QM/MM Studies of Contemporary and Novel Membrane Raft Fluorescent Probes

Abstract
:1. Introduction


2. Results and Discussion

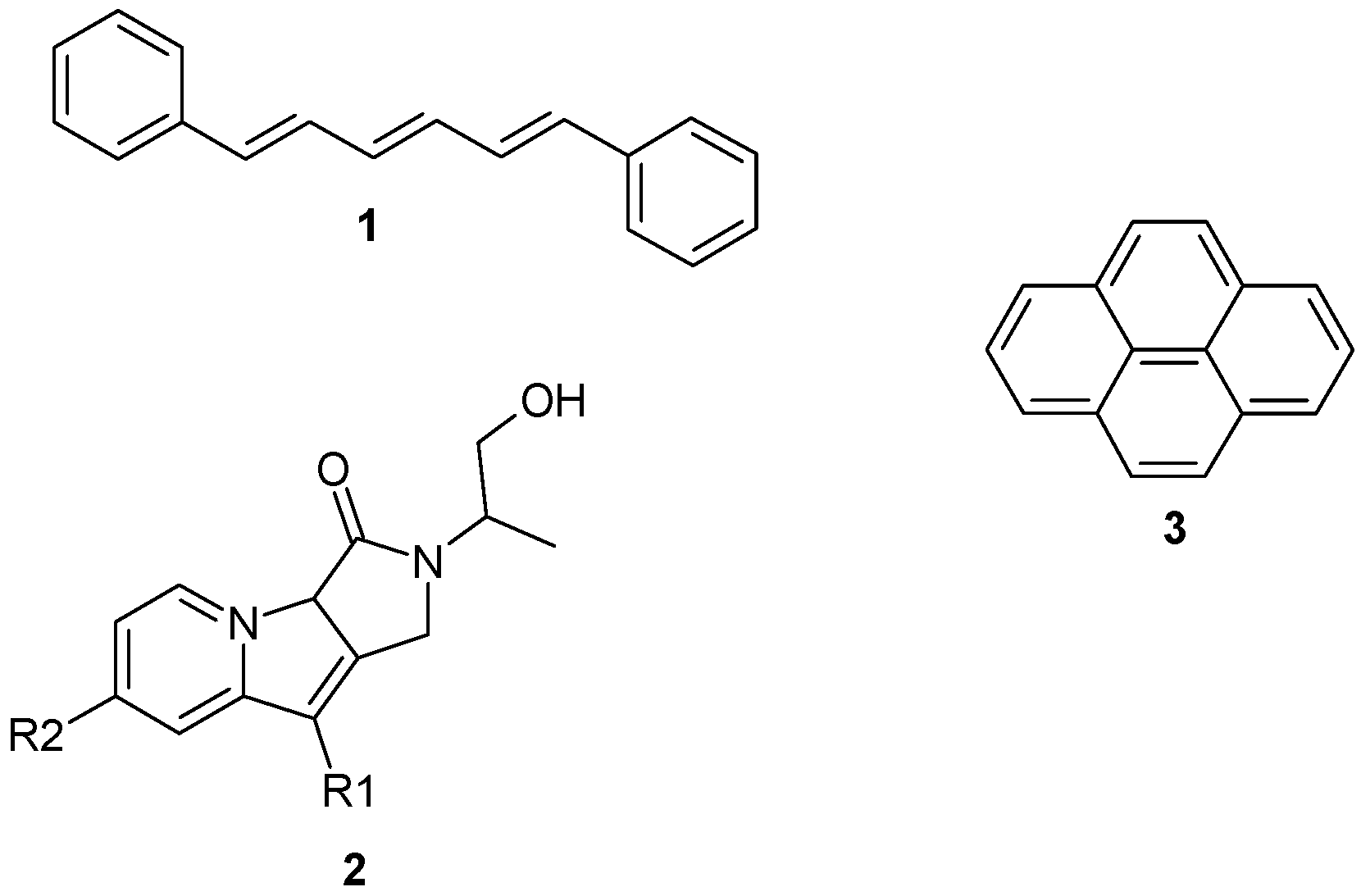{kind=link}
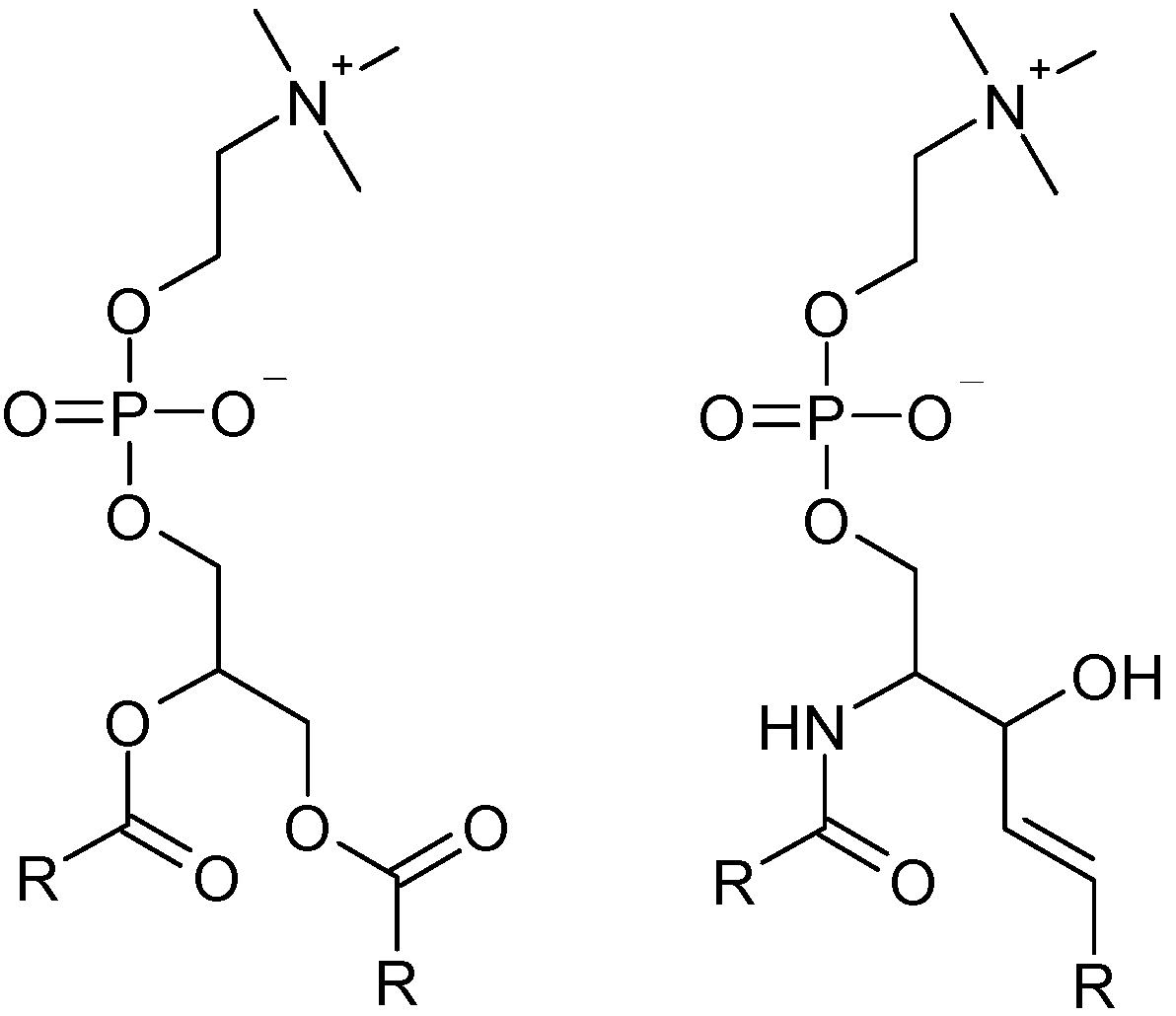{kind=link}
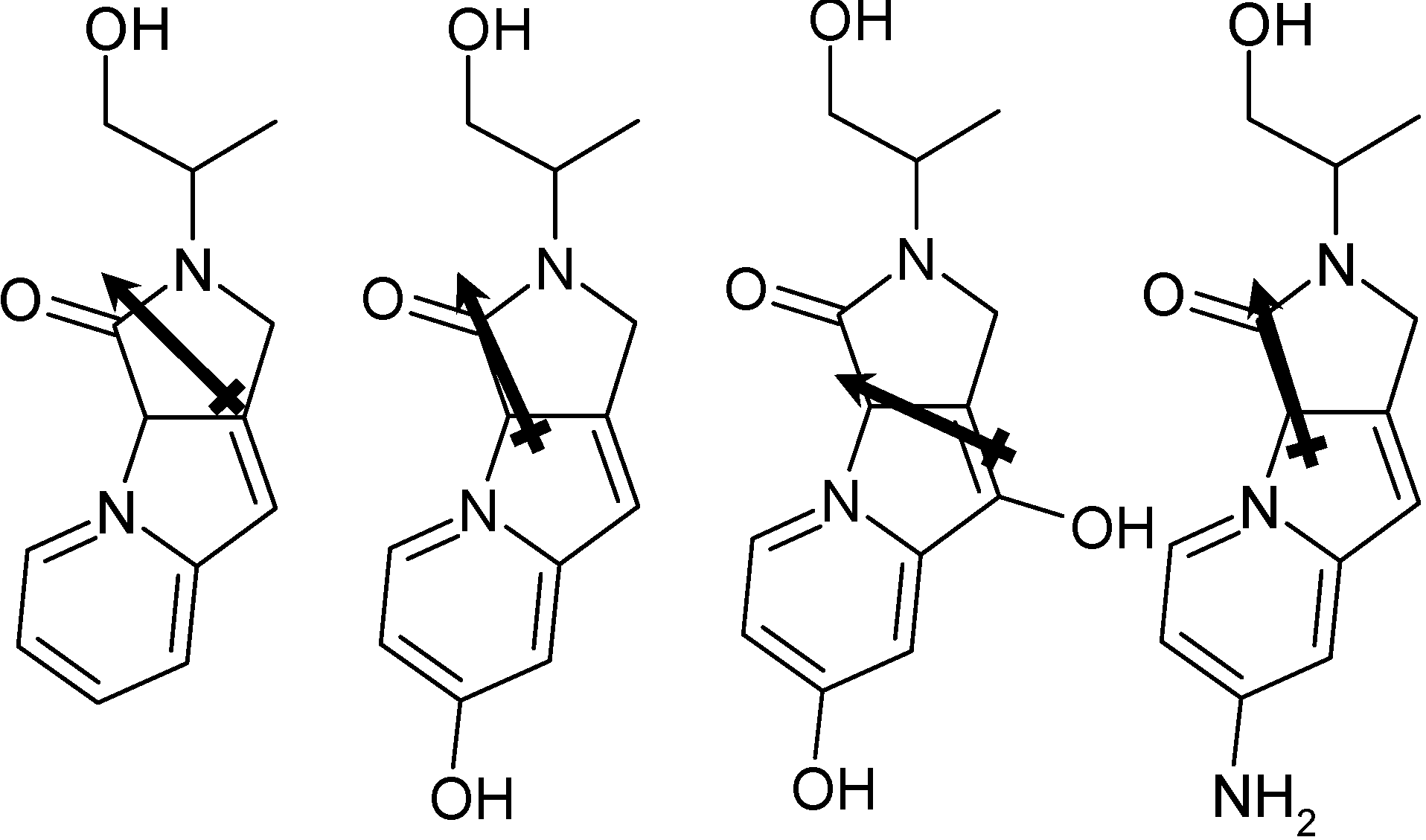{kind=link}
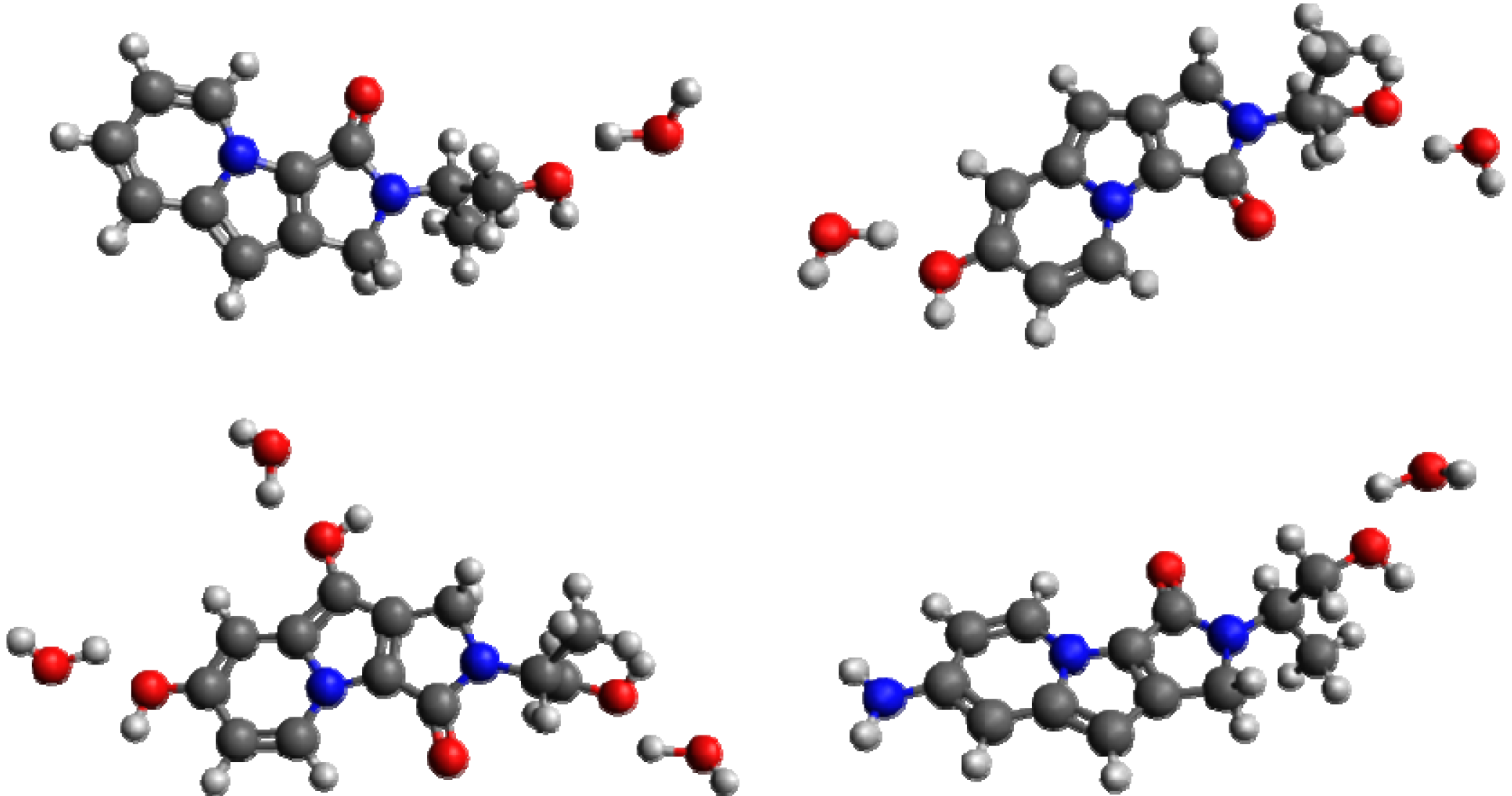{kind=link}
{kind=link}
| Molecule | ε | Vertical | 0-0 | Emission | Molecule | ε | Vertical | 0-0 | Emission |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 3.92 | 3.72 | 3.46 | 2c | 1 | 3.88 | 3.50 | 3.11 |
| 6.02 | 3.92 | 3.73 | 3.45 | 6.02 | 3.95 | 3.63 | 3.18 | ||
| 20.7 | 3.92 | 3.73 | 3.45 | 20.7 | 3.96 | 3.66 | 3.19 | ||
| 78.4 | 3.92 | 3.73 | 3.45 | 78.4 | 3.96 | 3.67 | 3.19 | ||
| 2a | 1 | 4.45 | 4.11 | 3.76 | 2d | 1 | 4.21 | 3.84 | 3.57 |
| 6.02 | 4.49 | 4.21 | 3.83 | 6.02 | 4.32 | 4.06 | 3.67 | ||
| 20.7 | 4.50 | 4.24 | 3.84 | 20.7 | 4.33 | 4.10 | 3.69 | ||
| 78.4 | 4.50 | 4.25 | 3.84 | 78.4 | 4.34 | 4.11 | 3.69 | ||
| 2b | 1 | 4.27 | 3.93 | 3.58 | 3 | 1 | 4.12 | 4.02 | 3.90 |
| 6.02 | 4.35 | 4.08 | 3.69 | 6.02 | 4.12 | 4.03 | 3.89 | ||
| 20.7 | 4.37 | 4.11 | 3.72 | 20.7 | 4.12 | 4.04 | 3.89 | ||
| 78.4 | 4.38 | 4.13 | 3.72 | 78.4 | 4.12 | 4.04 | 3.89 |
| Vertical Excitation | Emission | ||||
|---|---|---|---|---|---|
| PCM | Explicit Water + PCM | PCM | Explicit Water + PCM | QM/MM | |
| 2a | 4.50 | 4.42 | 3.84 | 3.73 | 3.83 |
| 2b | 4.38 | 4.31 | 3.72 | 3.63 | 3.49 |
| 2c | 3.96 | 4.09 | 3.19 | 3.10 | 2.91 |
| 2d | 4.34 | 4.26 | 3.69 | 3.59 | 3.57 |

| Molecule | DPPC | DPPC w/30 molar % Cholesterol |
|---|---|---|
| 1 | 3.45 | 3.44 |
| 2a | 3.74 | 3.75 |
| 2b | 3.57 | 3.57 |
| 2c | 2.99 | 3.47 |
| 2d | 3.58 | 3.37 |
| 3 | 3.91 | 3.91 |
3. Experimental Section
3.1. Gas-Phase Calculations

