A Tractable and Efficient One-Pot Synthesis of 5'-Azido-5'-deoxyribonucleosides

Abstract
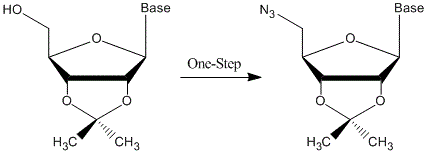
:
1. Introduction
2. Results and Discussion
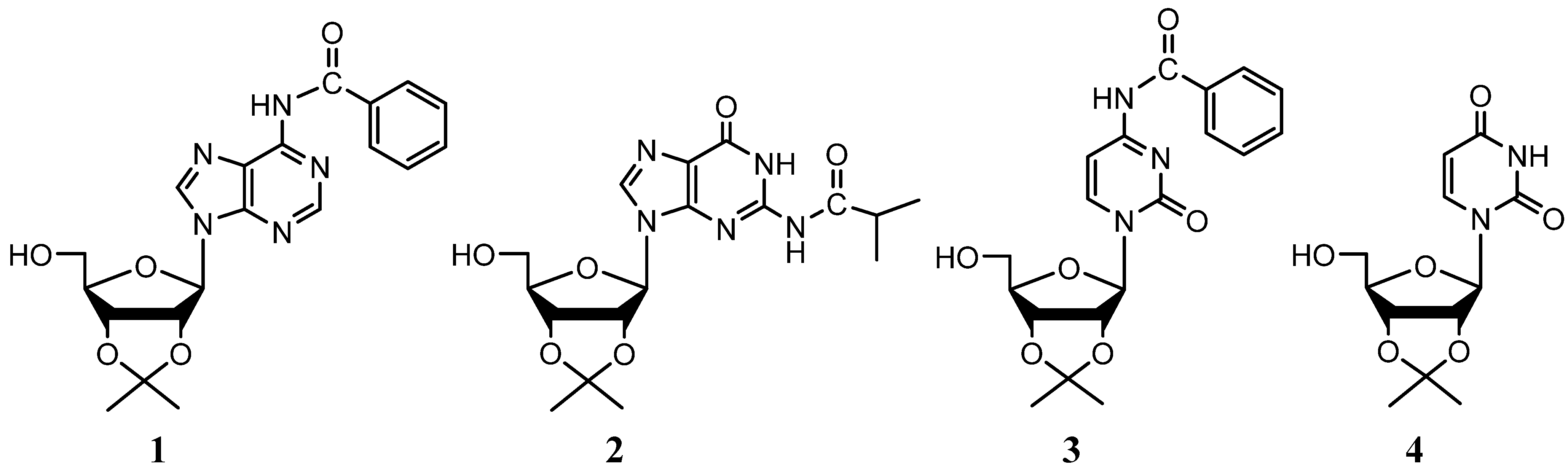
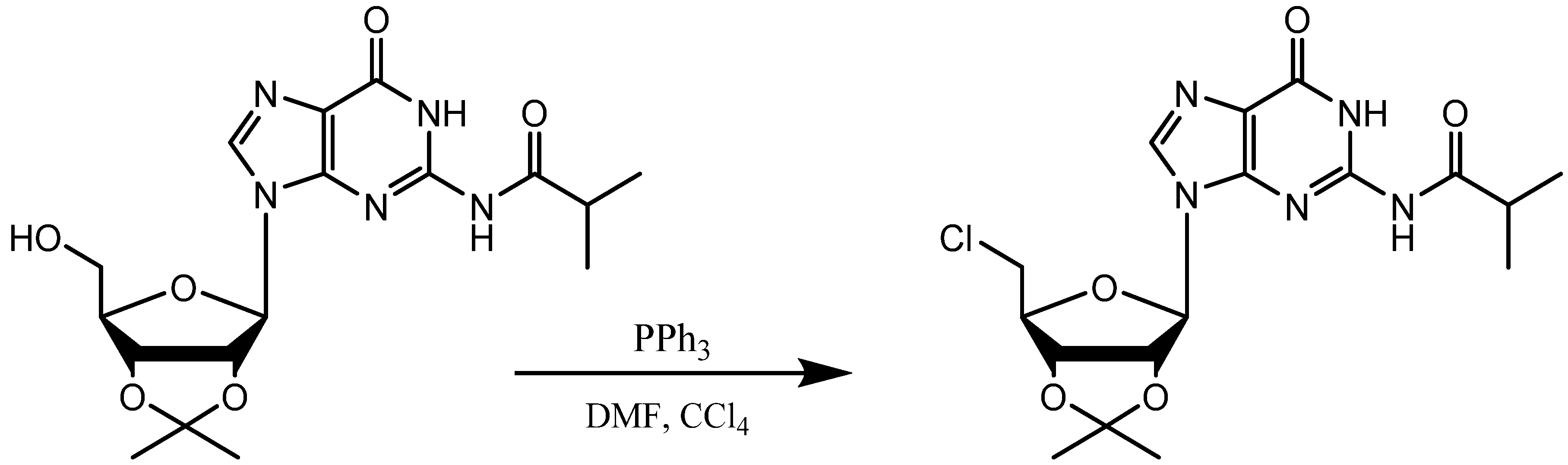
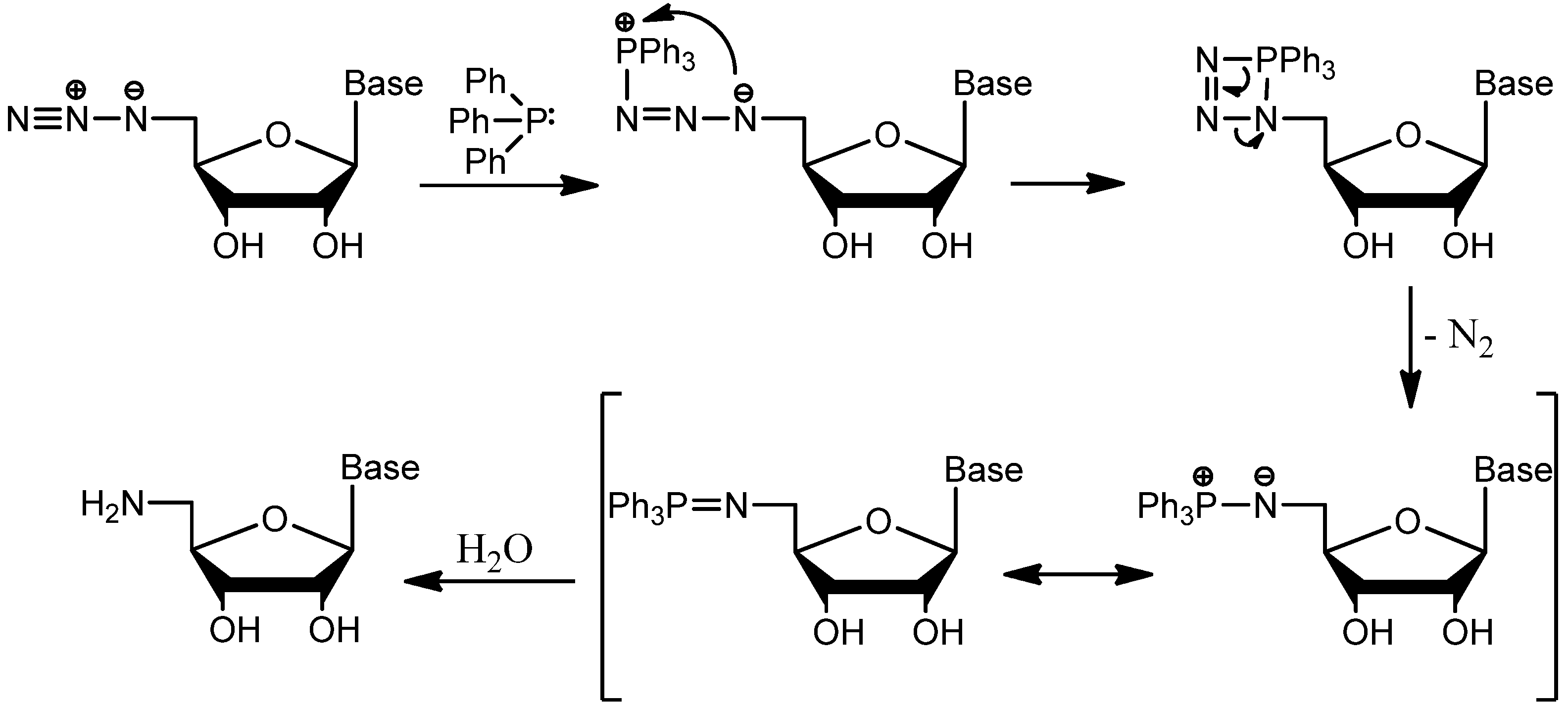

{kind=link}
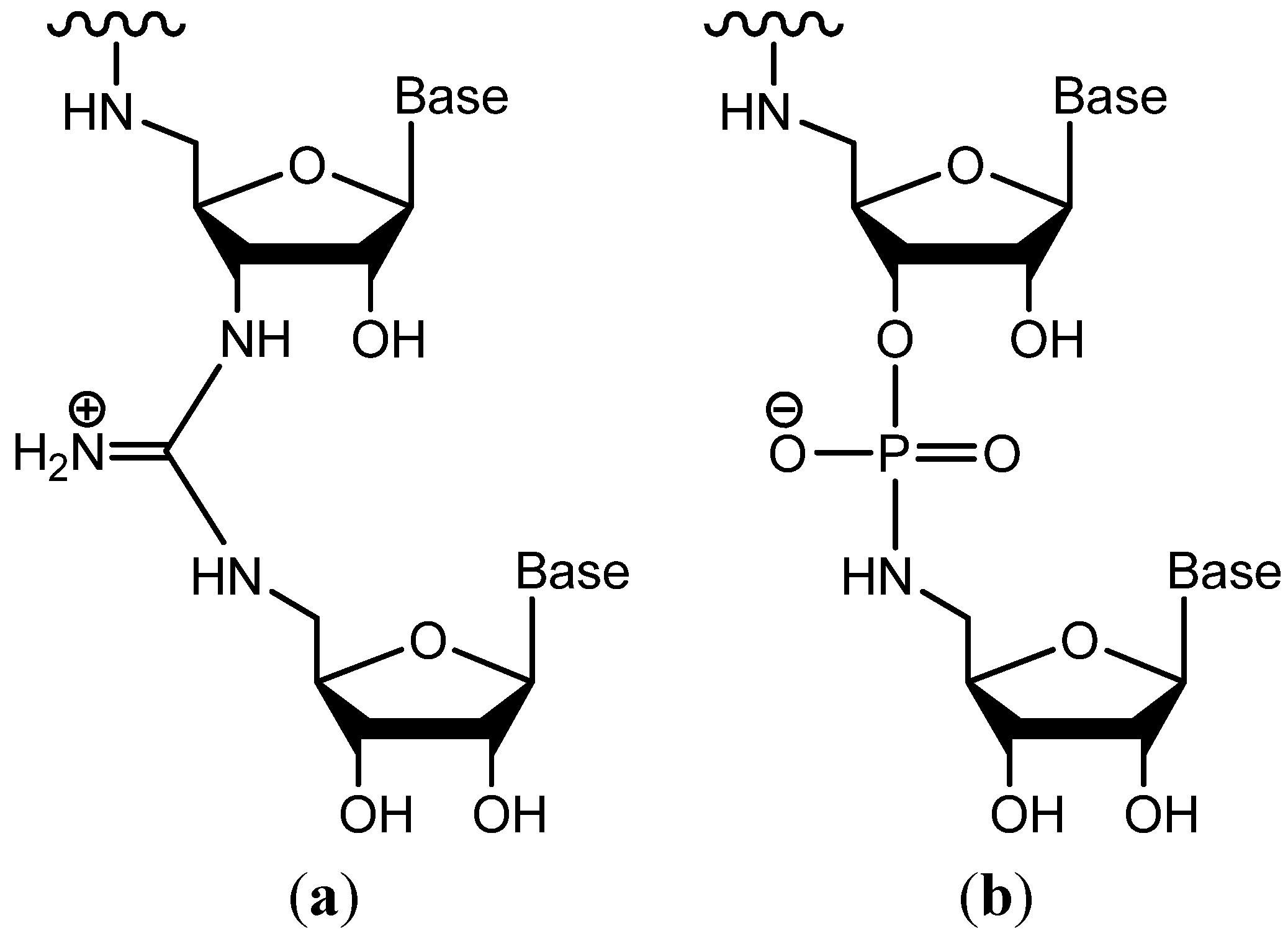
{kind=link}
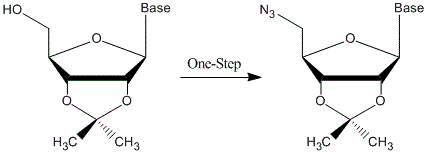
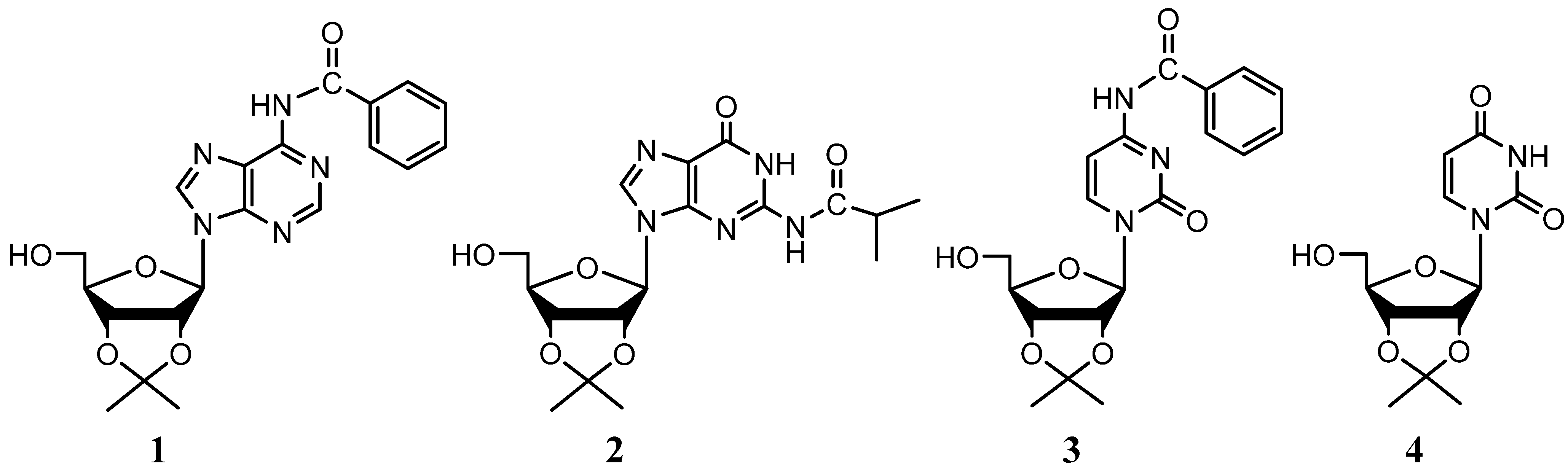{kind=link}
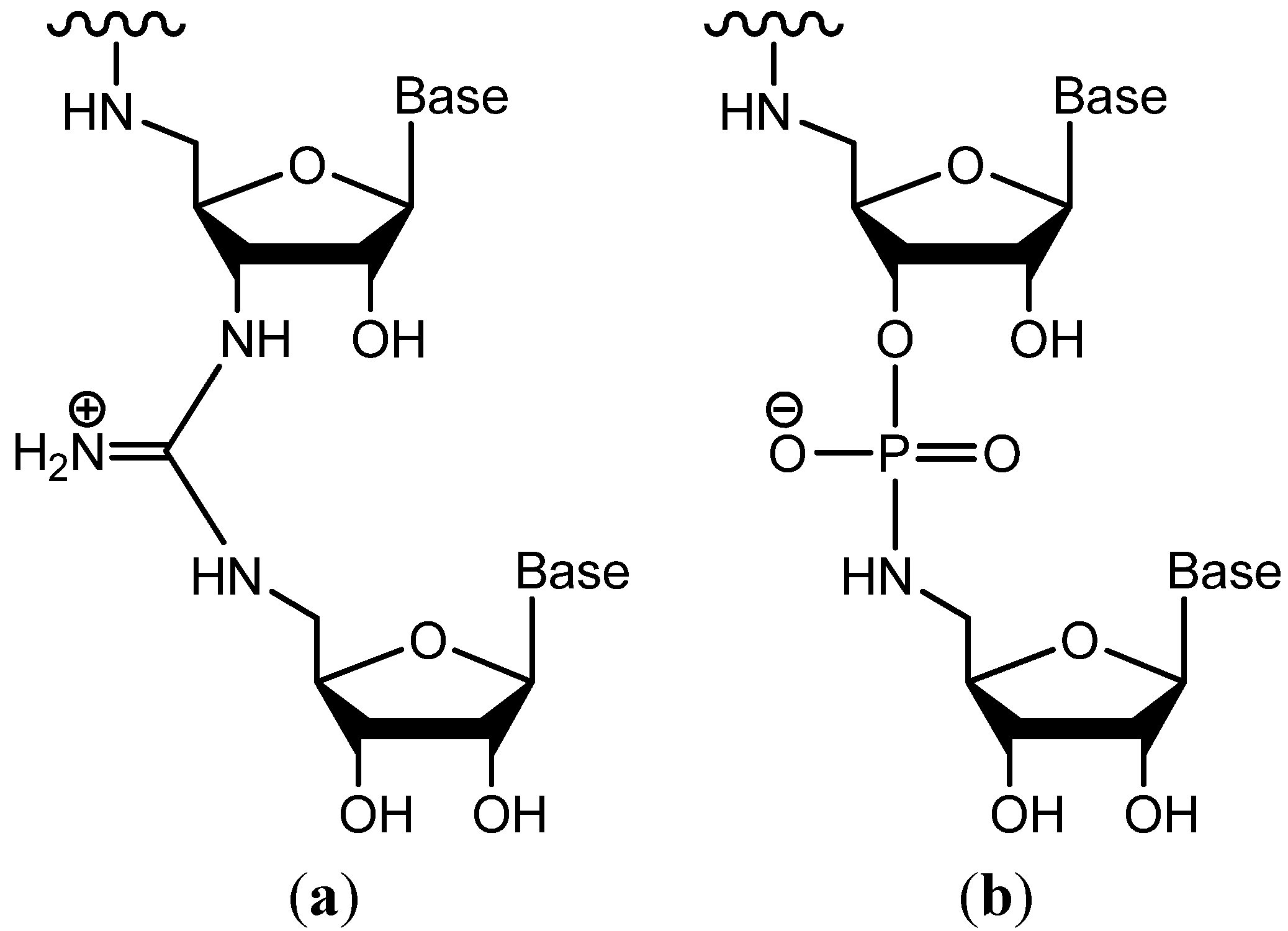{kind=link}
{kind=link}
{kind=link}
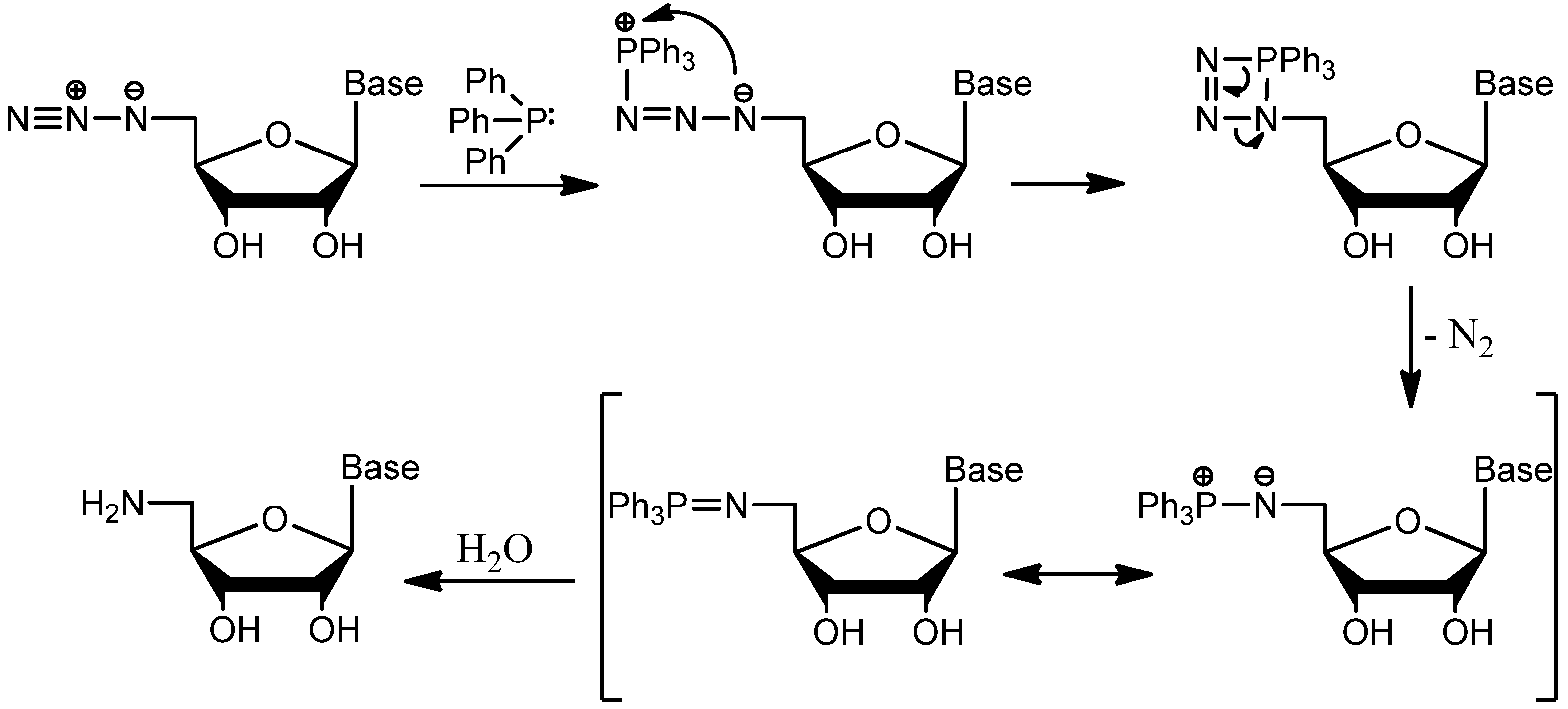{kind=link}
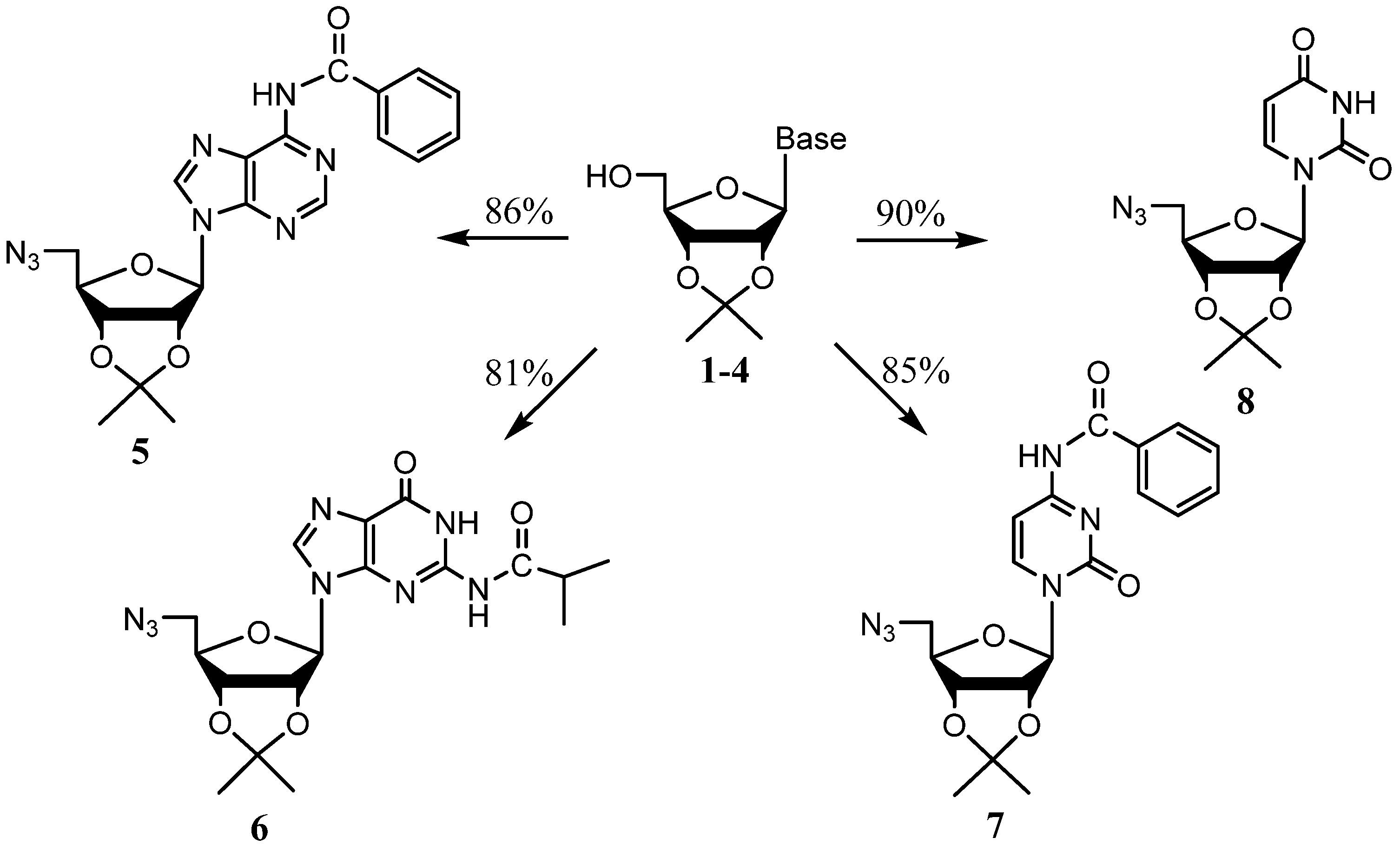
| Nucleoside | Synthetic Path | Overall yield (One-pot Yield) | Hata et al. yields (One step rxn.) |
|---|---|---|---|
| N6-Benzoyladenosine | 1→5 | 74.0% (86%) | 56% |
| N2-Isobutyrylguanosine | 2→6 | 68.0% (81%) | N/A |
| N4-Benzoylcytidine | 3→7 | 77.3% (85%) | 45% |
| Uridine | 4→8 | 79.2% (90%) | 92% |
3. Experimental
General Procedures
