Synthesis of Caffeic Acid Amides Bearing 2,3,4,5-Tetra-hydrobenzo[b][1,4]dioxocine Moieties and Their Biological Evaluation as Antitumor Agents

Abstract
:
1. Introduction

2. Results and Discussion
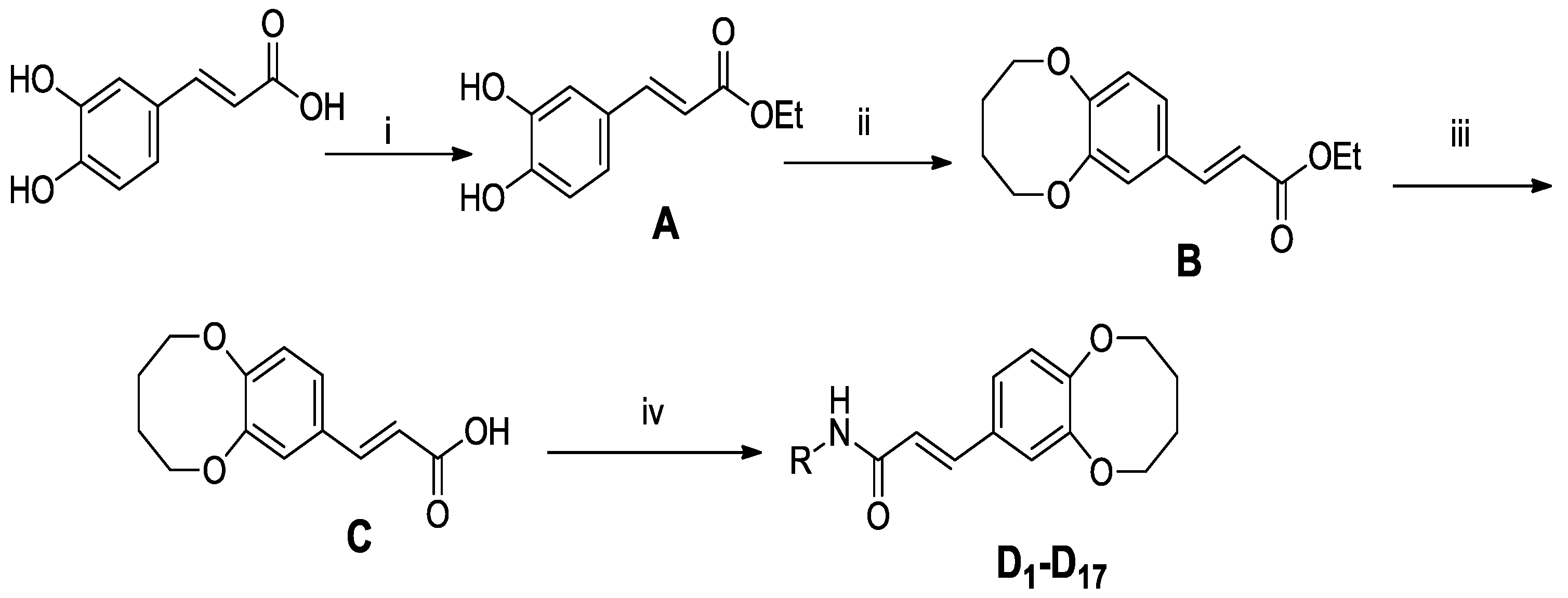
2.1. Chemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal parameters | Compound D9 |
|---|---|
| Empirical formula | C21H23NO4 |
| Molecular weight | 353.41 |
| Crystalsize (mm3) | 0.11 × 0.15 × 0.30 |
| Temperature (K) | 273(2) |
| Radiation | Mo-Kα (0.7103 Å) |
| Crystalsystem | Monoclinic |
| Space group | C 2/ c |
| a (Å) | 26.835(5) |
| b (Å) | 9.9597(16) |
| c (Å) | 18.698(3) |
| α (°) | 90.00 |
| β (°) | 133.339(4) |
| γ (°) | 90.00 |
| V (Å3) | 3635.0(10) |
| Z | 72 |
| Dc (g cm–3) | 1.415 |
| μ (mm–1) absort.coeff | 0.127 |
| F(000) | 1584 |
| θ rang (deg) | 2.09–25.97 |
| Reflectionscollected | 17808(Rint = 0.1279) |
| Indep. reflns | 3512 |
| Refns obs. [I > 2σ(I)] | 1499 |
| Data/restr./paras | 3512/0/236 |
| Goodness-of-fit on F2 | 0.984 |
| R1, wR2 (all data) | 0.1310/0.1965 |
| R1, wR2 [I > 2σ(I)] | 0.0575/0.0984 |
| Larg. peak/hole (e. Å) | 0.181/−0.202 |

