Pauson-Khand Reaction of Internal Dissymmetric Trifluoromethyl Alkynes. Influence of the Alkene on the Regioselectivity

Abstract
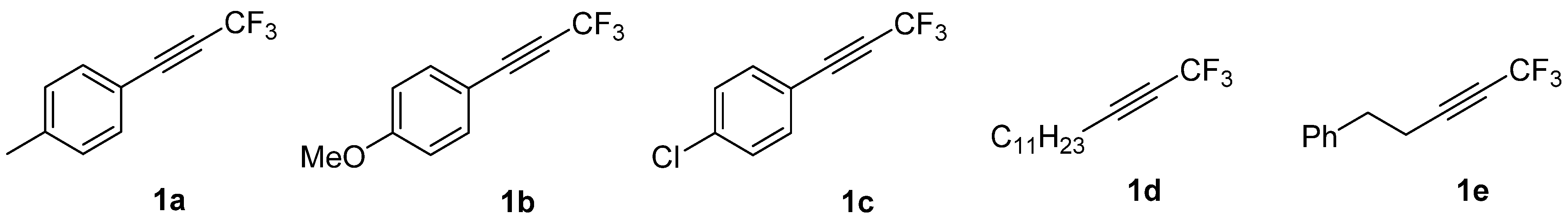
:1. Introduction
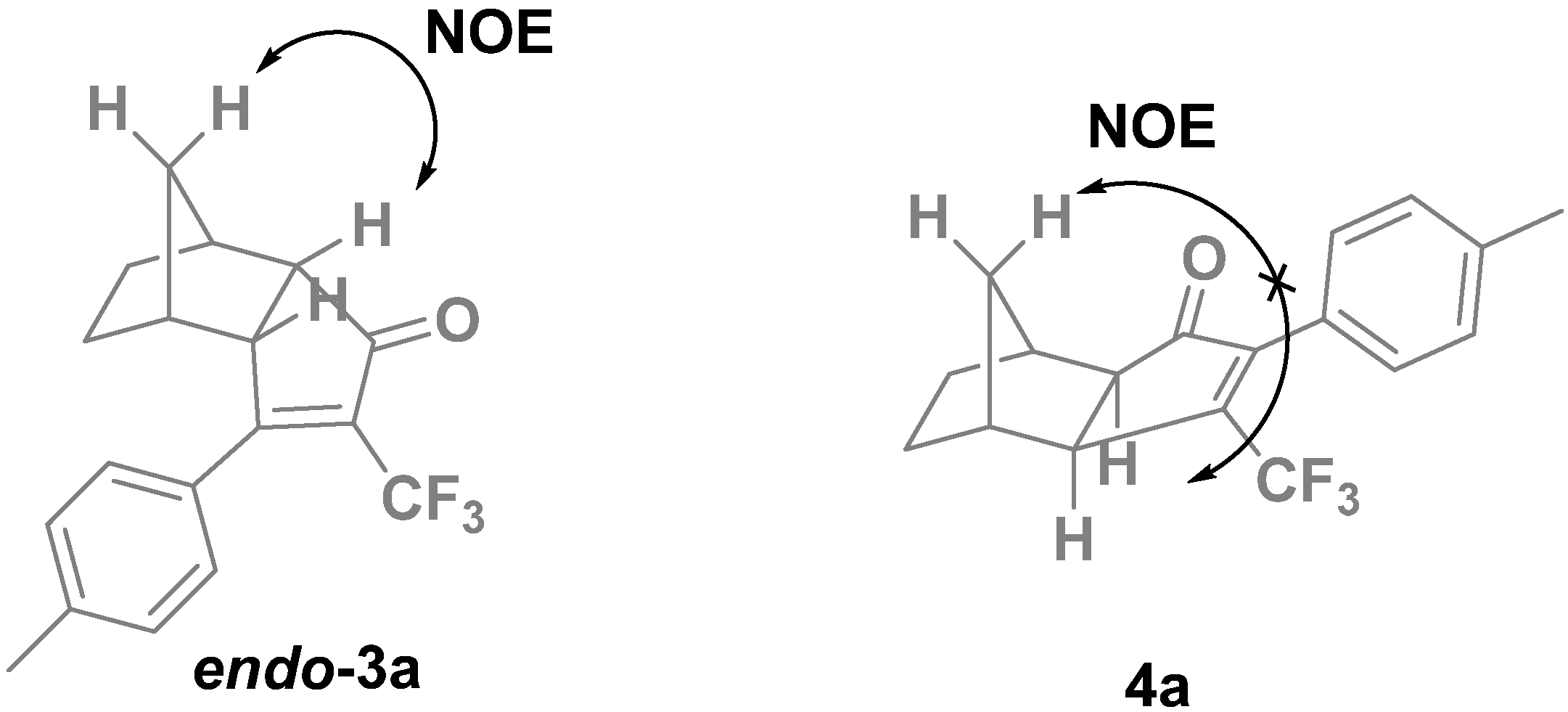
2. Results and Discussion




| Entry | SM | R | Product | Yield (%) | Ratio 3:4 |
|---|---|---|---|---|---|
| 1 | 2a | p-MePh | 3a, 4a | 87 | 86:14 |
| 2 | 2b | p-MeOPh | 3b, 4b | 51 | 86:14 |
| 3 | 2c | p-ClPh | 3c, 4c | 41 | 71:29 |
| 4 | 2d | C11H23 | 3d | 40 | >95:5 |

| Entry | SM | R | Conditions | Product | Yield |
|---|---|---|---|---|---|
| 1 | 1a | p-MePh | 6 bars ethylene, NMO b (10 equiv), 4 Å MS (activated a; 15x alkyne mass), rt, DCM | 5a | 18% |
| 2 | 1a | p-MePh | 6 bars ethylene, NMO b (10 equiv), rt, DCM | 5a | 25% |
| 3 | 1a | p-MePh | 6 bars ethylene, NMO b (10 equiv), 0 °C, DCM | 5a | 9% |
| 4 | 1a | p-MePh | 6 bars ethylene, 70 °C to 80 °C, toluene | 5a | 53% |
| 5 | 1b | p-MeOPh | 6 bars ethylene, 80 °C to 85 °C, toluene | 5b | 58% |
| 6 | 1c | p-ClPh | 6 bars ethylene, 80 °C to 85 °C, toluene | 5c | 56% |
| 7 | 1e | PhC2H4 | 6 bars ethylene, 80 °C to 85 °C, toluene | 5e | 23% |

{kind=link}
{kind=link}
| Entry | SM | R | Product | Yield (%) |
|---|---|---|---|---|
| 1 | 3a | p-MePh | 6a | 28 |
| 2 | 3b | p-MeOPh | 6b | 49 |
| 3 | 3c | p-ClPh | 6c | 30 |
| 4 | 3d | C11H23 | 6d | nd |
3. Experimental
3.1. General
3.2. General Conditions for the Pauson-Khand Reaction with Norbornene
3.3. General Conditions for the Des-trifluoromethylation Reaction
3.4. General Conditions for the Pauson-Khand Reaction with Ethylene
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ríos, R. The Pauson-Khand Reaction; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Vazquez-Romero, A.; Cardenas, L.; Blasi, E.; Verdaguer, X.; Riera, A. Synthesis of Prostaglandin and Phytoprostane B1 Via Regioselective Intermolecular Pauson-Khand Reactions. Org. Lett. 2009, 11, 3104–3107. [Google Scholar] [CrossRef]
- De Bruin, T.J.M.; Michel, C.; Vekey, K.; Greene, A.E.; Gimbert, Y.; Milet, A. First C-C bond formation in the Pauson-Khand reaction: Influence of carbon-carbon triple bond polarization on regiochemistry. J. Organomet. Chem. 2006, 691, 4281–4288. [Google Scholar] [CrossRef]
- De Bruin, T.J.M.; Milet, A.; Robert, F.; Gimbert, Y.; Greene, A.E. Theoretical Study of the Regiochemistry-Determining Step of the Pauson-Khand Reaction. J. Am. Chem. Soc. 2001, 123, 7184–7185. [Google Scholar] [CrossRef]