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vater, A.; Sell, S.; Kaczmarek, P.; Maasch, C.; Buchner, K.; Pruszynska-Oszmalek, E.; Kolodziejski, P.; Purschke, W.G.; Nowak, K.W.; Strowski, M.Z.; et al. A mixed mirror-image DNA/RNA aptamer inhibits glucagon and acutely improves glucose tolerance in models of type 1 and type 2 diabetes. J. Biol. Chem. 2013, 288, 21136–21147. [Google Scholar] [CrossRef]
- Duan, N.; Ding, X.; Wu, S.; Xia, Y.; Ma, X.; Wang, Z.; Chen, J. In vitro selection of a DNA aptamer targeted against Shigella dysenteriae. J. Microbiol. Methods 2013, 94, 170–174. [Google Scholar] [CrossRef]
- Pushpendra, S.; Arvind, P.; Anil, B. Nucleic Acids as Therapeutics. In From Nucleic Acids Sequences to Molecular Medicine; Erdmann, V.A., Barciszewski, J., Eds.; Springer: New York, NY, USA, 2012. [Google Scholar]
- Alvarez-Salas, L.M. Nucleic acids as therapeutic agents. Curr. Top. Med. Chem. 2008, 8, 1379–1404. [Google Scholar] [CrossRef]
- Jayasena, S.D. Aptamers: An emerging class of molecules that rival antibodies in diagnostics. Clin. Chem. 1999, 45, 1628–1650. [Google Scholar]