2.2. Anti-Proliferative Activity

| Compounds | R | IC50 (μM) | |||
|---|---|---|---|---|---|
| Hela | HepG2 | A431 | |||
| D1 |  | 13.64 | 3.47 | 16.58 | |
| D2 |  | 28.17 | 9.40 | 24.65 | |
| D3 |  | 19.62 | 10.21 | 8.96 | |
| D4 |  | 23.72 | 11.05 | 10.02 | |
| D5 |  | 21.40 | 10.50 | 9.31 | |
| D6 |  | 25.68 | 13.18 | 11.68 | |
| D7 |  | 8.71 | 3.16 | 12.56 | |
| D8 |  | 7.04 | 0.95 | 7.42 | |
| D9 |  | 6.75 | 0.79 | 7.03 | |
| D10 |  | 18.54 | 6.87 | 20.00 | |
| D11 |  | 16.70 | 4.94 | 18.95 | |
| D12 |  | 10.77 | 6.28 | 14.22 | |
| D13 |  | 24.30 | 15.23 | 14.05 | |
| D14 |  | 30.75 | 9.03 | 22.63 | |
| D15 |  | 5.15 | 1.48 | 17.80 | |
| D16 |  | 9.04 | 2.38 | 7.96 | |
| D17 |  | 19.52 | 7.90 | 21.56 | |
| C | - | 105 | 56 | 41 | |
| Caffeic acid | - | 544 | 225 | 93.5 | |
| Erlotinib | - | 0.08 | 0.04 | ||
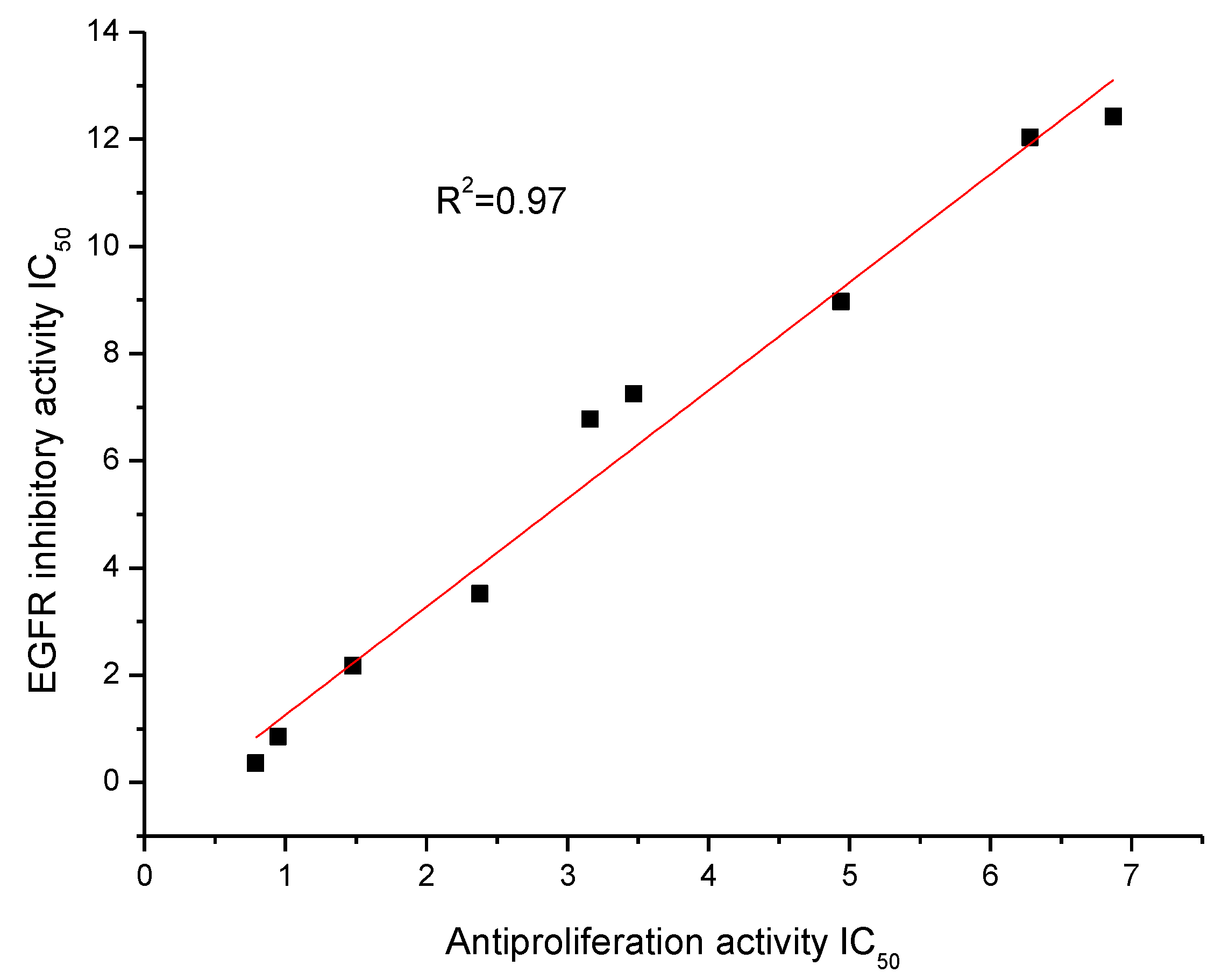
2.3. EGFR Inhibitory Activity
| Compounds | EGFR (IC50 (µM)) |
|---|---|
| D1 | 7.25 |
| D7 | 6.78 |
| D8 | 0.85 |
| D9 | 0.36 |
| D10 | 12.42 |
| D11 | 8.97 |
| D12 | 12.04 |
| D15 | 2.18 |
| D16 | 3.52 |
| Erlotinib | 0.03 |

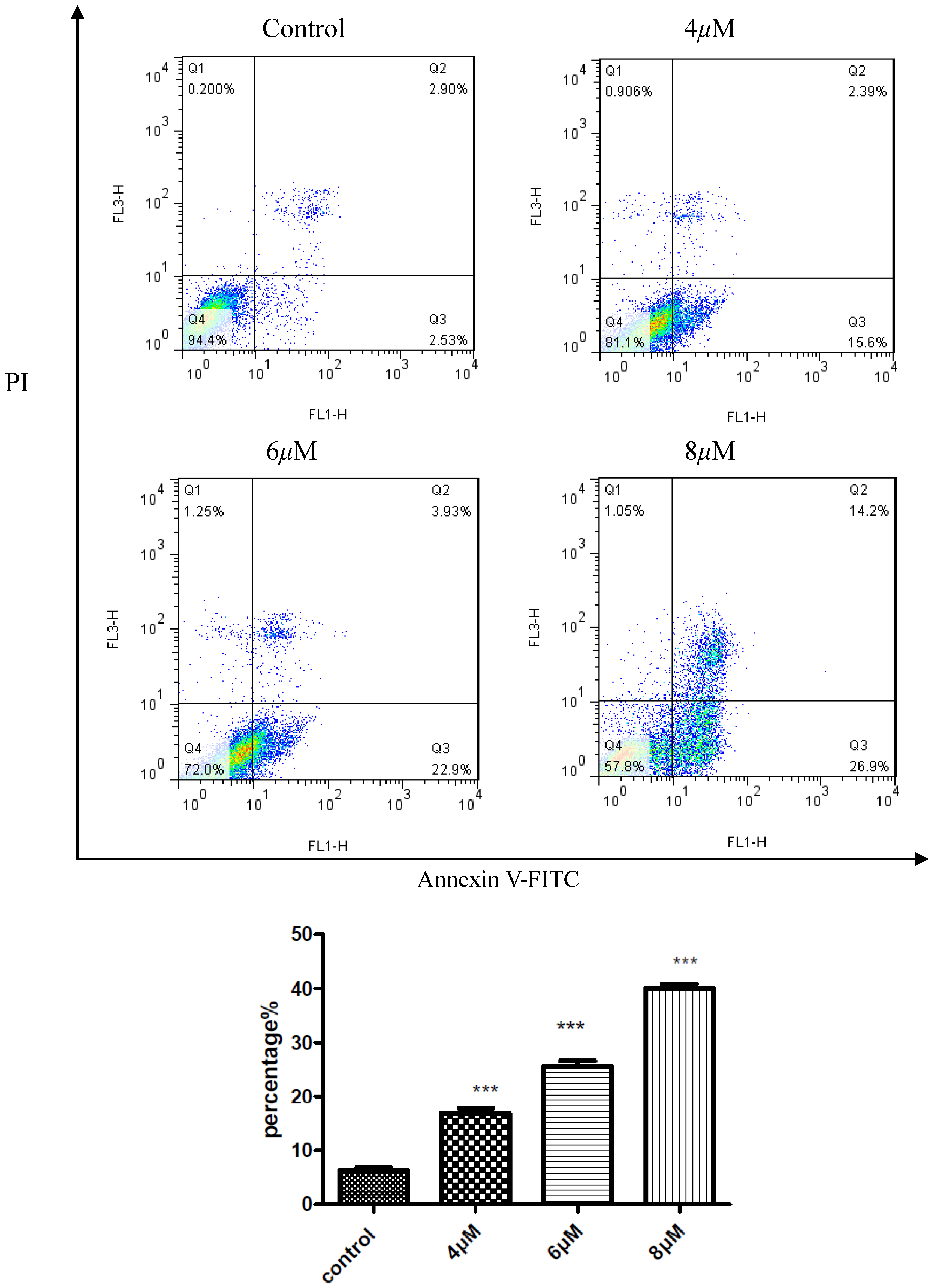
