibo, G. General method for rapid synthesis of multicomponent peptide mixtures. Int. J. Pept. Protein Res. 1991, 37, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Haggarty, S.J.; Schreiber, S.L. High-throughput screening of small molecules in miniaturized mammalian cell-based assays involving post-translational modifications. Chem. Biol. 1999, 6, 71–83. [Google Scholar] [CrossRef] [PubMed]
- MacBeath, G.; Koehler, A.N.; Schreiber, S.L. Printing small molecules as microarrays and detecting protein-ligand Interactions en masse. J. Am. Chem. Soc. 1999, 121, 7967–7968. [Google Scholar] [CrossRef]
- Vegas, A.J.; Fuller, J.H.; Koehler, A.N. Small-molecule microarrays as tools in ligand discovery. Chem. Soc. Rev. 2008, 37, 1385–1394. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Schreiber, S.L. Probing the role of homomeric and heteromeric receptor interactions in TGF-β signaling using small molecule dimerizers. Curr. Biol. 1998, 8, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.M.-C.; Neumann, C.S.; Nagayama, S.; Perlstein, E.O.; Schreiber, S.L. A library of spirooxindoles based on a stereoselective three-component coupling reaction. J. Am. Chem. Soc. 2004, 126, 16077–16086. [Google Scholar] [CrossRef] [PubMed]
- Sebahar, P.R.; Williams, R.M. The asymmetric total synthesis of (+)- and (−)-spirotryprostatin B. J. Am. Chem. Soc. 2000, 122, 5666–5667. [Google Scholar] [CrossRef]
- Sebahara, P.R.; Osadab, H.; Usuib, T.; Williams, R.M. Asymmetric, stereocontrolled total synthesis of (+)- and (−)-spirotryprostatin B via a diastereoselective azomethine ylide [1,3]-dipolar cycloaddition reaction. Tetrahedron 2002, 58, 6311–6322. [Google Scholar] [CrossRef]
- Spector, U.; Shocket, N.R.; Kashman, Y.; Groweiss, A. Latrunculins: Novel marine toxins that disrupt microfilament organization in cultured cells. Science 1983, 219, 493–395. [Google Scholar] [CrossRef] [PubMed]
- Ibbeson, B.M.; Laraia, L.; Alza, E.; O’Connor, C.J.; Tan, Y.S.; Davies, H.M.L.; McKenzie, G.; Venkitaraman, A.R.; Spring, D.R. Diversity-oriented synthesis as a tool for identifying new modulators of mitosis. Nat. Commun. 2014, 5, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Stafford, D.G.; Doan, B.D.; Houser, J.H. Tandem asymmetric cyclopropanation/Cope rearrangement. A highly diastereoselective and enantioselective method for the construction of 1,4-cycloheptadienes. J. Am. Chem. Soc. 1998, 120, 3326–3331. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Adsool, V.A.; Hale, C.R.H. An expedient procedure for the oxidative cleavage of olefinic bonds with PhI(OAc)2, NMO and catalytic OsO4. Org. Lett. 2010, 12, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Engel, T. Basic overview of chemoinformatics. J. Chem. Inf. Model. 2006, 46, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L. Chemoinformatic characterization of the chemical space and molecular diversity of compound libraries. In Diversity-Oriented Synthesis: Basics and Applications in Organic Synthesis, Drug Discovery and Chemical Biology, 1st ed.; Trabocchi, A., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2013; pp. 326–352. [Google Scholar]
- Varnek, A.; Baskin, I.I. Chemoinformatics as a theoretical chemistry discipline. Mol. Inform. 2011, 30, 20–32. [Google Scholar] [CrossRef]
- Dobson, C.M. Chemical space and biology. Nature 2004, 432, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S. Diversity-oriented synthesis: Exploring the intersections between chemistry and biology. Nat. Chem. Biol. 2005, 1, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Joliffe, I.T. Principal Component Analysis, 2nd ed.; Springer-Verlag: New York, NY, USA, 2002. [Google Scholar]