 . The first term on the right-hand side of equation 1 is the short-range component of the exchange and is evaluated with DFT, while the second term is the long-range component, which is evaluated with exact Hartree-Fock exchange.
. The first term on the right-hand side of equation 1 is the short-range component of the exchange and is evaluated with DFT, while the second term is the long-range component, which is evaluated with exact Hartree-Fock exchange.3.2. Solvated Calculations

3.3. QM/MM Water Calculations
3.4. QM/MM Membrane Calculations

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- The Nobel Prize in Chemistry 2013, Nobelprize.org. Nobel Media AB 2013. Available online: http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2013/ (accessed on 19 May 2014).
- Helms, V. Electronic excitations of biomolecules studied by quantum chemistry. Curr. Opin. Struct. Biol. 2002, 12, 169–175. [Google Scholar]
- Paulick, M.G.; Forstner, M.B.; Groves, J.T.; Bertozzi, C.R. A chemical approach to unraveling the biological function of the glycosylphosphatidylinositol anchor. Proc. Natl. Acad. Sci. USA 2007, 104, 20332–20337. [Google Scholar]
- Limon, A.; Reyes-Ruiz, J.M.; Eusebi, F.; Miledi, R. Properties of GluR3 receptors tagged with GFP at the amino or carboxyl terminus. Proc. Natl. Acad. Sci. USA 2007, 104, 15526–15530. [Google Scholar]
- Leake, M.C.; Chandler, J.H.; Wadhams, G.H.; Bai, F.; Berry, R.M.; Armitage, J.P. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature 2006, 443, 355–358. [Google Scholar]
- Prescott, M.; Battad, J.; Wilmann, P.; Rossjohn, J.; Devenish, R. Recent advances in all-protein chromophore technology. Biotechnol. Annu. Rev. 2006, 12, 31–66. [Google Scholar]
- Zhang, Z.; Alfonta, L.; Tian, F.; Bursulaya, B.; Uryu, S.; King, D.S.; Schultz, P.G. Selective incorporation of 5-hydroxytryptophan into proteins in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 8882–8887. [Google Scholar]
