Synthetic Applications of Intramolecular Thiol-Ene “Click” Reactions

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Thiyl Radicals in Organic Chemistry
3. The Intermolecular Thiol-Ene Reaction
4. The Intramolecular Thiol-Ene Reaction

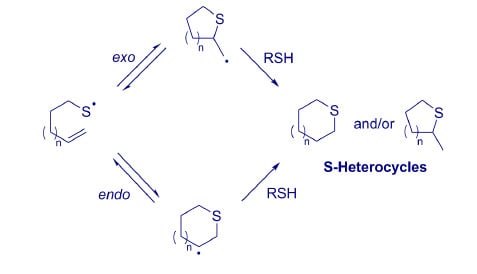
5. Regioselectivity of Intramolecular Thiol-ene Ring-Closing Reactions
5.1. 4-exo vs. 5-endo Systems

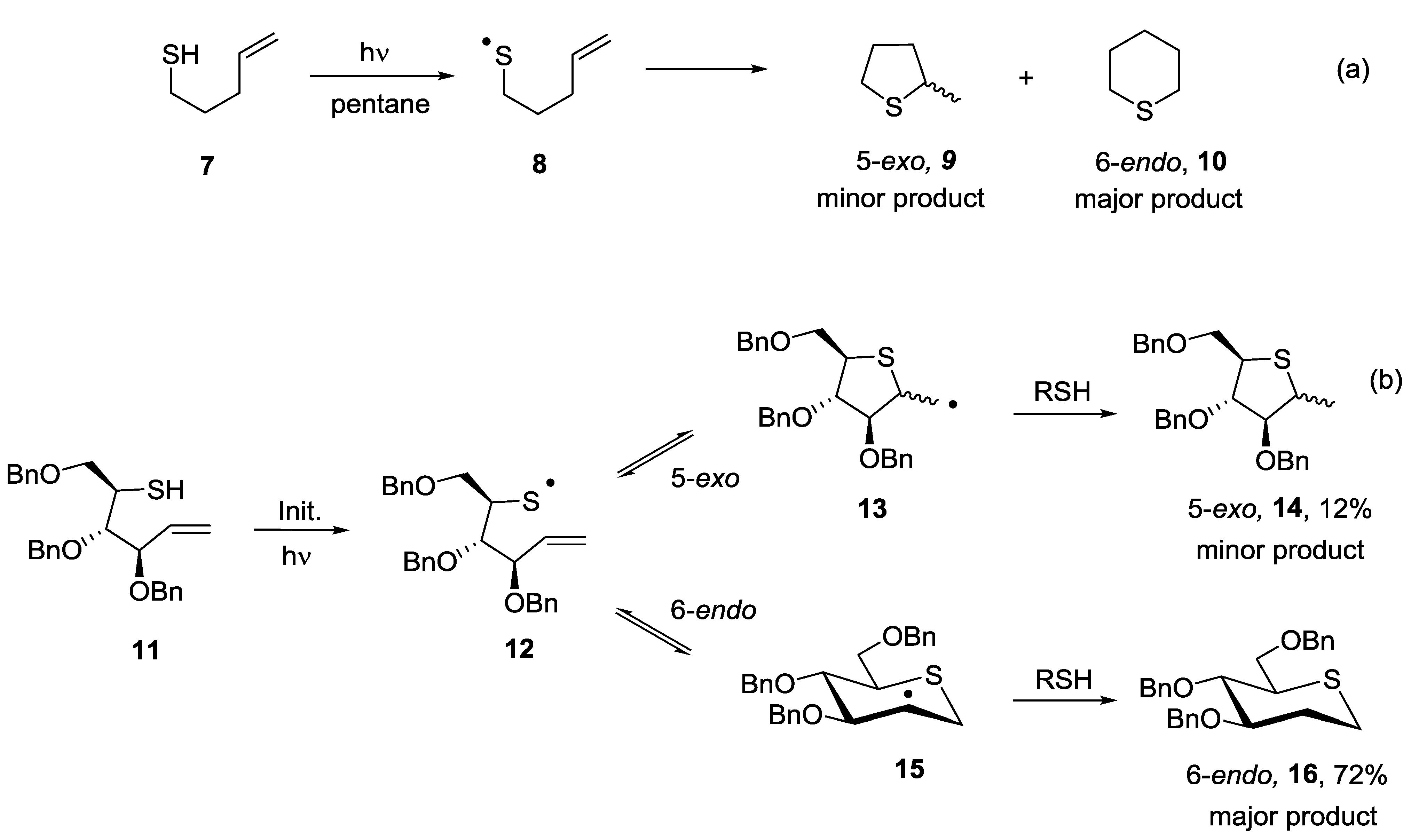
5.2. 5-exo vs. 6-endo Systems





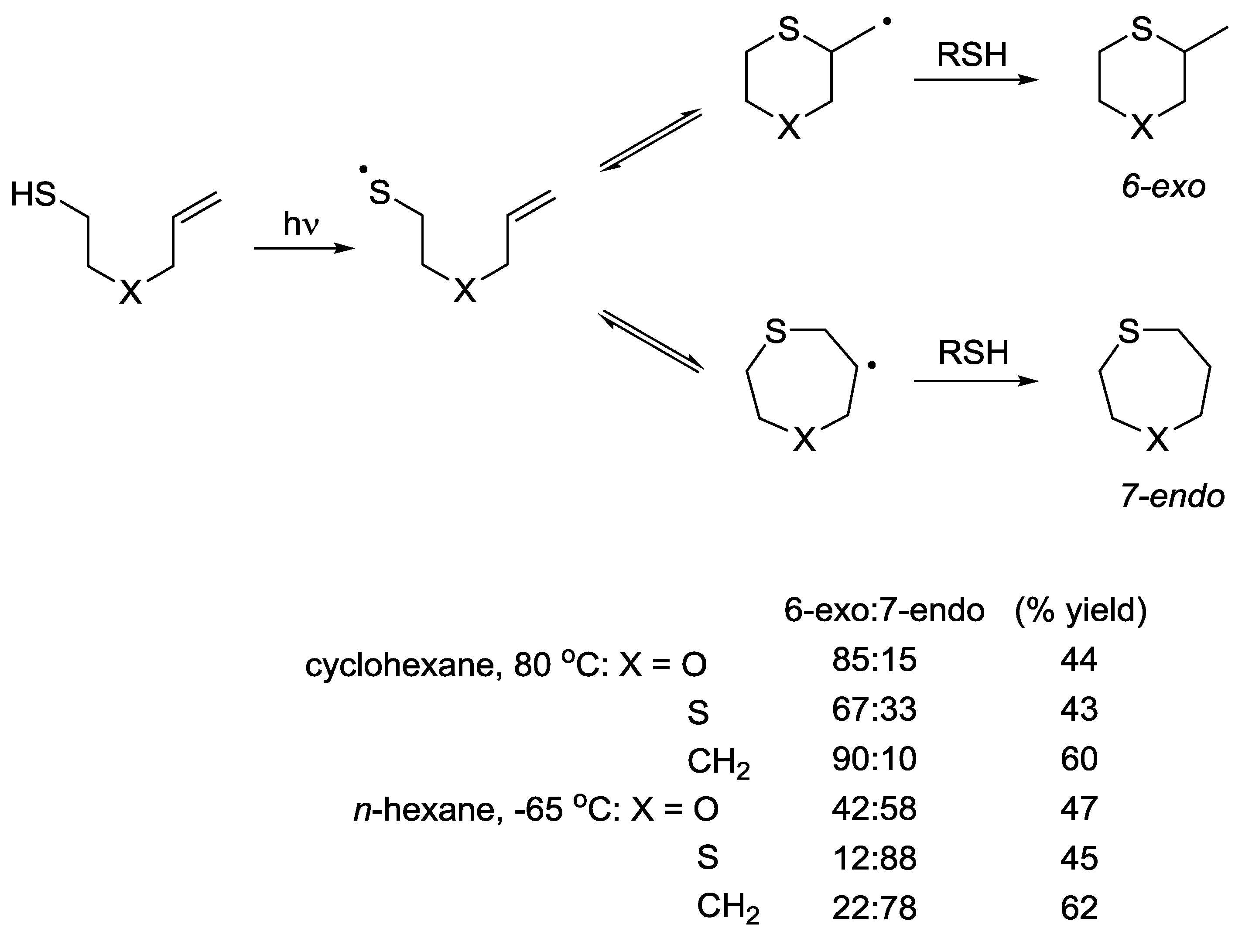
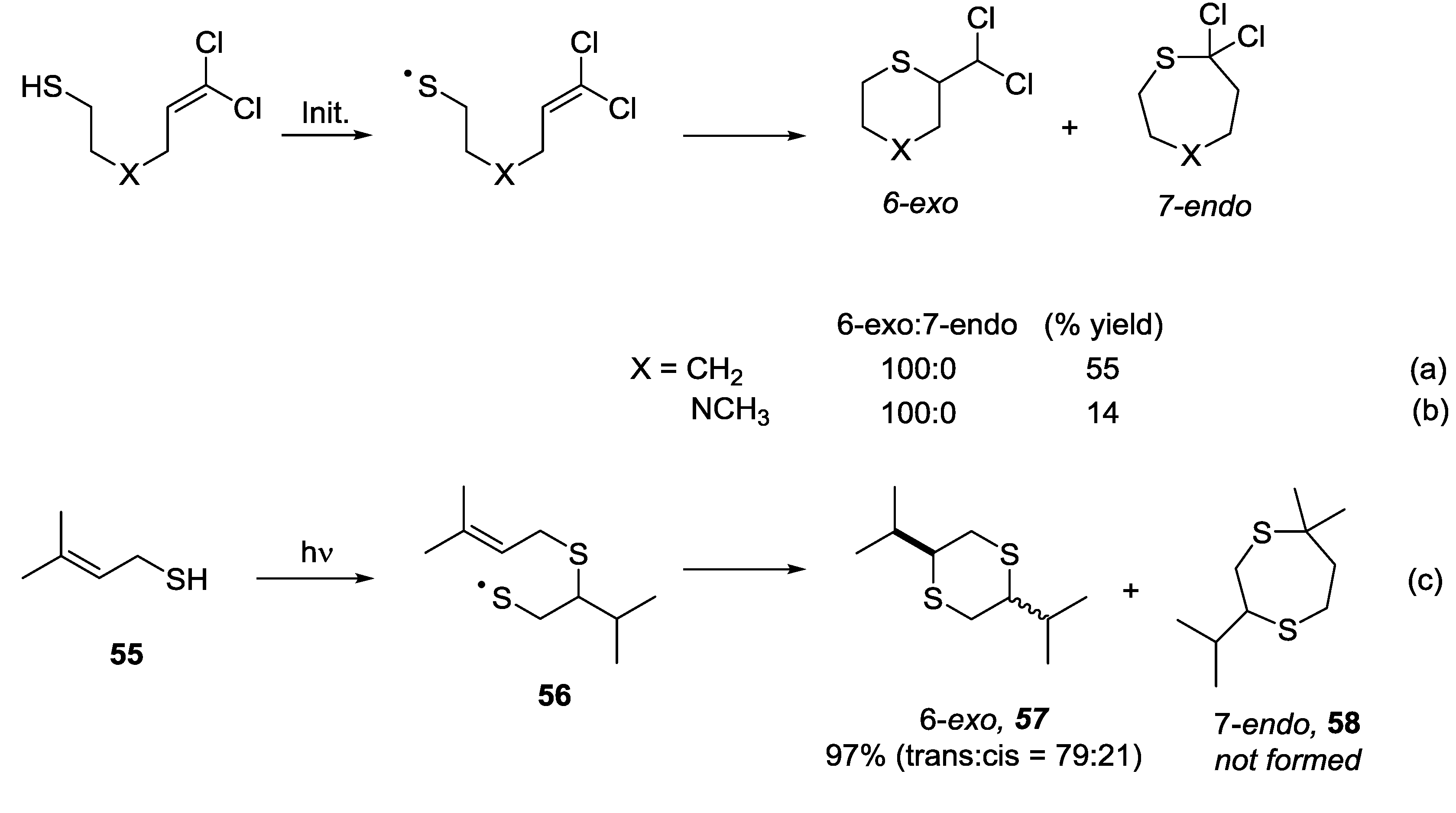
5.3. 6-exo vs. 7-endo Systems


5.4. 7-exo vs. 8-endo Systems

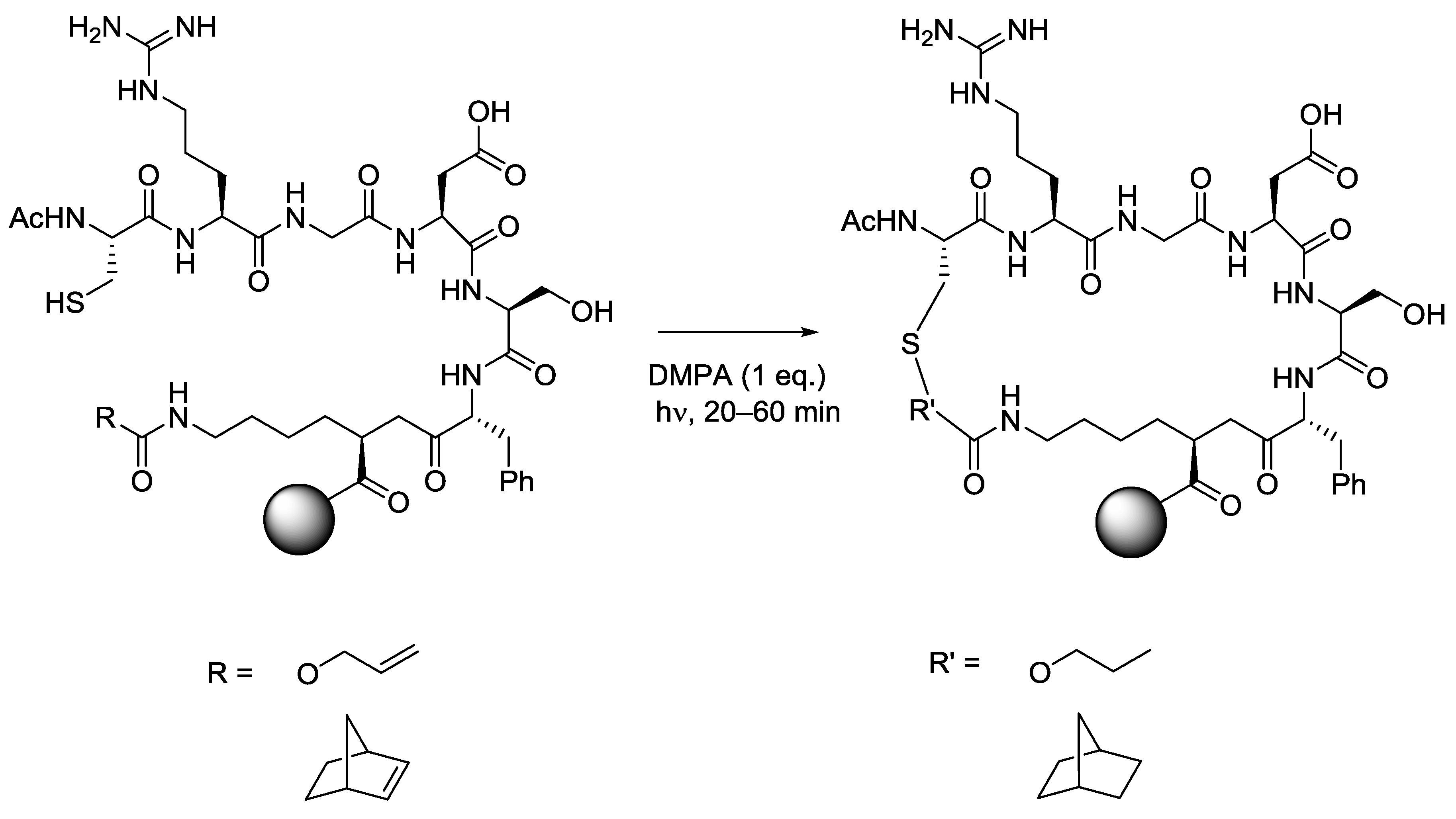
5.5. Macrocycles

6. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Naito, T. Heterocycle synthesis via radical reactions. Pure Appl. Chem. 2008, 80, 717–726. [Google Scholar]
- Xu, X.; Wan, X.; Geng, Y.; Zhang, J.; Xu, H. Recent progress in nitrogen-centered radical cyclizations. Youji Huaxue 2011, 31, 453–465. [Google Scholar]
- Portela-Cubillo, F.; Scanlan, E.M.; Scott, J.S.; Walton, J.C. From dioxime oxalates to dihydropyrroles and phenanthridines via iminyl radicals. Chem. Commun. 2008, 4189–4191. [Google Scholar] [CrossRef]
- Hartung, J.; Giese, B. A new route to alkoxy radicals—photochemical reactions with (alkylperoxy)cobaloximes. Chem. Ber. 1991, 124, 387–390. [Google Scholar] [CrossRef]