2.4. Apoptosis Assay

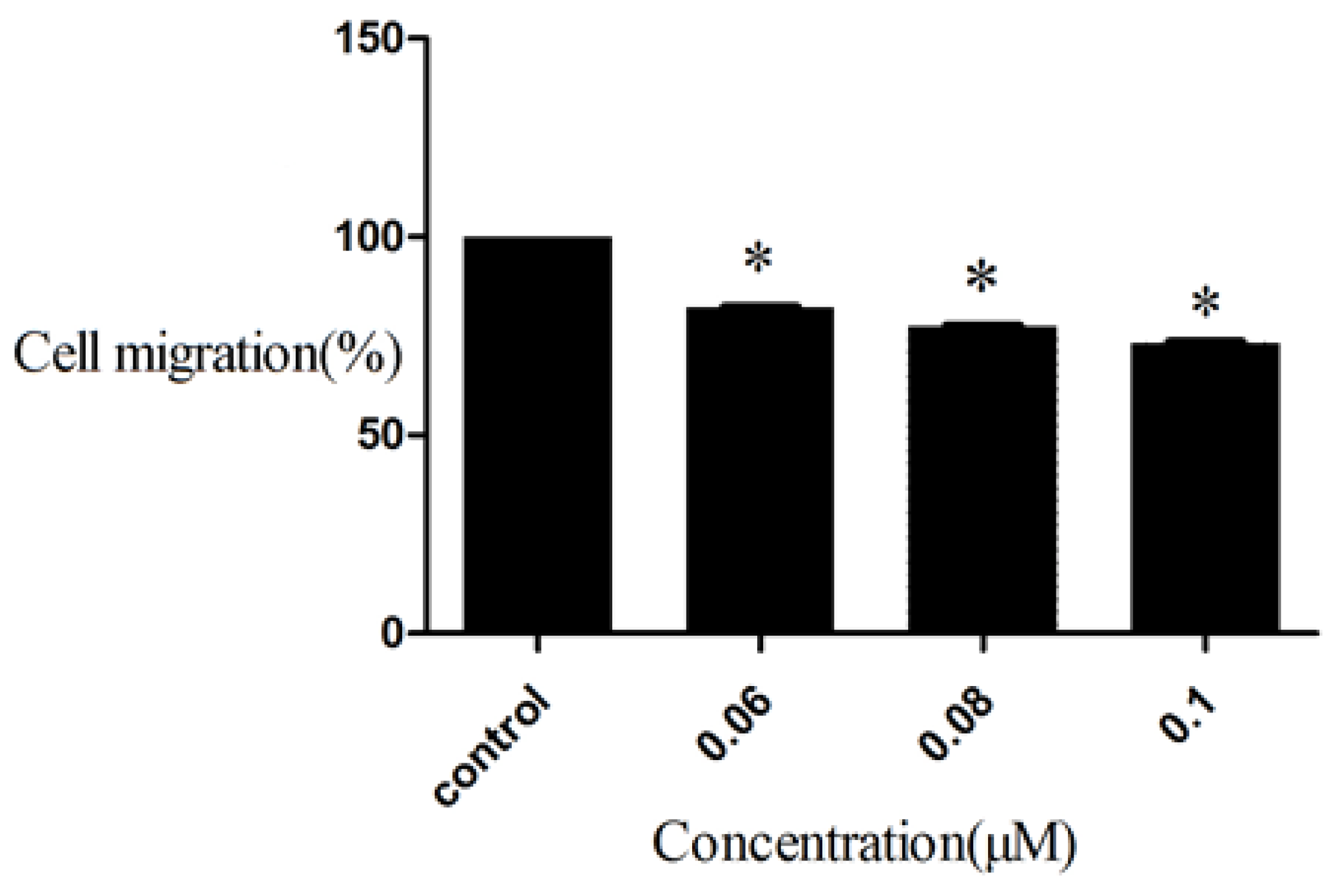
2.5. Inhibition to HepG2 Cell Migration of D9

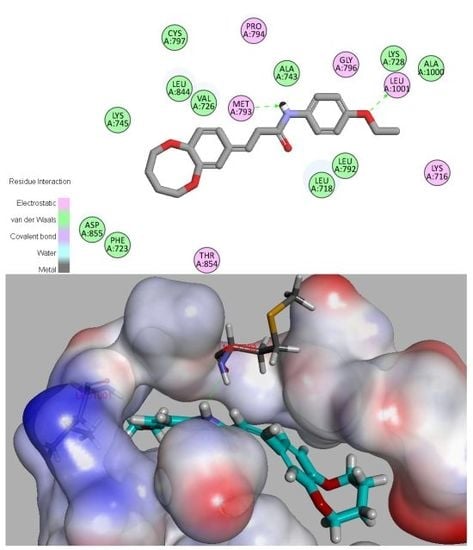
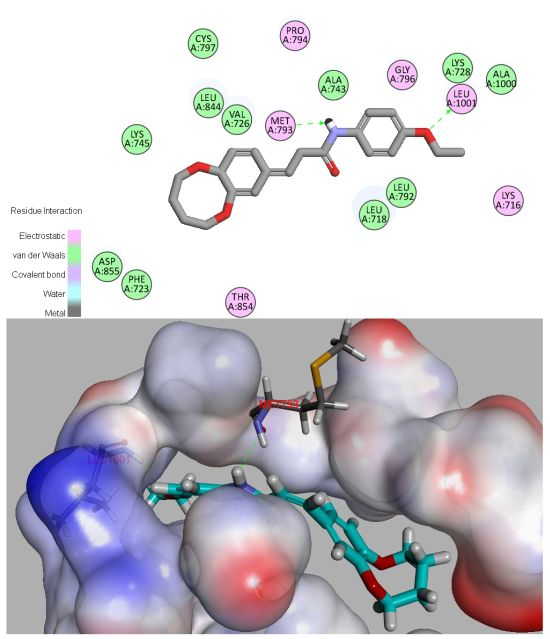
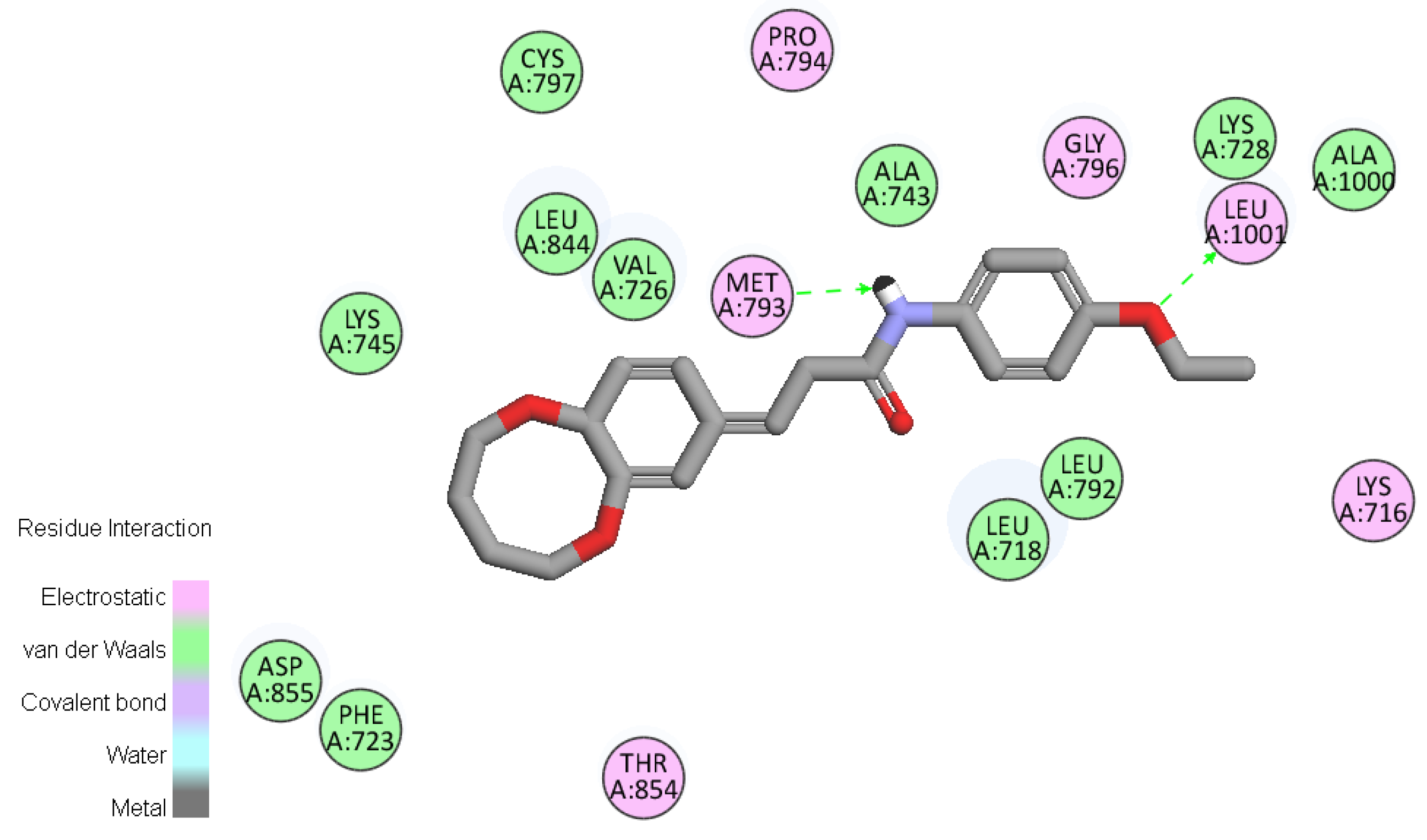
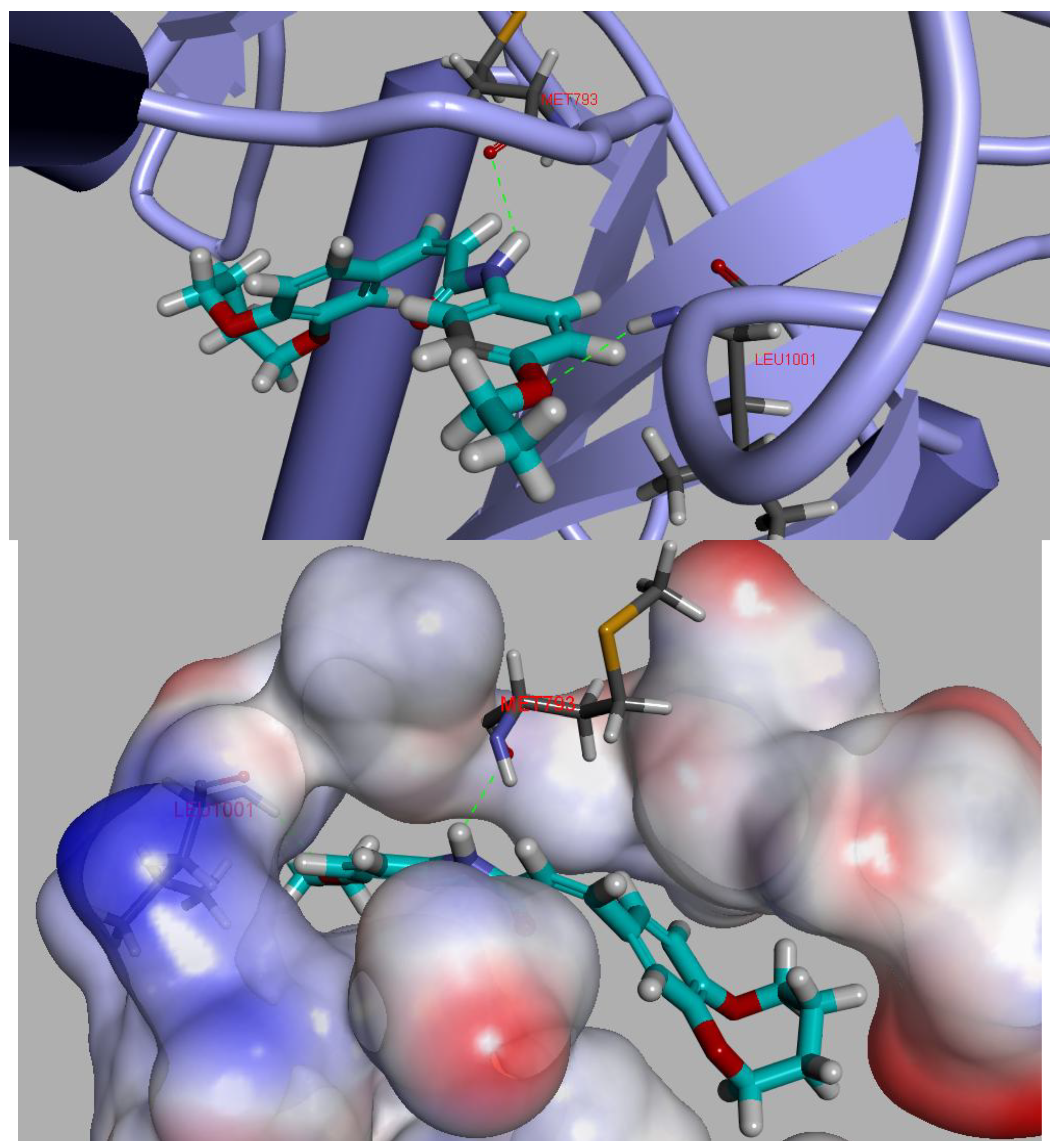
2.6. Molecular Docking


3. Experimental Section
3.1. Chemistry
3.1.1. Chemistry General Information
3.1.2. Experimental Procedure for the Synthesis of (E)-ethyl 3-(3,4-dihydroxyphenyl)acrylate (A)
3.1.3. Experimental Procedure for the Synthesis of (E)-ethyl 3-(2,3,4,5-tetrahydrobenzo[b][1,4]-dioxocin-8-yl)acrylate (B)
3.1.4. Experimental Procedure for the Synthesis of (E)-3-(2,3,4,5-tetrahydrobenzo[b][1,4]dioxocin-8-yl)acrylic acid (C)
3.1.5. General Experimental Procedure for the Synthesis of Compounds D1-D17
3.2. Cell Proliferation Assay
3.3. Apotosis Assay
3.4. General Procedure for Preparation, Purification EGFR, and Inhibitory Assay
3.5. Cell Migration Assay
3.6. Crystal Structure Determination
3.7. Molecular Docking Study
