Inhibition of GlcNAc-Processing Glycosidases by C-6-Azido-NAG-Thiazoline and Its Derivatives

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
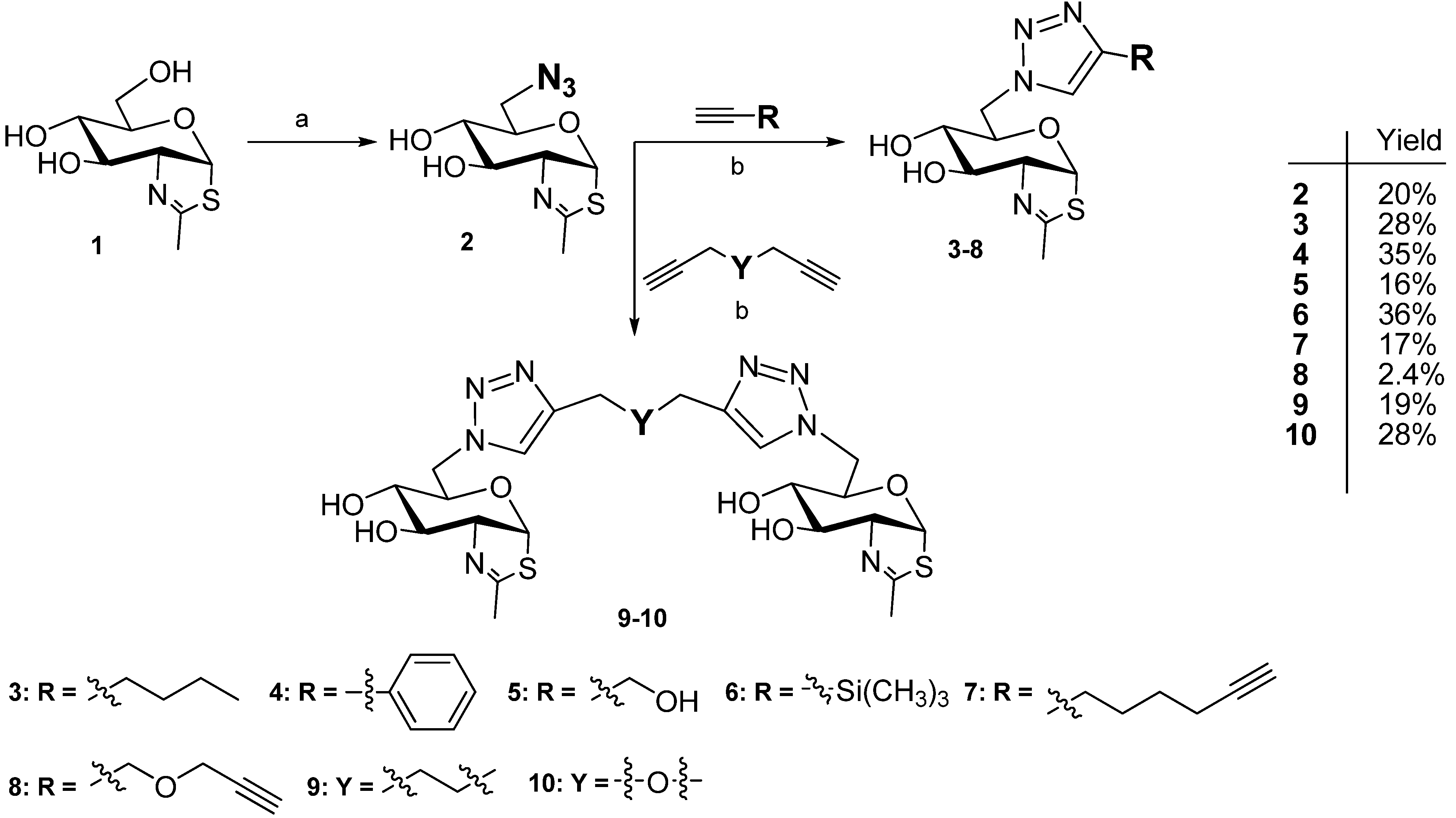
2.1. Synthesis of NAG-Thiazoline Derivatives 2–10

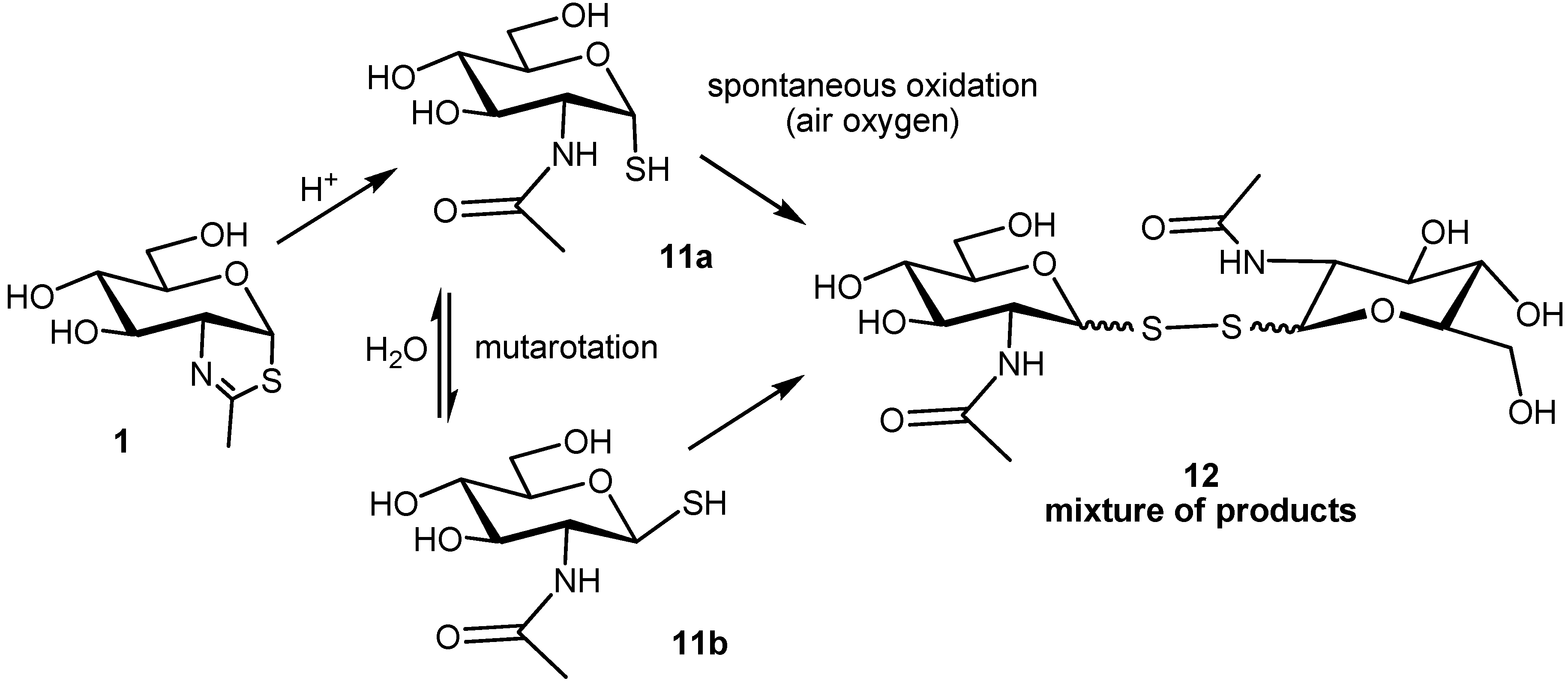
2.2. Stability of the NAG-Thiazoline Parent Structure 1

2.3. Inhibiton of GH20 and GH84 Glycosidases with NAG-Thiazoline Derivatives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KI [µM] | KI [µM] | ||
|---|---|---|---|
| Enzyme | NAG-thiazoline (1) | C-6-azido-NAG-thiazoline (2) | |
| β-N-acetylhexosaminidase | T. flavus | 42.7 ± 1.9 | 211.9 ± 16.3 |
| S. plicatus | 24 ± 5 | n.d. | |
| O-GlcNAcase | B. thetaiotaomicron | 0.029 ± 0.003 | 3.1 ± 0.3 |
| human | 0.18 ± 0.03 | 19.3 ± 11.9 | |
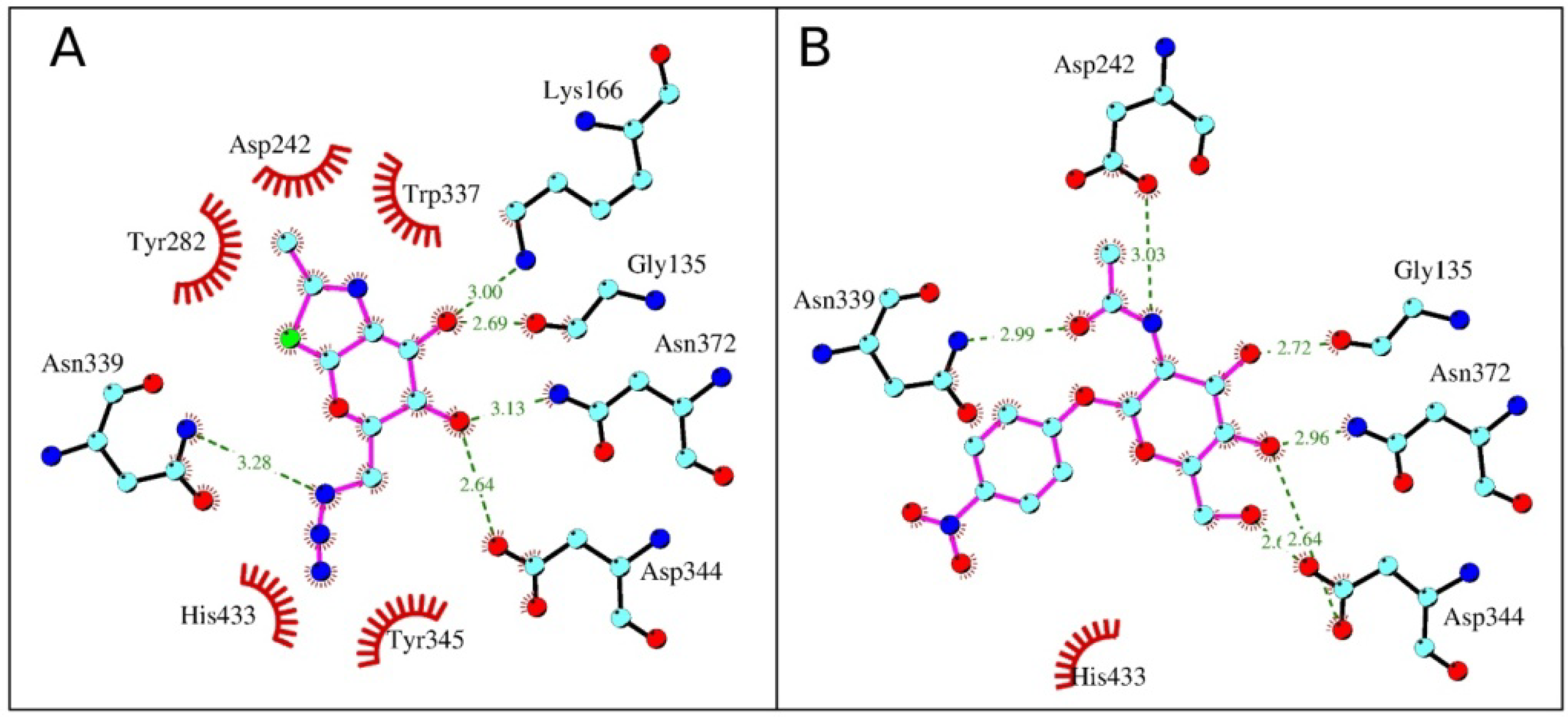
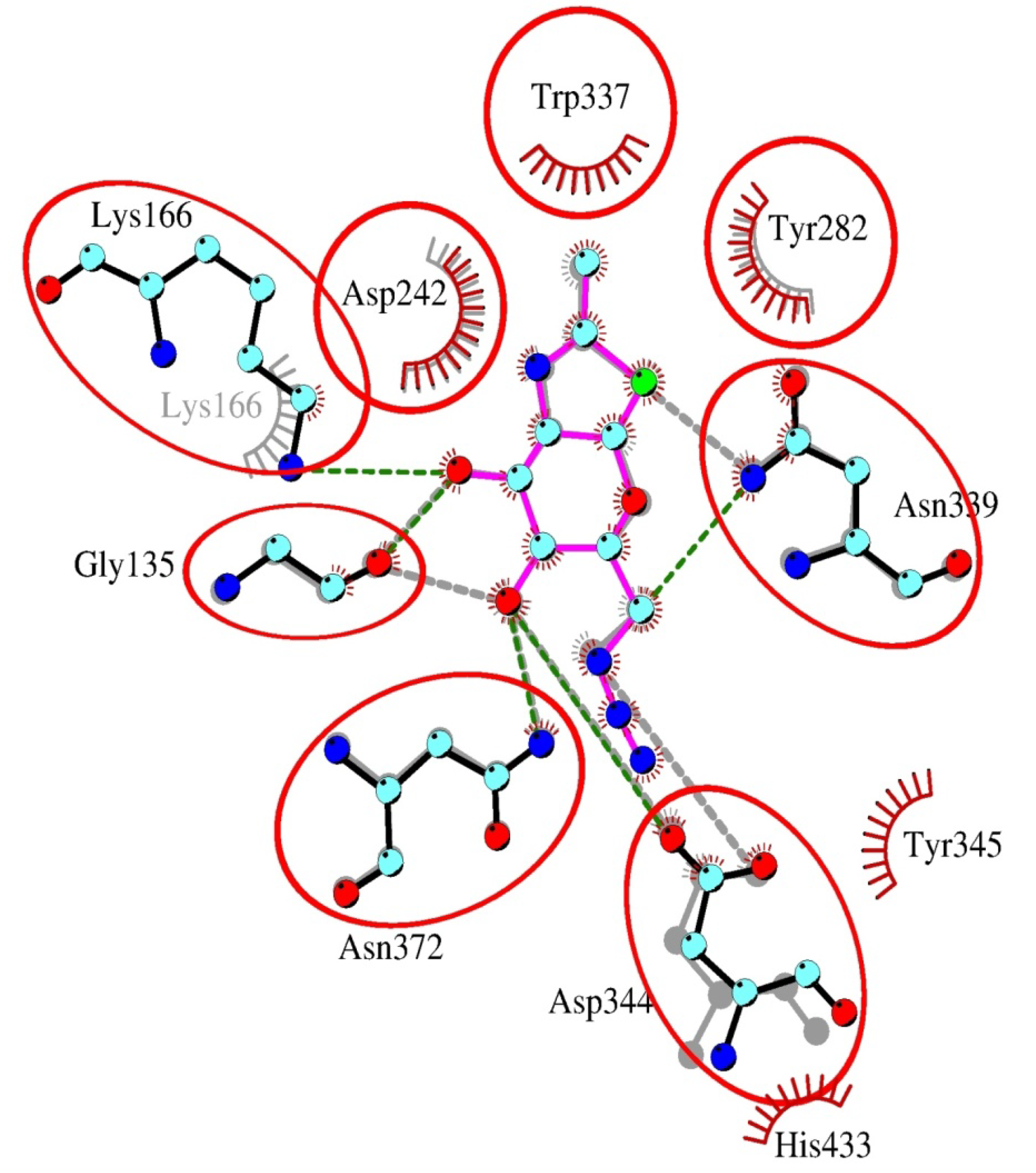
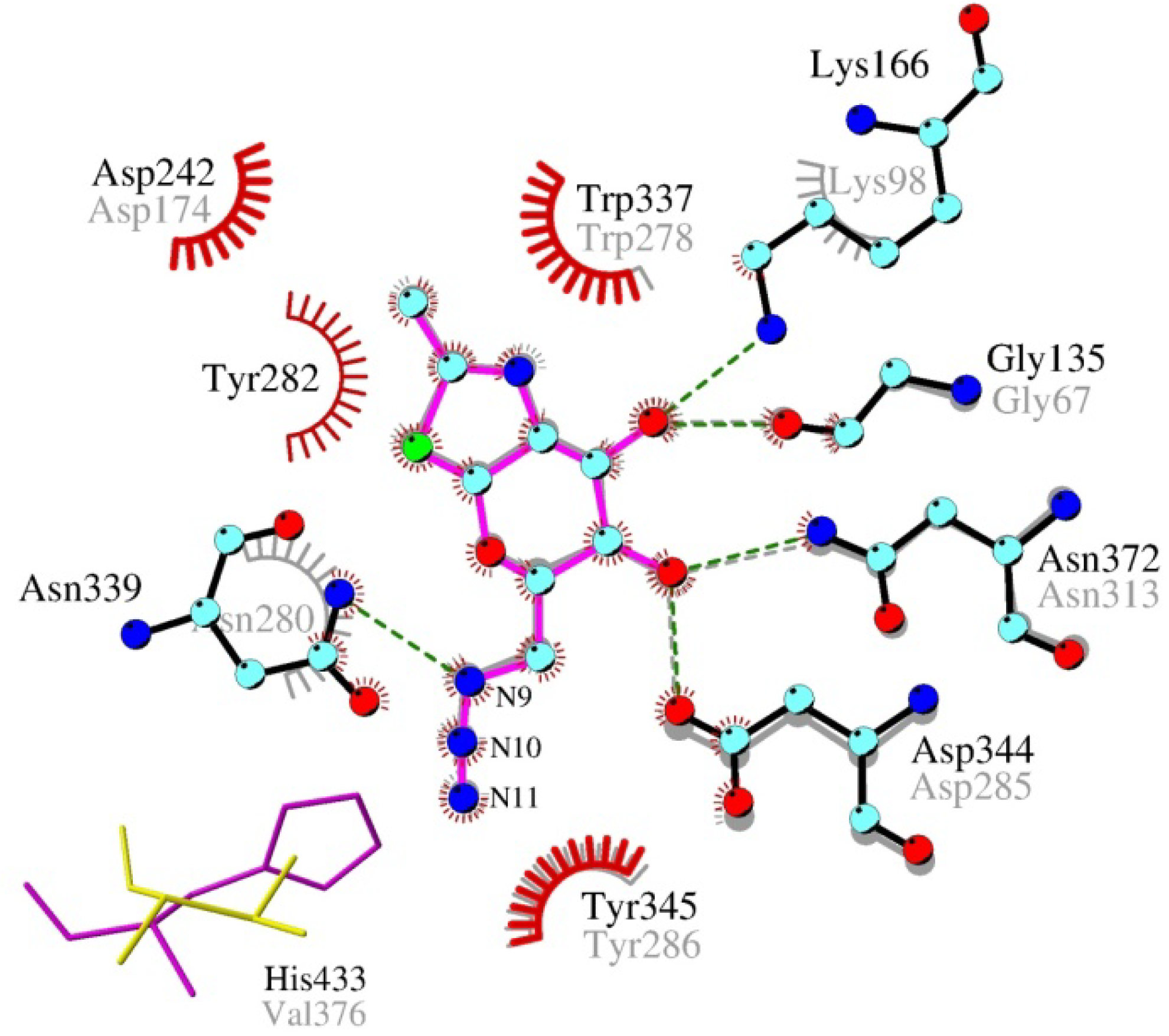