- Zard, S.Z. Iminyl radicals. A fresh look at a forgotten species (and some of its relatives). Synlett 1996, 1148–1154. [Google Scholar] [CrossRef]
- Bowman, W.R.; Clark, D.N.; Marmon, R.J. Synthesis of pyrrolizidines using aminyl radicals generated from sulfenamide precursors. Tetrahedron 1994, 50, 1295–1310. [Google Scholar] [CrossRef]
- Walling, C.; Pearson, M.S. Some radical reactions of trivalent phosphorus derivatives with mercaptans, peroxides, and olefins. A new radical cyclization. J. Am. Chem. Soc. 1964, 86, 2262–2266. [Google Scholar] [CrossRef]
- Denes, F.; Pichowicz, M.; Povie, G.; Renaud, P. Thiyl radicals in organic synthesis. Chem. Rev. 2014, 114, 2587–2693. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.E.; Bowman, C.N. Thiol-Ene Click Chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef]
- Denisov, E.; Chatgilialoglu, C.; Shestakov, A.; Denisova, T. Rate constants and transition-state geometry of reactions of alkyl, alkoxyl, and peroxyl radicals with thiols. Int. J. Chem. Kinet. 2009, 41, 284–293. [Google Scholar] [CrossRef]
- Nguyen, V.-H.; Nishino, H.; Kajikawa, S.; Kurosawa, K. Mn(III)-based reactions of alkenes and alkynes with thiols. An approach toward substituted 2,3-dihydro-1,4-oxathiins and simple route to (E)-vinyl sulfides. Tetrahedron 1998, 54, 11445–11460. [Google Scholar]