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA: Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Harari, P.M.; Huang, S. Head and neck cancer as a clinical model for molecular targeting of therapy: Combining EGFR blockade with radiation. Intern. J. Rad. Oncol. Biol. Phys. 2001, 49, 427–433. [Google Scholar] [CrossRef]
- Song, J.I.; Grandis, J.R. STAT signaling in head and neck cancer. Oncogene 2000, 19, 2489–2495. [Google Scholar] [CrossRef]
- Leek, R.D.; Hunt, N.C.; Landers, R.J.; Lewis, C.E.; Royds, J.A.; Harris, A.L. Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J. Pathol. 2000, 190, 430–436. [Google Scholar] [CrossRef]
- Richard, J.; Sainsbury, C.; Needham, G.; Farndon, J.; Malcolm, A.; Harris, A. Epidermal-growth-factor receptor status as predictor of early recurrence of and death from breast cancer. Lancet 1987, 329, 1398–1402. [Google Scholar] [CrossRef]
- Sanz Ortega, J.; Steinberg, S.M.; Moro, E.; Saez, M.; Lopez, J.A.; Sierra, E.; Sanz Esponera, J.; Merino, M.J. Comparative study of tumor angiogenesis and immunohistochemistry for p53, c-ErbB2, c-myc and EGFr as prognostic factors in gastric cancer. Histol. Histopathol. 2000, 15, 455–462. [Google Scholar]