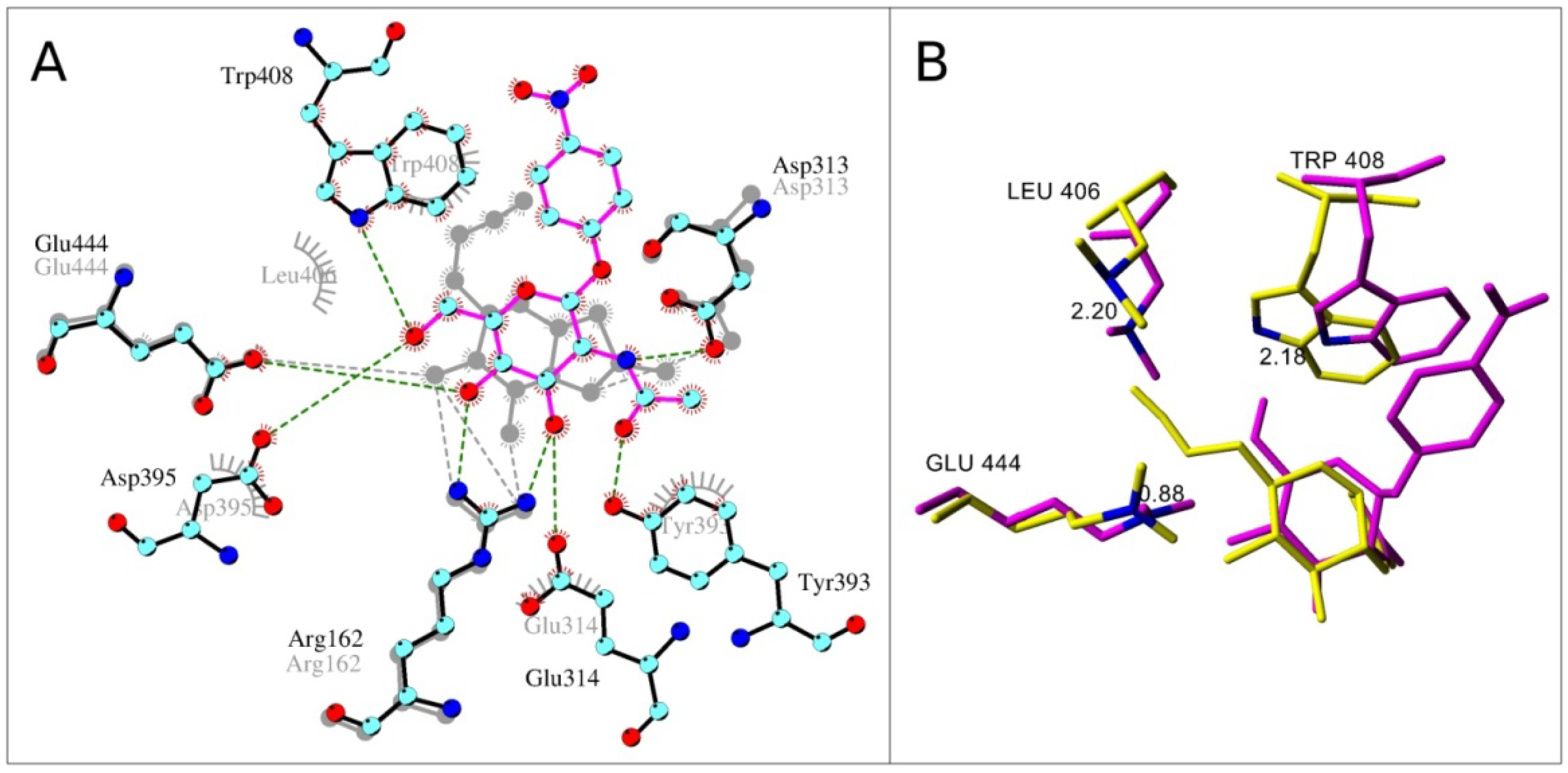
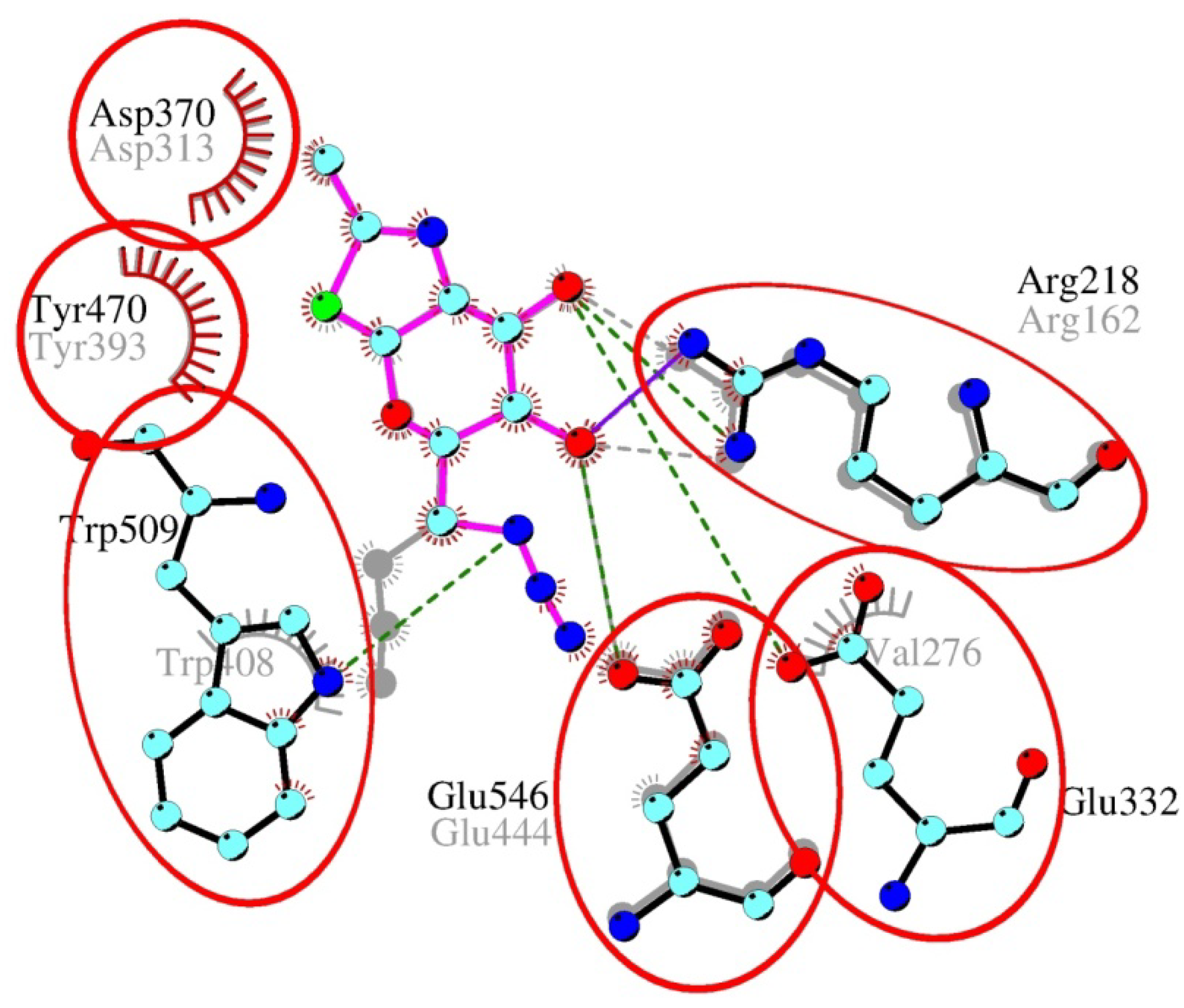
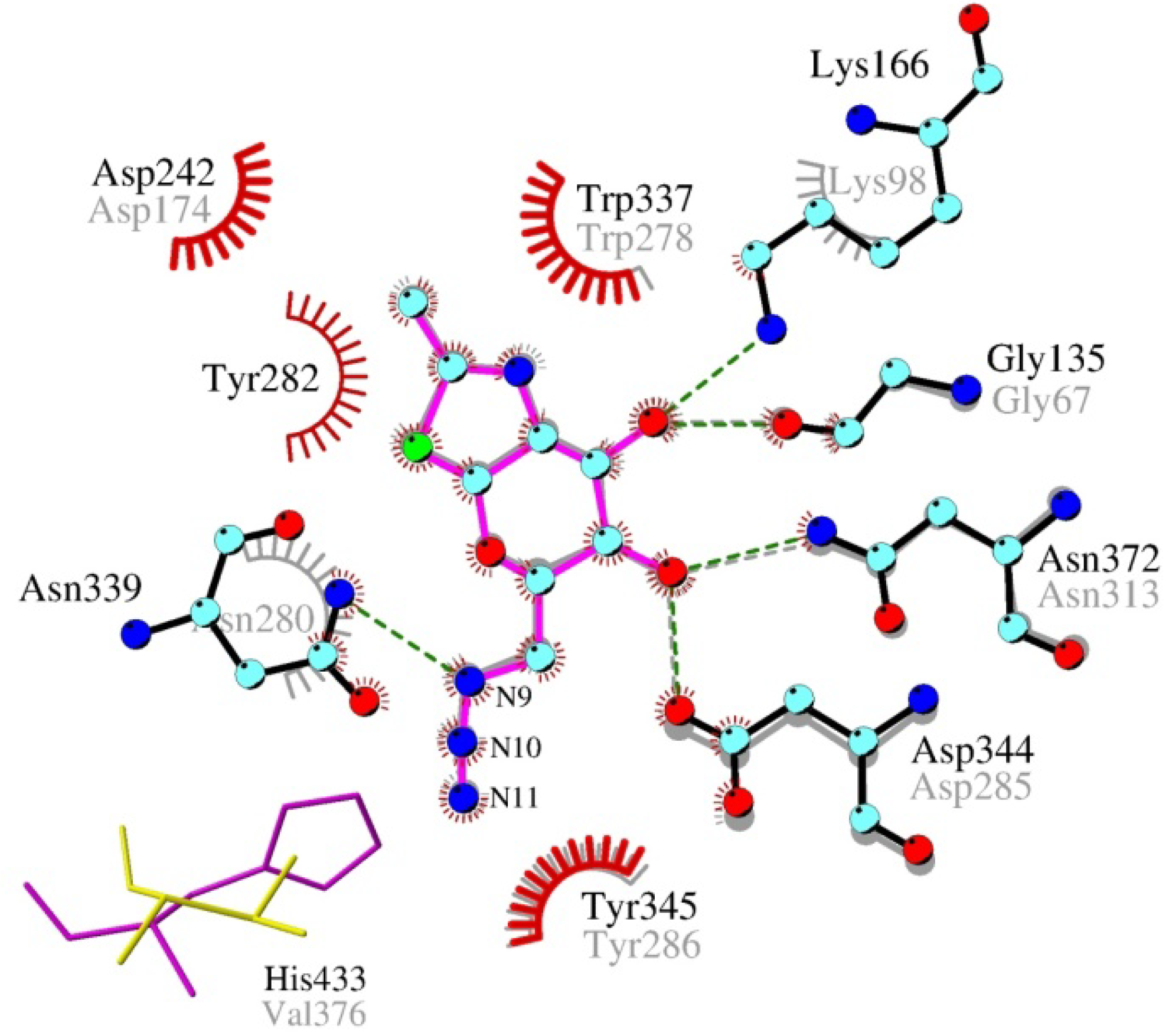
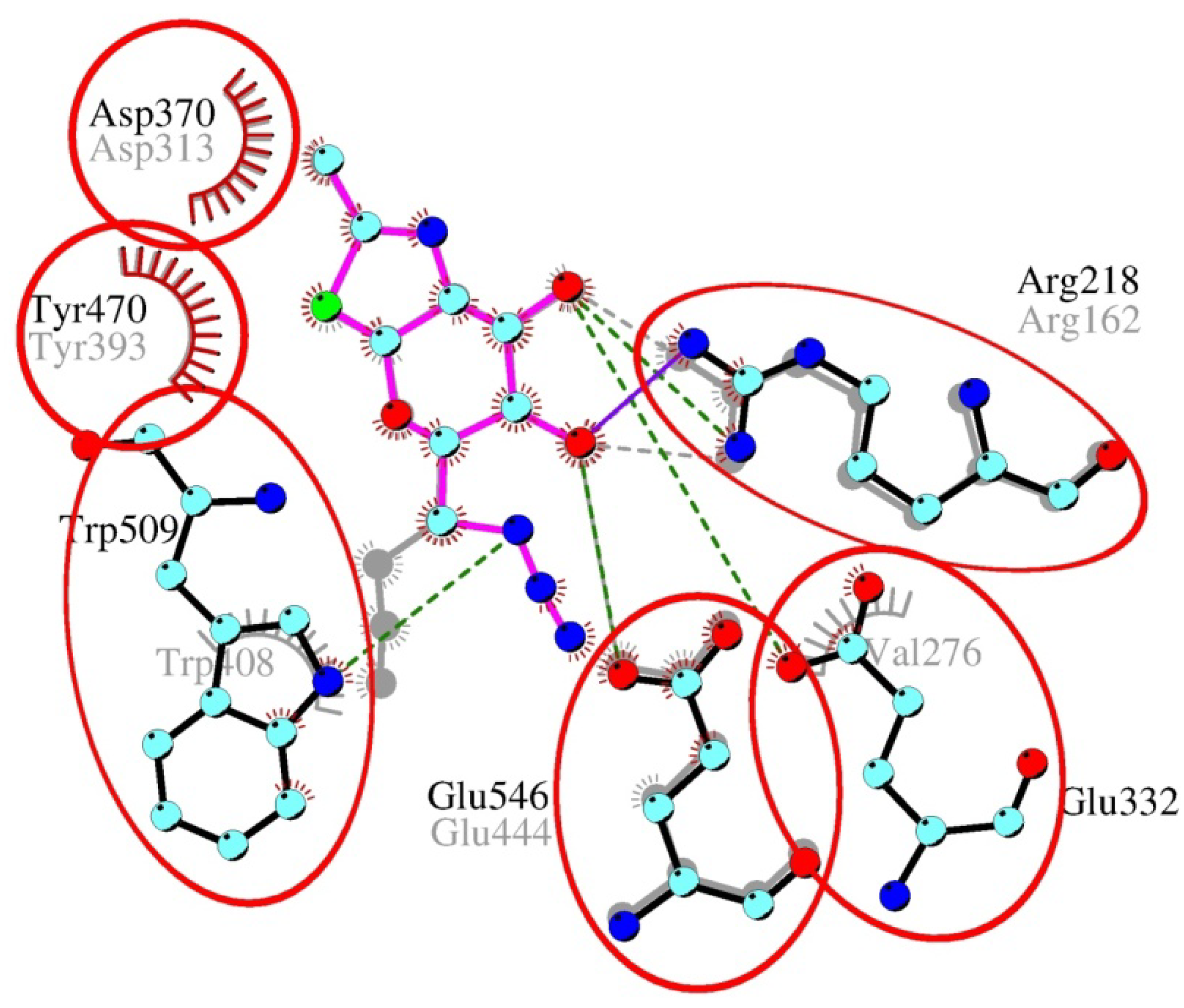
2.4. Interactions and Calculated Binding Energies of C-6-azido-NAG-thiazoline (2) in The Active Site of Enzymes
| Enzyme | NAG-thiazoline (1) [kcal/mol] | C-6-azido-NAG-thiazoline (2) [kcal/mol] | pNP-β-GlcNAc [kcal/mol] | |
|---|---|---|---|---|
| β-N-acetylhexosaminidase | S. plicatus | −9.22 (6−7) | −7.36 (3−5) | −9.48 (5−8) |
| T. flavus | −7.63 (3−6) | −7.71 (3−4) | −10.19 (6−9) | |
| O-GlcNAcase | B. thetaiotaomicron | −7.14 (4−5) | −8.65 (4−5) | −7.75 (6−7) |
| human | −6.42 (3−4) | −7.16 (3−4) | −7.45 (5−6) | |



3. Experimental
3.1. General Methods
3.2. NMR Spectroscopy
3.3. Mass Spectrometry
3.4. Synthesis of NAG-Thiazoline Derivatives 2–10
3.5. Stability of NAG-Thiazoline 1
3.6. Analysis of Decomposition Products of NAG-Thiazoline 1