- Kolberg, M.; Strand, K.R.; Graff, P.; Kristoffer Andersson, K. Structure, function, and mechanism of ribonucleotide reductases. Biochim. Biophys. Acta Proteins Proteomics 2004, 1699, 1–34. [Google Scholar] [CrossRef]
- Torrents, E.; Aloy, P.; Gibert, I.; Rodriguez-Trelles, F. Ribonucleotide reductases: Divergent evolution of an ancient enzyme. J. Mol. Evol. 2002, 55, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Markey, L.; Giordani, S.; Scanlan, E.M. Native chemical ligation, thiol-ene Click: A methodology for the synthesis of functionalized peptides. J. Org. Chem. 2013, 78, 4270–4277. [Google Scholar] [CrossRef] [PubMed]
- Floyd, N.; Vijayakrishnan, B.; Koeppe, J.R.; Davis, B.G. Thiyl glycosylation of olefinic proteins: S-Linked glycoconjugate synthesis. Angew. Chem. Int. Ed. 2009, 48, 7798–7802. [Google Scholar] [CrossRef]
- Dondoni, A.; Massi, A.; Nanni, P.; Roda, A. A New ligation strategy for peptide and protein glycosylation: Photoinduced thiol-ene coupling. Chem. Eur. J. 2009, 15, 11444–11449. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A. The emergence of thiol-ene coupling as a click process for materials and bioorganic chemistry. Angew. Chem. Int. Ed. 2008, 47, 8995–8997. [Google Scholar] [CrossRef]