- Mei, H.; Bing, T.; Yang, X.; Qi, C.; Chang, T.; Liu, X.; Cao, Z.; Shangguan, D. Functional-group specific aptamers indirectly recognizing compounds with alkyl amino group. Anal. Chem. 2012, 84, 7323–7329. [Google Scholar]
- Tiley, L.S.; Malim, M.H.; Tewary, H.K.; Stockley, P.G.; Cullen, B.R. Identification of a high-affinity RNA-binding site for the human immunodeficiency virus type 1 Rev protein. Proc. Natl. Acad. Sci. USA 1992, 89, 758–762. [Google Scholar] [CrossRef]
- Germer, K.; Leonard, M.; Zhang, X. RNA aptamers and their therapeutic and diagnostic applications. Int. J. Biochem. Mol. Biol. 2013, 4, 27–40. [Google Scholar]
- Wang, K.; Wu, L.; Qui, Z.; Yan, X.; Li, X.; Chen, H.; Zhang, P.; Zhang, J. Synthesis and antitumor activity of novel ribonucleosides with C-5 OH replaced by a diaminopyrimidinyl group. Bioorg. Med. Chem. Lett. 2011, 21, 916–919. [Google Scholar] [CrossRef]
- Wang, T.; Lee, H.J.; Tosh, D.K.; Kim, H.O.; Pal, S.; Choi, S.; Lee, Y.; Moon, H.R.; Zhao, L.X.; Lee, K.M.; et al. Design, synthesis, and molecular modeling studies of 5'-deoxy-5'-ureidoadenosine: 5'-Ureido group as multiple hydrogen bonding donor in the active site of S-adenosylhomocysteine hydrolase. Bioorg. Med. Chem. Lett. 2007, 17, 4456–4459. [Google Scholar] [CrossRef]