3.7. Enzymes
3.8. β-N-Acetylhexosaminidase and O-GlcNAcase Activity and Inhibition Assays
3.9. Molecular Modeling and Docking
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflictts of Interest
References
- Knapp, S.; Vocadlo, D.; Gao, Z.; Kirk, B.; Lou, J.; Withers, S.G. NAG-Thiazoline, an N-acetyl-β-hexosaminidase inhibitor that implicates acetamido participation. J. Am. Chem. Soc. 1996, 118, 6804–6805. [Google Scholar] [CrossRef]
- Slámová, K.; Bojarová, P.; Petrásková, L.; Křen, V. β-N-Acetylhexosaminidase: What’s in a name…? Biotechnol. Adv. 2010, 28, 682–693. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; He, Y.; Whitworth, G.E.; Stubbs, K.A.; McEachern, E.J.; Davies, G.J.; et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008, 4, 483–490. [Google Scholar] [CrossRef]
- Mahuran, D.J. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim. Biophys. Acta 1999, 1455, 105–138. [Google Scholar]
- Macauley, M.S.; Whitworth, G.E.; Debowski, A.W.; Chin, D.; Vocadlo, D.J. O-GlcNAcase uses substrate-assisted catalysis: Kinetic analysis and development of highly selective mechanism-inspired inhibitors. J. Biol. Chem. 2005, 280, 25313–25322. [Google Scholar] [CrossRef]
- Knapp, S.; Abdo, M.; Ajayi, K.; Huhn, R.A.; Emge, T.J.; Kim, E.J.; Hanover, J.A. Tautomeric modification of GlcNAc-thiazoline. Org. Lett. 2007, 9, 2321–2324. [Google Scholar] [CrossRef]
- Amorelli, B.; Yang, C.; Rempel, B.; Withers, S.G.; Knapp, S. N-Acetylhexosaminidase inhibitory properties of C-1 homologated GlcNAc- and GalNAc-thiazolines. Bioorg. Med. Chem. Lett. 2008, 18, 2944–2947. [Google Scholar] [CrossRef]
- Krejzová, J.; Šimon, P.; Vavříková, E.; Slámová, K.; Pelantová, H.; Riva, S.; Spiwok, V.; Křen, V. Enzymatic synthesis of new C-6-acylated derivatives of NAG-thiazoline and evaluation of their inhibitor activities towards fungal β-N-acetylhexosaminidase. J. Mol. Catal. B-Enzym. 