- Tedder, J.M.; Walton, J.C. The kinetics and orientation of free-radical addition to olefins. Acc. Chem. Res. 1976, 9, 183–191. [Google Scholar] [CrossRef]
- Surzur, J.M. Radical cyclizations by intramolecular additions. React. Intermed. 1982, 2, 121–295. [Google Scholar]
- Bastien, G.; Crozet, M.P.; Flesia, E.; Surzur, J.M. Radical alkylating heterocyclization by photolysis of ethylenic sulfides. II. Study of the reaction mechanism. Bull. Soc. Chim. Fr. 1979, 606–613. [Google Scholar]
- Surzur, J.M.; Crozet, M.P.; Dupuy, C. Radical cyclization of ethylenic mercaptans. Comptes Rendus Seances l'Academie Sci. Ser. C Sci. Chim. 1967, 264, 610–613. [Google Scholar]
- Malone, A.; Scanlan, E.M. Applications of thiyl radical cyclizations for the synthesis of thiosugars. Org. Lett. 2013, 15, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Malone, A.; Scanlan, E.M. Applications of 5-exo-trig thiyl radical cyclizations for the synthesis of thiosugars. J. Org. Chem. 2013, 78, 10917–10930. [Google Scholar] [CrossRef] [PubMed]
- Nouguier, R.; Surzur, J.M. Synthesis of 6-thiabicyclo[3. 2.1]octanes by photolysis of 1-methylthiocyclohex-3-ene. Tetrahedron 1976, 32, 2001–2003. [Google Scholar]