- Hogue, C.W.V.; Rashquinha, I.; Szabo, A.G.; MacManus, J.P. A new intrinsic fluorescent probe for proteins Biosynthetic incorporation of 5-hydroxytryptophan into oncomodulin. FEBS Lett. 1992, 310, 269–272. [Google Scholar]
- Ross, J.B.A.; Senear, D.F.; Waxman, E.; Kombo, B.B.; Rusinova, E.; Huang, Y.T.; Laws, W.R.; Hasselbacher, C.A. Spectral enhancement of proteins: Biological incorporation and fluorescence characterization of 5-hydroxytryptophan in bacteriophage lambda cI repressor. Proc. Natl. Acad. Sci. USA 1992, 89, 12023–12027. [Google Scholar]
- Botchway, S.W.; Barba, I.; Jordan, R.; Harmston, R.; Haggie, P.M.; William, S.P.; Fulton, A.M.; Parker, A.W.; Brindle, K.M. A novel method for observing proteins in vivo using a small fluorescent label and multiphoton imaging. Biochem. J. 2005, 390, 787–790. [Google Scholar]
- Robinson, D.; Besley, N.A.; O’Shea, P.; Hirst, J.D. Di-8-ANEPPS emission spectra in phospholipid/cholesterol membranes: A theoretical study. J. Phys. Chem. B 2011, 115, 4160–4167. [Google Scholar]
- Robinson, D.; Besley, N.A. Modelling the spectroscopy and dynamics of plastocyanin. Phys. Chem. Chem. Phys. 2010, 12, 9667–9676. [Google Scholar]
- Besley, N.A.; Robinson, D. Theoretical simulation of the spectroscopy and dynamics of a red copper protein. Faraday Discuss. 2011, 148, 55–70. [Google Scholar]
- Briggs, E.A.; Besley, N.A.; Robinson, D. QM/MM excited state molecular dynamics and fluorescence spectroscopy of BODIPY. J. Phys. Chem. A 2013, 117, 2644–2650. [Google Scholar]
- Bouevitch, O.; Lewis, A.; Pinevsky, I.; Wuskell, J.P.; Loew, L.M. Probing membrane potential with nonlinear optics. Biophys. J. 1993, 65, 672–679. [Google Scholar]
- Campagnola, P.J.; Wei, M.D.; Lewis, A.; Loew, L.M. High-Resolution nonlinear optical imaging of live cells by second harmonic generation. Biophys. J. 1999, 77, 3341–3349. [Google Scholar]