- Kopp, F.; Stratton, C.F.; Akella, L.B.; Tan, D.S. Diversity-oriented synthesis approach to macrocycles via oxidative ring expansion. Nat. Chem. Biol. 2012, 8, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Sauer, W.H.B.; Schwarz, M.K. Molecular shape diversity of combinatorial libraries: A prerequisite for broad bioactivity. J. Chem. Inf. Comput. Sci. 2003, 43, 987–1003. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.W.; Ramek, A.; Wang, Y.; Kaya, T.; Wilson, J.A.; Clemons, P.A.; Young, D.M. Route to three-dimensional fragments using diversity-oriented synthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 6799–6804. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Young, D.W.; Bender, A.; Hoyt, J.; McWhinnie, E.; Chirn, G.W.; Tao, C.Y.; Tallarico, J.A.; Labow, M.; Jenkins, J.L.; Mitchison, T.J.; et al. Integrating high-content screening and ligand-target prediction to identify mechanism of action. Nat. Chem. Biol. 2008, 4, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Samanta, A.; Ghosh, K.; Zhai, D.; Xu, W.; Su, D.; Leong, C.; Chang, T.Y. Target identification: A challenging step in forward chemical genetics. Interdiscip. Bio Cent. 2011, 3, 1–18. [Google Scholar]
- Resnick, M.A.; Cox, B.S. Yeast as an honorary mammal. Mutat. Res. 2000, 451, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C. Chemical genomics in yeast. Genome Biol. 2004, 5. [Google Scholar] [CrossRef]
- Giaever, G.; Flaherty, P.; Kumm, J.; Proctor, M.; Nislow, C.; Jaramillo, D.F.; Chu, A.M.; Jordan, M.I.; Arkin, A.P.; Davis, R.W. Chemogenomic profiling: Identifying the functional interactions of small molecules in yeast. Proc. Natl. Acad. Sci. USA 2004, 101, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Lum, P.Y.; Armour, C.D.; Stepaniants, S.B.; Cavet, G.; Wolf, M.K.; Butler, J.S.; Hinshaw, J.C.; Garnier, P.; Prestwich, G.D.; Leonardson, A.; et al. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell 2004, 116, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Parsons, A.B.; Brost, R.L.; Ding, H.; Li, Z.; Zhang, C.; Sheikh, B.; Brown, G.W.; Kane, P.M.; Hughes, T.R.; Boone, C. Integration of chemical-genetics and genetics interaction data links bioactive compounds to cellular target pathways. Nat. Biotechnol. 2004, 22, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Parsons, A.B.; Lopez, A.; Givoni, I.E.; Williams, D.E.; Gray, C.A.; Porter, J.; Chua, G.; Sopko, R.; Brost, R.L.; Ho, C.H.; et al. Exploring the mode-of-action of bioactive compounds by chemical-genetics profiling in yeast. Cell 2006, 126, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, I.; Trabocchi, A.; Marchi, E.; Guarna, A.; Cavalieri, D. A systems biology approach to dissection of the effects of small bicyclic peptidomimetics on a panel of Saccharomyces cerevisiae mutants. J. Biol. Chem. 2010, 285, 23477–23485. [Google Scholar] [CrossRef] [PubMed]
- Trabocchi, A.; Stefanini, I.; Morvillo, M.; Ciofi, L.; Cavalieri, D.; Guarna, A. Chemical genetics approach to identify new small molecule modulators of cell growth by phenotypic screening of Saccharomyces cerevisiae strains with a library of morpholine-derived compounds. Org. Biol. Chem. 2010, 8, 5552–5557. [Google Scholar] [CrossRef]
- Trabocchi, A.; Cavalieri, D.; Guarna, A. Chemical genetics approach to drug discovery by diversity-oriented synthesis (DOS) of peptidomimetics. Pure Appl. Chem. 2011, 83, 687–698. [Google Scholar] [CrossRef]
- Gante, J. Peptidomimetics—Tailored enzyme inhibitors. Angew. Chem. Int. Ed. 1994, 33, 1699–1720. [Google Scholar] [CrossRef]
- Giannis, A.; Kolter, T. Peptidomimetics for receptor ligands discovery, development and medical perspectives. Angew. Chem. Int. Ed. 1993, 32, 1244–1267. [Google Scholar] [CrossRef]
- Trabocchi, A.; Menchi, G.; Guarna, F.; Machetti, F.; Scarpi, D.; Guarna, A. Design, synthesis and applications of 3-aza-6,8-dioxa-bicyclo[3.2.1]octane-based scaffolds for peptidominetic chemistry. Synlett 2006, 3, 331–353. [Google Scholar]
- Guarna, A.; Guidi, A.; Machetti, F.; Menchi, G.; Occhiato, E.G.; Scarpi, D.; Sisi, S.; Trabocchi, A. Synthesis and reactivity of bicycles derived from tartaric acids and α-amino acids: A novel class of conformationally constrained dipeptide isosteres based upon enantiopure 3-aza-6,8-dioxabicyclo [3.2.1]octane-7-carboxylic acid. J. Org. Chem. 1999, 64, 7347–7364. [Google Scholar] [CrossRef]
- Guarna, A.; Bucelli, I.; Machetti, F.; Menchi, G.; Occhiato, E.G.; Scarpi, D.; Trabocchi, A. Synthesis of new enantiopure bicycles γ/δ-amino acid (BTKa), derived from tartaric acid and α-aminoacetophenone. Tetrahedron 2002, 58, 9865–9870. [Google Scholar] [CrossRef]