- Makisumi, Y.; Murabayashi, A. Mechanism of the cyclization reaction of the intermediate, 3-mercapto-4-methallylquinoline, in the thio-Claisen rearrangement. Tetrahedron Lett. 1969, 10, 2453–2456. [Google Scholar] [CrossRef]
- Makisumi, Y.; Murabayashi, A. Thio-Claisen rearrangement of allyl 3-quinolyl sulfides. Tetrahedron Lett. 1969, 10, 2449–2452. [Google Scholar] [CrossRef]
- Meyers, C.Y.; Rinaldi, C.; Bonoli, L. Formation of thiochroman as a major product in the Claisen rearrangement of allyl phenyl sulfide. J. Org. Chem. 1963, 28, 2440–2442. [Google Scholar] [CrossRef]
- Kwart, H.; Evans, R.E. The thio-Claisen rearrangement. The mechanism of thermal rearrangement of allyl aryl sulfides. J. Org. Chem. 1966, 31, 413–419. [Google Scholar]
- Maki, Y.; Sako, M. Photochemical formation of 3-methylenecepham. Tetrahedron Lett. 1976, 17, 4291–4294. [Google Scholar] [CrossRef]
- Maki, Y.; Sako, M. Concentration-dependent photoreaction of benzothiazolyldithioazetidinones: novel photochemical formation of penam derivatives. J. Chem. Soc. Chem. Commun. 1978, 836–838. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Schiesser, C.H. Regio- and stereoselectivity of alkenyl radical ring closure: A theoretical study. Tetrahedron 1985, 41, 3925–3941. [Google Scholar] [CrossRef]
- Spellmeyer, D.C.; Houk, K.N. Force-field model for intramolecular radical additions. J. Org. Chem. 1987, 52, 959–974. [Google Scholar] [CrossRef]