- Millard, A.C.; Jin, L.; Lewis, A.; Loew, L.M. Direct measurement of the voltage sensitivity of second-harmonic generation from a membrane dye in patch-clamped cells. Opt. Lett. 2003, 28, 1221–1223. [Google Scholar]
- Fluhler, E.; Burnham, V.G.; Loew, L.M. Spectra, membrane binding, and potentiometric responses of new charge shift probes. Biochemistry 1985, 24, 5749–5755. [Google Scholar]
- Zhang, J.; Davidson, R.M.; Wei, M.D.; Loew, L.M. Membrane electric properties by combined patch clamp and fluorescence ratio imaging in single neurons. Biophys. J. 1998, 74, 48–53. [Google Scholar]
- Loew, L.M. Potentiometric dyes: Imaging electrical activity of cell membranes. Pure Appl. Chem. 1996, 68, 1405–1409. [Google Scholar]
- Millard, A.C.; Jin, L.; Wei, M.D.; Wuskell, J.P.; Lewis, A.; Loew, L.M. Sensitivity of second harmonic generation from styryl dyes to transmembrane potential. Biophys. J. 2004, 86, 1169–1176. [Google Scholar]
- Montana, V.; Farkas, D.L.; Loew, L.M. Dual-Wavelength ratiometric fluorescence measurements of membrane potential. Biochemistry 1989, 28, 4536–4539. [Google Scholar]
- Goff, G.L.; Vitha, M.F.; Clarke, R.J. Orientational polarisability of lipid membrane surfaces. Biochim. Biophys. Acta 2007, 1768, 562–570. [Google Scholar]
- Rusu, C.F.; Lanig, H.; Othersen, O.G.; Kryschi, C.; Clark, T. Monitoring biological membrane-potential changes: A CI QM/MM study. J. Phys. Chem. B 2008, 112, 2445–2455. [Google Scholar]
- Benniston, A.C.; Copley, G. Lighting the way ahead with boron dipyrromethene (BODIPY) dyes. Phys. Chem. Chem. Phys. 2009, 11, 4124–4131. [Google Scholar]
- Treibs, A.; Kreuzer, F.H. Difluorboryl complexes of di-and tripyrrylmethene. Justus Liebigs. Ann. Chem. 1968, 718, 208–223. [Google Scholar]