- Ferrandina, G.; Ranelletti, F.O.; Lauriola, L.; Fanfani, F.; Legge, F.; Mottolese, M.; Nicotra, M.R.; Natali, P.G.; Zakut, V.H.; Scambia, G. Cyclooxygenase-2 (COX-2), epidermal growth factor receptor (EGFR), and Her-2/neu expression in ovarian cancer. Gynecol. Oncol. 2002, 85, 305–310. [Google Scholar] [CrossRef]
- Yarden, Y. The EGFR family and its ligands in human cancer: Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37, 3–8. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Gee, J.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Cohen, R.B. Epidermal growth factor receptor as a therapeutic target in colorectal cancer. Clin. Color. Cancer 2003, 2, 246–251. [Google Scholar] [CrossRef]
- Ravn, H.; Brimer, L. Structure and antibacterial activity of plantamajoside, a caffeic acid sugar ester from Plantago major subs major. Phytochemistry 1988, 27, 3433–3437. [Google Scholar] [CrossRef]
- Shin, K.; Kim, I.; Park, Y.; Ha, J.; Choi, J.; Park, H.; Lee, Y.S.; Lee, K. Anti-inflammatory effect of caffeic acid methyl ester and its mode of action through the inhibition of prostaglandin E2, nitric oxide and tumor necrosis factor-α production. Biochem. Pharmacol. 2004, 68, 2327–2336. [Google Scholar] [CrossRef]