- RajanBabu, T.V. From carbohydrates to optically active carbocycles I: Stereochemical control in sugar hex-5-enyl radical cyclization. J. Am. Chem. Soc. 1987, 109, 609–611. [Google Scholar] [CrossRef]
- RajanBabu, T.V.; Fukunaga, T.; Reddy, G.S. Stereochemical control in hex-5-enyl radical cyclizations: From carbohydrates to carbocycles 3. J. Am. Chem. Soc. 1989, 111, 1759–1769. [Google Scholar] [CrossRef]
- RajanBabu, T.V. Stereochemistry of intramolecular free-radical cyclization reactions. Acc. Chem. Res. 1991, 24, 139–145. [Google Scholar]
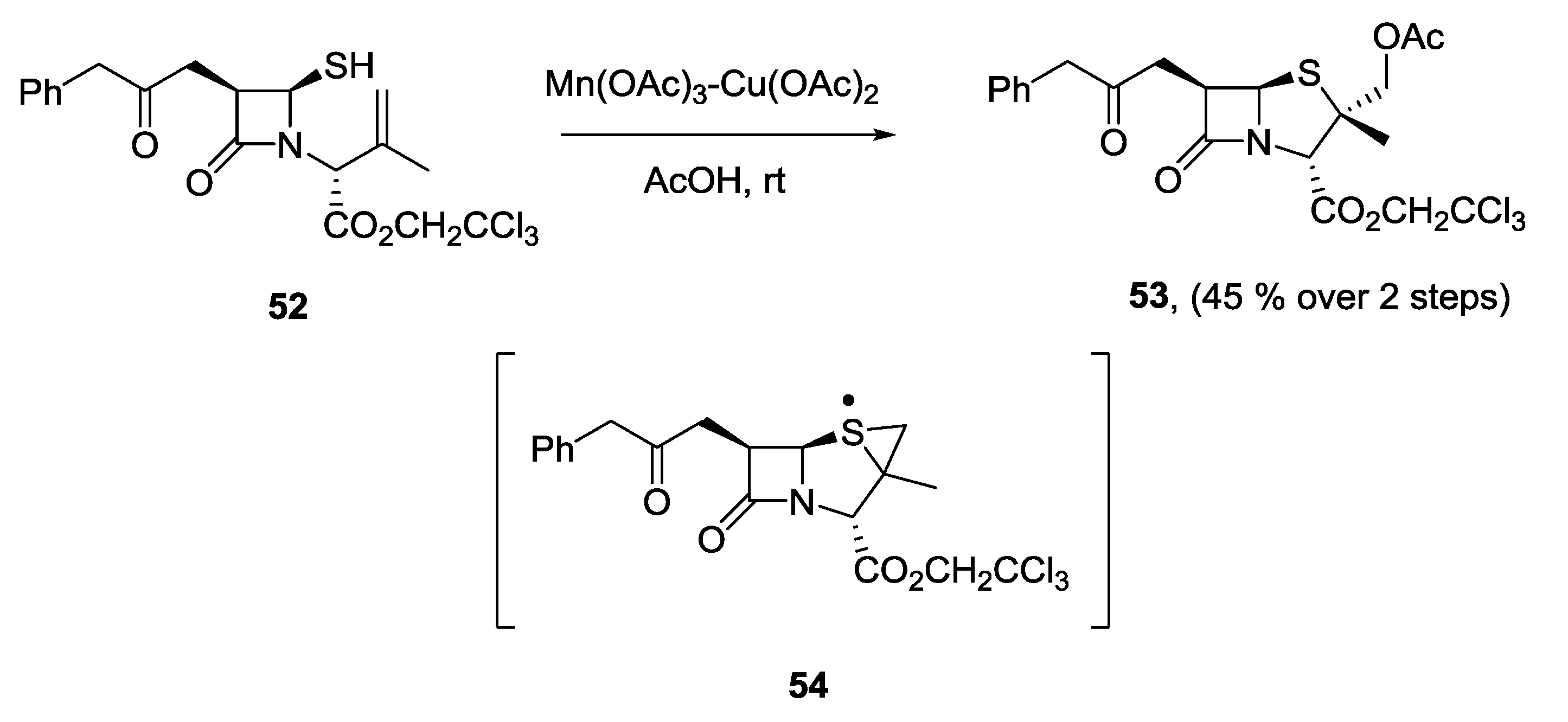
- Cabri, W.; Candiani, I.; Bedeschi, A.; Santi, R. Metal-promoted thiyl radical cyclizations in β-lactam antibiotics. Tetrahedron Lett. 1992, 33, 4783–4786. [Google Scholar] [CrossRef]
- Cabri, W.; Borghi, D.; Arlandini, E.; Sbraletta, P.; Bedeschi, A. Metal-catalyzed and -promoted cyclization for the synthesis of cephalosporins: A possible DAOC/DAC synthetase biomimetic process. Tetrahedron 1993, 49, 6837–6848. [Google Scholar] [CrossRef]
- Cabri, W.; Candiani, I.; Bedeschi, A. Iron(III)-copper(II) and manganese(III)-copper(II) promoted cyclizations: A new stereoselective approach towards α-methyl substituted penicillin derivatives. J. Chem. Soc. Chem. Commun. 1994, 597–598. [Google Scholar] [CrossRef]