- Wolfe, J.L.; Kawate, T.; Belenky, A.; Stanton, V., Jr. Synthesis and polymerase incorporation of 5'-amino-2',5'-dideoxy-5'-N-triphosphate nucleotides. Nucleic Acids Res. 2002, 30, 3739–3747. [Google Scholar] [CrossRef]
- Pathak, T. Azidonucleosides: Synthesis, reactions, and biological properties. Chem. Rev. 2002, 102, 1623–1667. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef]
- Hong, V.; Steinmetz, N.F.; Manchester, M.; Finn, M.G. Labeling live cells by copper-catalyzed alkyne—Azide click chemistry. Bioconjug. Chem. 2010, 21, 1912–1916. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Edit. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef]
- Avti, P.; Maysinger, D.; Kakkar, A. Alkyne-azide “click” chemistry in designing nanocarriers for applications in biology. Molecules 2013, 18, 9531–9549. [Google Scholar] [CrossRef]
- Van Berkel, S.S.; van Eldijk, M.B.; van Hest, J.C.M. Staudinger ligation as a method for bioconjugation. Angew. Chem. Int. Edit. 2011, 50, 8806–8827. [Google Scholar] [CrossRef]
- Köhn, M.; Breinbauer, R. The staudinger ligation—A gift to chemical biology. Angew. Chem. Int. Edit. 2004, 43, 3106–3116. [Google Scholar] [CrossRef]
- Kalia, J.; Abbott, N.L.; Raines, R.T. General method for site-specific protein immobilization by staudinger ligation. Bioconjug. Chem. 2007, 18, 1064–1069. [Google Scholar] [CrossRef]
- Saxon, E.; Armstrong, J.I.; Bertozzi, C.R. A “traceless” staudinger ligation for the chemoselective synthesis of amide bonds. Org. Lett. 2000, 2, 2141–2143. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Brase, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic azides: An exploding diversity of a unique class of compounds. Angew. Chem. Int. Edit. 2005, 44, 5188–5240. [Google Scholar] [CrossRef]
- Scriven, E.F.V.; Turnbull, K. Azides: Their preparation and synthetic uses. Chem. Rev. 1988, 88, 297–368. [Google Scholar] [CrossRef]
- Bae, I.; Han, H.; Chang, S. Highly efficient one-pot synthesis of N-sulfonylamidines by Cu-catalyzed three-component coupling of sulfonyl azide, alkyne, and amine. J. Am. Chem. Soc. 2005, 127, 2038–2039. [Google Scholar] [CrossRef]