- Ulrich, G.; Ziessel, R.; Harriman, A. The chemistry of fluorescent BODIPY dyes: Versatility unsurpassed. Angew. Chem. Int. Ed. 2008, 47, 1184–1201. [Google Scholar]
- Partridge, W.P.; Laurendeau, N.M.; Johnson, C.C.; Steppel, R.N. Performance of pyrromethene 580 and 597 in a commercial Nd: YAG-Pumped dye-laser system. Opt. Lett. 1994, 19, 1630–1632. [Google Scholar]
- García-Moreno, I.; Amat-Guerri, F.; Liras, M.; Costela, A.; Infantes, L.; Sastre, R.; López Arbeloa, F.; Bañuelos Prieto, J.; López Arbeloa, I. Structural changes in the BODIPY dye PM567 enhancing the laser action in liquid and solid media. Adv. Funct. Mater. 2007, 17, 3088–3098. [Google Scholar]
- Jagtap, K.K.; Maity, D.K.; Ray, A.K.; Dasgupta, K.; Ghosh, S.H. High efficiency dye laser with low fluorescence yield pyrromethene dyes: Experimental and theoretical studies. Appl. Phys. B-Laser Opt. 2011, 103, 917–924. [Google Scholar]
- Loudet, A.; Burgess, K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in neurobiology: Probing their chemistry and biology with molecular imaging. Chem. Rev. 2008, 108, 1517–1549. [Google Scholar]
- Boens, N.; Leen, V.; Dehaen, W. Fluorescent indicators based on BODIPY. Chem. Soc. Rev. 2012, 41, 1130–1172. [Google Scholar]
- Santra, M.; Moon, H.; Park, M.H.; Lee, T.W.; Kim, Y.K.; Ahn, K.H. Dramatic substituent effects on the photoluminescence of boron complexes of 2-(benzothiazol-2-yl)phenols. Chem. Eur. J. 2012, 18, 9886–9893. [Google Scholar]
- Bozdemir, O.A.; Cakmak, S.E.; Ekiz, O.O.; Dana, A.; Akkaya, E.U. Towards unimolecular luminescent solar concentrators: Bodipy-Based dendritic energy-transfer cascade with panchromatic absorption and monochromatized emission. Angew. Chem. Int. Ed. 2011, 50, 10907–10912. [Google Scholar]
- Kamkaew, A.; Lim, S.H.; Lee, H.B.; Kiew, L.V.; Chung, L.Y.; Burgess, K. BODIPY dyes in photodynamic therapy. Chem. Soc. Rev. 2013, 42, 77–88. [Google Scholar]