- Gordon, E.M.; Cimarusti, C.M. On the transformation of penicillins to 3-methyl or 3-methylene cephams. Tetrahedron Lett. 1977, 18, 3425–3428. [Google Scholar] [CrossRef]
- Crozet, M.P.; Surzur, J.M.; Dupuy, C. Reversibility of the intramolecular addition of the thiyl radical. Tetrahedron Lett. 1971, 2031–2034. [Google Scholar] [CrossRef]
- Surzur, J.M.; Crozet, M.P. Formation of thiazolidines by ultraviolet irradiation of 2-aminoethanethiols. C. R. Acad. Sci. Paris Ser. C 1969, 268, 2109–2112. [Google Scholar]
- Kaafarani, M.; Crozet, M.P.; Surzur, J.M. Free radical heterocyclization of unsaturated aminothiols. I. General aspects of the formation of thiazolidine compounds. Bull. Soc. Chim. Fr. 1981, 449–457. [Google Scholar]
- Petrova, R.G.; Slepushkin, V.V.; Freidlina, R.K. Homolytic cyclization of unsaturated thiols containing α,α-dichlorovinyl groups. Dokl. Akad. Nauk SSSR 1972, 202, 857–860. [Google Scholar]
- Petrova, R.G.; Maiorova, T.D.; Freidlina, R.K. Homolytic cyclization of N-methyl-N-3,3-(dichloroallyl)-β-aminoethanethiol. Izv. Akad. Nauk SSSR Seriya Khimicheskaya 1973, 2540–2543. [Google Scholar]
- Takabe, K.; Katagiri, T.; Tanaka, J. Photodimerization of prenyl mercaptan. Tetrahedron Lett. 1970, 4805–4806. [Google Scholar] [CrossRef]