2013, 87, 128–134. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Shan, X.; Maculey, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. J. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef]
- Vocadlo, D.; McEachern, E.W.O. Selective Glycosidase Inhibitors and Uses Thereof. Patent 2008/025170 A1, 6 March 2008. [Google Scholar]
- Fialová, P.; Weignerová, L.; Rauvolfová, J.; Přikrylová, V.; Pišvejcová, A.; Ettrich, R.; Kuzma, M.; Sedmera, P.; Křen, V. Hydrolytic and transglycosylation reactions of N-acyl modified substrates catalysed by β-N-acetylhexosaminidases. Tetrahedron 2004, 60, 693–701. [Google Scholar] [CrossRef]
- Slámová, K.; Bojarová, P.; Gerstorferová, D.; Fliedrová, B.; Hofmeisterová, J.; Fiala, M.; Pompach, P.; Křen, V. Sequencing, cloning and high-yield expression of a fungal β-N-acetylhexosaminidase in Pichia pastoris. Protein Expr. Purif. 2012, 82, 212–217. [Google Scholar] [CrossRef]
- Mark, B.L.; Vocadlo, D.J.; Knapp, S.; Triggs-Raine, B.L.; Withers, S.G.; James, M.N.G. Crystallographic evidence for substrate-assisted catalysis in a bacterial β-hexosaminidase. J. Biol. Chem. 2001, 276, 10330–10337. [Google Scholar]
- Gaussian 03, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2004.
- Wang, J.; Wang, W.; Kollmann, P.; Case, D. Antechamber, an accessory software package for molecular mechanical calculation. J. Comput. Chem. 2005, 25, 1157–1174. [Google Scholar]
- Tews, I.; Perrakis, A.; Oppenheim, A.; Dauter, Z.; Wilson, K.S.; Vorgias, C.E. Bacterial chitobiase structure provides insight into catalytic mechanism and the basis of Tay-Sachs disease. Nat. Struct. Biol. 1996, 3, 638–648. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Dennis, R.J.; Taylor, E.J.; Macauley, M.S.; Stubbs, K.A.; Turkenburg, J.P.; Hart, S.J.; Black, G.N.; Vocadlo, D.J.; Davies, G.J. Structure and mechanism of a bacterial β-glucosaminidase having O-GlcNAcase activity. Nat. Struct. Mol. Biol. 2006, 13, 365–371. [Google Scholar] [CrossRef]
- Slámová, K.; Kulik, N.; Fiala, M.; Krejzová-Hofmeisterová, J.; Ettrich, R.; Křen, V. Expression, characterization and homology modelling of a novel eukaryotic GH84 β-N-acetylglucosaminidase fom Penicillium chrysogenum. Protein Expr. Purif. 2014, 95, 204–210. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Essman, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1 and 2 are available from the authors at the Institute of Microbilology, Prague, Czech Republic.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krejzová, J.; Šimon, P.; Kalachova, L.; Kulik, N.; Bojarová, P.; Marhol, P.; Pelantová, H.; Cvačka, J.; Ettrich, R.; Slámová, K.; et al. Inhibition of GlcNAc-Processing Glycosidases by C-6-Azido-NAG-Thiazoline and Its Derivatives. Molecules 2014, 19, 3471-3488. https://doi.org/10.3390/molecules19033471
Krejzová J, Šimon P, Kalachova L, Kulik N, Bojarová P, Marhol P, Pelantová H, Cvačka J, Ettrich R, Slámová K, et al. Inhibition of GlcNAc-Processing Glycosidases by C-6-Azido-NAG-Thiazoline and Its Derivatives. Molecules. 2014; 19(3):3471-3488. https://doi.org/10.3390/molecules19033471
Chicago/Turabian StyleKrejzová, Jana, Petr Šimon, Lubica Kalachova, Natallia Kulik, Pavla Bojarová, Petr Marhol, Helena Pelantová, Josef Cvačka, Rüdiger Ettrich, Kristýna Slámová, and et al. 2014. "Inhibition of GlcNAc-Processing Glycosidases by C-6-Azido-NAG-Thiazoline and Its Derivatives" Molecules 19, no. 3: 3471-3488. https://doi.org/10.3390/molecules19033471