- Chen, J.H.; Ho, C. Antioxidant activities of caffeic acid and its related hydroxycinnamic acid compounds. J. Agric. Food Chem. 1997, 45, 2374–2378. [Google Scholar] [CrossRef]
- Yamada, J.; Tomita, Y. Antimutagenic activity of caffeic acid and related compounds. Biosci. Biotechnol. Biochem. 1996, 60, 328–329. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Nishizawa, M.; Yamagishi, T.; Tanaka, T.; Nonaka, G.; Cosentino, L.M.; Snider, J.V.; Lee, K. Anti-AIDS agents, 18. Sodium and potassium salts of caffeic acid tetramers from Arnebia euchroma as anti-HIV agents. J. Nat. Prod. 1995, 58, 392–400. [Google Scholar] [CrossRef]
- Chen, J.; Shao, Y.; Huang, M.; Chin, C.; Ho, C. Inhibitory effect of caffeic acid phenethyl ester on human leukemia HL-60 cells. Cancer Lett. 1996, 108, 211–214. [Google Scholar] [CrossRef]
- Liao, H.; Chen, Y.; Liu, J.; Hsu, M.; Shieh, H.; Liao, H.; Shieh, C.; Shiao, M.; Chen, Y. Inhibitory effect of caffeic acid phenethyl ester on angiogenesis, tumor invasion, and metastasis. J. Agric. Food Chem. 2003, 51, 7907–7912. [Google Scholar] [CrossRef]