- Robert, F.; Milet, A.; Gimbert, Y.; Konya, D.; Greene, A. E. Regiochemistry in the Pauson-Khand Reaction: Has a Trans Effect Been Overlooked? J. Am. Chem. Soc. 2001, 123, 5396–5400. [Google Scholar] [CrossRef]
- Fager-Jokela, E.; Muuronen, M.; Patzschke, M.; Helaja, J. Electronic Regioselectivity of Diarylalkynes in Cobalt-Mediated Pausond-Khand Reaction: An Experimental and Computational Study with Para- and Meta-Substituted Diarylalkynes and Norbornene. J. Org. Chem. 2012, 77, 9134–9147. [Google Scholar] [CrossRef]
- Moulton, B.E.; Whitwood, A.C.; Duhme-Klair, A.K.; Lynam, J.M.; Fairlamb, I.J.S. Regiochemistry in cobalt-mediated intermolecular Pauson-Khand reactions of unsymmetrical internal heteroaromatic alkynes with norbornene. J. Org. Chem. 2011, 76, 5320–5334. [Google Scholar] [CrossRef]
- Kizirian, J.; Aiguabella, N.; Pesquer, A.; Fustero, S.; Bello, P.; Verdaguer, X.; Riera, A. Regioselectivity in Intermolecular Pauson-Khand Reactions of Dissymmetric Fluorinated Alkynes. Org. Lett. 2010, 12, 5620–5623. [Google Scholar] [CrossRef]
- Aiguabella, N.; del Pozo, C.; Verdaguer, X.; Fustero, S.; Riera, A. Synthesis and Application of β-Substituted Pauson-Khand Adducts: Trifluoromethyl as a Removable Steering Group. Angew. Chem. Int. Ed. 2013, 52, 5355–5359. [Google Scholar]
- Peer, K. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Liu, H.; Gu, Z.; Jiang, X. Direct Trifluoromethylation of the C-H Bond. Adv. Synth. Catal. 2013, 355, 617–626. [Google Scholar] [CrossRef]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef]
- Arimitsu, S.; Bottom, R.L.; Hammond, G.B. Selective enhancement effects of silver salts on the transition metal-catalyzed synthesis of gem-difluorinated heterocyclics using 2,2-difluorohomopropargyl amides. J. Fluorine Chem. 2008, 129, 1047–1051. [Google Scholar] [CrossRef]
- Ferry, A.; Billard, T.; Langlois, B.R. Synthesis of alpha-Trifluoromethylated nitrogen bicycles. Synlett 2005, 1027–1029. [Google Scholar]
- Harthong, S.; Billard, T.; Langlois, B.R. Towards the syntheses of alpha-trifluoromethylated oxygenated heterocycles and their precursors. Synthesis 2005, 2253–2263. [Google Scholar]
- Ishizaki, M.; Suzuki, D.; Hoshino, O. Scope and limitation of intramolecular Pauson-Khand reaction of fluorine-containing enynes. J. Fluorine Chem. 2001, 111, 81–90. [Google Scholar] [CrossRef]
- Nadano, R.; Fuchibe, K.; Ikeda, M.; Takahashi, H.; Ichikawa, J. Rapid and Slow Generation of 1-Trifluoromethylvinyllithium: Syntheses and Applications of CF3-Containing Allylic Alcohols, Allylic Amines, and Vinyl Ketones. Chem. Asian J. 2010, 5, 1875–1883. [Google Scholar] [CrossRef]