- Erten-Ela, S.; Yilmaz, M.D.; Icli, B.; Dede, Y.; Icli, S.; Akkaya, E.U. A panchromatic boradiazaindacene (BODIPY) sensitizer for dye- sensitized solar cells. Org. Lett. 2008, 10, 3299–3302. [Google Scholar]
- Catalán, J. Toward a generalized treatment of the solvent effect based on four empirical scales: Dipolarity (SdP, a new scale), polarizability (SP), acidity (SA), and basicity (SB) of the medium. J. Phys. Chem. B 2009, 113, 5951–5960. [Google Scholar]
- Filarowski, A.; Kluba, M.; Ciesĺ ik-Boczula, K.; Koll, A.; Kochel, A.; Pandey, L.; de Borggraeve, W.M.; Van der Auweraer, M.; Catalán, J.; Boens, N. Generalized solvent scales as a tool for investigating solvent dependence of spectroscopic and kinetic parameters. Application to fluorescent BODIPY dyes. Photochem. Photobiol. Sci 2010, 9, 996–1008. [Google Scholar]
- Boens, N.; Leen, V.; Dehaen, W.; Wang, L.; Robeyns, K.; Qin, W.; Tang, X.; Beljonne, D.; Tonnelé, C.; Paredes, J.M.; et al. Visible absorption and fluorescence spectroscopy of conformationally constrained, annulated BODIPY dyes. J. Phys. Chem. A 2012, 116, 9621–9631. [Google Scholar]
- López Arbeloa, F.; López Arbeloa, T.; López Arbeloa, I.; García-Moreno, I.; Costela, A.; Sastre, R.; Amat-Guerri, F. Photophysical and lasing properties of pyrromethene 567 dye in liquid solution: Environment effects. Chem. Phys. 1998, 236, 331–341. [Google Scholar]
- Boens, N.; Jiao, L.; Zhang, P.; Yu, C.; Wang, J.; Briggs, E.A.; Besley, N.A.; Robinson, D. Structural control of the photophysical properties of meso-alkyl BODIPY dyes. University of Leuven: Leiven, Belgium, in preparation (unpublished work); 2014. [Google Scholar]
- Gurtovenko, A.A.; Anwar, J.; Vattulainen, I. Defect-Mediated trafficking across cell membranes: Insights from in silico modeling. Chem. Rev. 2010, 110, 6077–6103. [Google Scholar]