- Lv, P.; Li, H.; Xue, J.; Shi, L.; Zhu, H. Synthesis and biological evaluation of novel luteolin derivatives as antibacterial agents. Eur. J. Med. Chem. 2009, 44, 908–914. [Google Scholar] [CrossRef]
- Sun, J.; Yang, Y.; Li, W.; Zhang, Y.; Wang, X.; Tang, J.; Zhu, H. Synthesis, biological evaluation and molecular docking studies of 1,3,4-thiadiazole derivatives containing 1,4-benzodioxan as potential antitumor agents. Bioorg. Med. Chem. Lett. 2011, 21, 6116–6121. [Google Scholar] [CrossRef]
- Yun, C.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Qian, Y.; Corum, L.; Meng, Q.; Blenis, J.; Zheng, J.Z.; Shi, X.; Flynn, D.C.; Jiang, B. PI3K induced actin filament remodeling through Akt and p70S6K1: implication of essential role in cell migration. Am. J. Physiol.-Cell. Ph. 2004, 286, C153–C163. [Google Scholar]
- Qiu, X.; Janson, C.A.; Smith, W.W.; Head, M.; Lonsdale, J.; Konstantinidis, A.K. Refined structures of β-ketoacyl-acyl carrier protein synthase III. J. Mol. Biol. 2001, 307, 341–356. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the seventeen compounds are available from the authors.