- Nadano, R.; Ichikawa, J. A facile synthesis of N-[2-(trifluoromethyl)allyl]amides and their transformation into angularly trifluoromethylated bicyclic cyclopentenones. Chem. Lett. 2007, 36, 22–23. [Google Scholar] [CrossRef]
- Konno, T.; Kida, T.; Tani, A.; Ishihara, T. A novel synthesis of fluorine-containing cyclopentenones via Pauson-Khand reaction. J. Fluorine Chem. 2012, 144, 147–156. [Google Scholar] [CrossRef]
- Zhang, K.; Qiu, X.; Huang, Y.; Qing, F. An Improved Copper-Mediated Oxidative Trifluoromethylation of Terminal Alkynes. Eur. J. Org. Chem. 2012, 58–61. [Google Scholar] [CrossRef]
- Chu, L.; Qing, F. Copper-Mediated Aerobic Oxidative Trifluoromethylation of Terminal Alkynes with Me3SiCF3. J. Am. Chem. Soc. 2010, 132, 7262–7263. [Google Scholar] [CrossRef]
- Revés, M.; Lledó, A.; Ji, Y.; Blasi, E.; Riera, A.; Verdaguer, X. Tetramethylnorbornadiene, a versatile alkene for cyclopentenone synthesis through intermolecular Pauson-Khand reactions. Org. Lett. 2012, 14, 3534–3537. [Google Scholar] [CrossRef]
- Lledó, A.; Fuster, A.; Revés, M.; Verdaguer, X.; Riera, A. The Pauson-Khand reaction of medium sized trans-cycloalkenes. Chem. Commun. 2013, 49, 3055–3057. [Google Scholar] [CrossRef]
- Perez-Serrano, L.; Blanco-Urgoiti, J.; Casarrubios, L.; Dominguez, G.; Perez-Castells, J. Synthesis of Tricyclic Aromatic Compounds by the Intramolecular Pauson-Khand Reaction Promoted by Molecular Sieves. J. Org. Chem. 2000, 65, 3513–3519. [Google Scholar] [CrossRef]
- Perez-Serrano, L.; Casarrubios, L.; Dominguez, G.; Perez-Castells, J. Pauson-Khand Reaction Induced by Molecular Sieves. Org. Lett. 1999, 1, 1187–1188. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aiguabella, N.; Arce, E.M.; Del Pozo, C.; Verdaguer, X.; Riera, A. Pauson-Khand Reaction of Internal Dissymmetric Trifluoromethyl Alkynes. Influence of the Alkene on the Regioselectivity. Molecules 2014, 19, 1763-1774. https://doi.org/10.3390/molecules19021763
Aiguabella N, Arce EM, Del Pozo C, Verdaguer X, Riera A. Pauson-Khand Reaction of Internal Dissymmetric Trifluoromethyl Alkynes. Influence of the Alkene on the Regioselectivity. Molecules. 2014; 19(2):1763-1774. https://doi.org/10.3390/molecules19021763
Chicago/Turabian StyleAiguabella, Nuria, Elsa M. Arce, Carlos Del Pozo, Xavier Verdaguer, and Antoni Riera. 2014. "Pauson-Khand Reaction of Internal Dissymmetric Trifluoromethyl Alkynes. Influence of the Alkene on the Regioselectivity" Molecules 19, no. 2: 1763-1774. https://doi.org/10.3390/molecules19021763