- Robinson, D.; Besley, N.A.; O’Shea, P.; Hirst, J.D. Water order profiles on phospholipid/cholesterol membrane bilayer surfaces. J. Comput. Chem. 2011, 32, 2613–2618. [Google Scholar]
- Mihailescu, M.; Vaswani, R.G.; Jardón-Valadez, E.; Castro-Román, F.; Freites, J.A.; ZWorcester, D.L.; Chamberlin, A.R.; Tobias, D.J.; White, S.H. Acyl-Chain methyl distributions of liquid-ordered and disordered membranes. Biophys. J. 2011, 100, 1455–1462. [Google Scholar]
- O’Connor, J.W.; Klauda, J.B. Lipid membranes with a majority of cholesterol: Applications to the ocular lens and aquaporin 0. J. Phys. Chem. B 2011, 115, 6455–6464. [Google Scholar]
- Zidar, J.; Merzel, F.; Hodoscek, M.; Rebolj, K.; Sepcic, K.; Macek, P.; Janezic, D. Liquid-Ordered phase formation in cholesterol/sphingomyelin bilayers: All-atom molecular dynamics simulations. J. Phys. Chem. B 2009, 113, 15795–15802. [Google Scholar]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell. Biol. 2000, 1, 31–39. [Google Scholar]
- Robinson, D. A polarizable force-field for cholesterol and sphingomyelin. J. Chem. Theory Comput. 2013, 9, 2498–2503. [Google Scholar]
- Chowdhary, J.; Harder, E.; Lopes, P.E.M.; Huang, L.; MacKerell, A.D., Jr.; Roux, B. A polarizable force field of dipalmitoylphosphatidylcholine based on the classical drude model for molecular dynamics simulations of lipids. J. Phys. Chem. B 2013, 117, 9142–9160. [Google Scholar]
- Lu, Z.; Zhang, Y. Interfacing ab initio quantum mechanical method with classical drude osillator polarizable model for molecular dynamics simulation of chemical reactions. J. Chem. Theory Comput. 2008, 4, 1237–1248. [Google Scholar]