- Trabocchi, A.; Potenza, D.; Occhiato, E.G.; Guarna, A. Synthesis and conformational analysis of small peptides containing 6-endo-BT(t)L scaffolds as reverse turn mimetics. J. Org. Chem. 2002, 67, 7483–7492. [Google Scholar] [CrossRef] [PubMed]
- Calugi, C.; Guarna, A.; Trabocchi, A. Identification of constrained peptidomimetic chemotypes as HIV protease inhibitors. Eur. J. Med. Chem. 2014, 84, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Trabocchi, A.; Mannino, C.; Machetti, F.; de Bernardis, F.; Arancia, S.; Cauda, R.; Cassone, A.; Guarna, A. Identification of inhibitors of drug-resistant Candida albicans strains from a library of bicyclic peptidomimetic compounds. J. Med. Chem. 2010, 53, 2502–2509. [Google Scholar] [CrossRef] [PubMed]
- Scarpi, D.; Cirelli, D.; Matrone, C.; Castronovo, G.; Rosini, P.; Occhiato, E.G.; Romano, F.; Bartali, L.; Clemente, A.M.; Bottegoni, G.; et al. Low molecular weight, non-peptidic agonists of TrkA receptor with NGF-mimetic activity. Cell Death Dis. 2012, 3. [Google Scholar] [CrossRef]
- Gaisne, M.; Becam, A.M.; Verdiere, J.; Herbert, C.J. A ‘natural’ mutation in Saccharomyces cerevisiae strains derived from S288c affects the complex regulatory gene HAP1 (CYP1). Curr. Genet. 1999, 36, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Beltrame, L.; Rizzetto, L.; Paola, R.; Rocca-Serra, P.; Gambineri, L.; Battaglia, C.; Cavalieri, D. Using pathway signatures as means of identifying similarities among microarray experiments. PLoS One 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, I.; de Filippo, C.; Cavalieri, D. Yeast as a model in high-throughput screening of small-molecule libraries. In Diversity-Oriented Synthesis: Basics and Applications in Organic Synthesis, Drug Discovery and Chemical Biology, 1st ed.; Trabocchi, A., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2013; pp. 455–482. [Google Scholar]
- Lalli, C.; Trabocchi, A.; Sladojevich, F.; Menchi, G.; Guarna, A. Diversity-oriented synthesis of morpholine-containing molecular scaffolds. Chem. Eur. J. 2009, 15, 7871–7875. [Google Scholar] [CrossRef] [PubMed]
- Ciofi, L.; Morvillo, M.; Sladojevich, F.; Guarna, A.; Trabocchi, A. Skeletal diversity by sequential one-pot and stepwise routes using morpholine ester scaffolds. Tetrahedron Lett. 2010, 51, 6282–6285. [Google Scholar] [CrossRef]
- Levin, J.I.; Chen, J.M.; Laakso, L.M.; Du, M.; Du, X.; Venkatesan, A.M.; Sandanayaka, V.; Zask, A.; Xu, J.; Xu, W.; et al. Acetylenic TACE inhibitors. Part 2: SAR of six-membered cyclic sulfonamide hydroxamates. Bioorg. Med. Chem. Lett. 2005, 15, 4345–4349. [Google Scholar] [CrossRef] [PubMed]
- Almstead, N.G.; Bradley, R.S.; Pikul, S.; De, B.; Natchus, M.G.; Taiwo, Y.O.; Gu, F.; Williams, L.E.; Hynd, B.A.; Janusz, M.J.; et al. Design, synthesis, and biological evaluation of potent thiazine- and thiazepine-based matrix metalloproteinase inhibitors. J. Med. Chem. 1999, 42, 4547–4562. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.B.; Carvalho, I. Diketopiperazines: biological activity and synthesis. Tetrahedron 2007, 63, 9923–9932. [Google Scholar] [CrossRef]
- Nicholson, B.; Lloyd, G.K.; Miller, B.R.; Palladino, M.A.; Kisob, Y.; Hayashib, Y.; Neuteboom, S.T.B. NPI-2358 is a tubulin-depolymerizing agent: In vitro evidence for activity as a tumor vascular-disrupting agent. Anticancer Drugs 2006, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenci, E.; Guarna, A.; Trabocchi, A. Diversity-Oriented Synthesis as a Tool for Chemical Genetics. Molecules 2014, 19, 16506-16528. https://doi.org/10.3390/molecules191016506
Lenci E, Guarna A, Trabocchi A. Diversity-Oriented Synthesis as a Tool for Chemical Genetics. Molecules. 2014; 19(10):16506-16528. https://doi.org/10.3390/molecules191016506
Chicago/Turabian StyleLenci, Elena, Antonio Guarna, and Andrea Trabocchi. 2014. "Diversity-Oriented Synthesis as a Tool for Chemical Genetics" Molecules 19, no. 10: 16506-16528. https://doi.org/10.3390/molecules191016506