- Kitamura, M.; Koga, T.; Yano, M.; Okauchi, T. Direct synthesis of organic azides from alcohols using 2-azido-1,3-dimethylimidazolinium hexafluorophosphate. Synlett 2012, 23, 1335–1338. [Google Scholar] [CrossRef]
- Dobosh, B.J. The Synthesis of Azides from Alcohols Using Sulfonyl Azides. Masters Thesis, Youngstown State University, Youngstown, OH, USA, August 2008. [Google Scholar]
- Rokhum, L.; Bez, G. A practical one-pot synthesis of azides directly from alcohols. J. Chem. Sci. 2012, 124, 687–691. [Google Scholar] [CrossRef]
- Hata, T.; Yamamoto, I.; Sekine, M. A simple method for the synthesis of 5'-azido-5'-deoxyribonucleosides. Chem. Lett. 1975, 977–980. [Google Scholar] [CrossRef]
- Reddy, G.V.S.; Rao, G.V.; Subramanyam, R.V.K.; Iyengar, D.S. A new novel and practical one pot methodology for conversion of alcohols to amines. Synth. Commun. 2000, 301, 2233–2237. [Google Scholar]
- Hamid, M.H.S.A. Conversion of Alcohols into Amines by Borrowing Hydrogen. Ph.D. Thesis, University of Bath, Bath, UK, November 2008. [Google Scholar]