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yuan, J.-W.; Qiu, H.-Y.; Wang, P.-F.; Makawana, J.A.; Yang, Y.-A.; Zhang, F.; Yin, Y.; Lin, J.; Wang, Z.-C.; Zhu, H.-L. Synthesis of Caffeic Acid Amides Bearing 2,3,4,5-Tetra-hydrobenzo[b][1,4]dioxocine Moieties and Their Biological Evaluation as Antitumor Agents. Molecules 2014, 19, 7269-7286. https://doi.org/10.3390/molecules19067269
Yuan J-W, Qiu H-Y, Wang P-F, Makawana JA, Yang Y-A, Zhang F, Yin Y, Lin J, Wang Z-C, Zhu H-L. Synthesis of Caffeic Acid Amides Bearing 2,3,4,5-Tetra-hydrobenzo[b][1,4]dioxocine Moieties and Their Biological Evaluation as Antitumor Agents. Molecules. 2014; 19(6):7269-7286. https://doi.org/10.3390/molecules19067269
Chicago/Turabian StyleYuan, Ji-Wen, Han-Yue Qiu, Peng-Fei Wang, Jigar A. Makawana, Yong-An Yang, Fei Zhang, Yong Yin, Jie Lin, Zhong-Chang Wang, and Hai-Liang Zhu. 2014. "Synthesis of Caffeic Acid Amides Bearing 2,3,4,5-Tetra-hydrobenzo[b][1,4]dioxocine Moieties and Their Biological Evaluation as Antitumor Agents" Molecules 19, no. 6: 7269-7286. https://doi.org/10.3390/molecules19067269