- Koenig, K.E.; Weber, W.P. Addition of hydrogen sulfide to α,ω-dienes to yield medium sized heterocycles. 1,1-Dimethyl-1-sila-5-thiacyclooctane. Tetrahedron Lett. 1973, 3151–3152. [Google Scholar]
- Koenig, K.E.; Felix, R.A.; Weber, W.P. Unsaturated organosilicon heterocycles. J. Org. Chem. 1974, 39, 1539–1542. [Google Scholar] [CrossRef]
- Aimetti, A.A.; Shoemaker, R.K.; Lin, C.-C.; Anseth, K.S. On-resin peptide macrocyclization using thiol-ene click chemistry. Chem. Commun. 2010, 46, 4061–4063. [Google Scholar] [CrossRef]
- Aimetti, A.A.; Feaver, K.R.; Anseth, K.S. Synthesis of cyclic, multivalent Arg-Gly-Asp using sequential thiol-ene/thiol-yne photoreactions. Chem. Commun. 2010, 46, 5781–5783. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scanlan, E.M.; Corcé, V.; Malone, A. Synthetic Applications of Intramolecular Thiol-Ene “Click” Reactions. Molecules 2014, 19, 19137-19151. https://doi.org/10.3390/molecules191119137
Scanlan EM, Corcé V, Malone A. Synthetic Applications of Intramolecular Thiol-Ene “Click” Reactions. Molecules. 2014; 19(11):19137-19151. https://doi.org/10.3390/molecules191119137
Chicago/Turabian StyleScanlan, Eoin M., Vincent Corcé, and Aoife Malone. 2014. "Synthetic Applications of Intramolecular Thiol-Ene “Click” Reactions" Molecules 19, no. 11: 19137-19151. https://doi.org/10.3390/molecules191119137