- Patil, D.D.; Mhaske, D.K.; Wadhawa, G.C.; Patare, M.A. One pot conversion of alcohol to amine using sodium azide with zinc and zinc chloride. J. Pharm. Res. Opin. 2011, 1, 170–171. [Google Scholar]
- Hata, T.; Yamamoto, I.; Sekine, M. A new method for the synthesis of 5'-amino-nucleosides and their phosphoramidate derivatives. Chem. Lett. 1976, 601–604. [Google Scholar] [CrossRef]
- Suresh, S. Synthesis of Protected 5'-Aminouridine for Modification of Solid-Support in Synthesis of Modified siRNA. Masters Thesis, Northeastern University, Boston, MA, USA, June 2008. [Google Scholar]
- Kierzek, R.; Li, Y.; Turner, D.H.; Bevilacqua, P.C. 5'-Amino pyrene provides a sensitive, nonperturbing florescent probe of RNA secondary and tertiary structure formation. J. Am. Chem. Soc. 1993, 115, 4985–4992. [Google Scholar] [CrossRef]
- Yamamoto, I.; Sekine, M.; Hata, T. One-step synthesis of 5'-azido-nucleosides. J. Chem. Soc. Perkin Trans. 1980, 1, 306–310. [Google Scholar] [CrossRef]
- Awad, A.M.; Collazo, M.J.; Carpio, K.; Flores, C.; Bruice, T.C. A convenient synthesis of the cytidyl 3'-terminal monomer for solid-phase synthesis of RNG oligonucleotides. Tetrahedron Lett. 2012, 53, 3792–3794. [Google Scholar] [CrossRef]
- Samuels, E.R.; McNary, J.; Aguilar, M.; Awad, A.M. Effective Synthesis of 3'-deoxy-3'-azido nucleosides for antiviral and antisense ribonucleic guanidine (RNG) applications. Nucleos. Nucleot. Nucl. 2013, 32, 109–123. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Peterson, T.V.; Streamland, T.U.B.; Awad, A.M. A Tractable and Efficient One-Pot Synthesis of 5'-Azido-5'-deoxyribonucleosides. Molecules 2014, 19, 2434-2444. https://doi.org/10.3390/molecules19022434
Peterson TV, Streamland TUB, Awad AM. A Tractable and Efficient One-Pot Synthesis of 5'-Azido-5'-deoxyribonucleosides. Molecules. 2014; 19(2):2434-2444. https://doi.org/10.3390/molecules19022434
Chicago/Turabian StylePeterson, Theodore V., Tobin U. B. Streamland, and Ahmed M. Awad. 2014. "A Tractable and Efficient One-Pot Synthesis of 5'-Azido-5'-deoxyribonucleosides" Molecules 19, no. 2: 2434-2444. https://doi.org/10.3390/molecules19022434