- Boulanger, E.; Thiel, W. Toward QM/MM simulation of enzymatic reactions with the drude oscillator polarizable force field. J. Chem. Theory Comput. 2014, 10, 1795–1809. [Google Scholar]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussman, J.; Ochsenfeld, C.; Brown, S.T. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar]
- Krylov, A.I.; Gill, P.M.W. Q-Chem: An engine for innovation. WIREs Comput. Mol. Sci. 2013, 3, 317–326. [Google Scholar]
- Woodcock, H.L.; Hodošček, M.; Gilbert, A.T.B.; Gill, P.M.W.; Schaefer, H.F.; Brooks, B.R. Interfacing Q-Chem and CHARMM to perform QM/MM reaction path calculations. J. Comput. Chem. 2007, 28, 1485–1502. [Google Scholar]
- Adamo, C.; Scuseria, G.E.; Barone, V. Accurate excitation energies from time-dependent density functional theory: Assessing the PBE0 model. J. Chem. Phys. 1999, 111, 2889–2899. [Google Scholar]
- Henderson, T.M.; Janesko, B.G.; Scuseria, G.E. Generalized gradient approximation model exchange holes for range-separated hybrids. J. Chem. Phys. 2008, 128, 194105. [Google Scholar]
- Liu, J.; Liang, W. Analytical second derivatives of excited-state energy within the time-dependent density functional theory coupled with a conductor-like polarizable continuum model. J. Chem. Phys. 2013, 138, 024101. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam, a fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar]
- Jo, S.; Kim, T.; Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One 2007, 2, e880. [Google Scholar]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blake, H.L.; Robinson, D. QM/MM Studies of Contemporary and Novel Membrane Raft Fluorescent Probes. Molecules 2014, 19, 10230-10241. https://doi.org/10.3390/molecules190710230
Blake HL, Robinson D. QM/MM Studies of Contemporary and Novel Membrane Raft Fluorescent Probes. Molecules. 2014; 19(7):10230-10241. https://doi.org/10.3390/molecules190710230
Chicago/Turabian StyleBlake, Hannah L., and David Robinson. 2014. "QM/MM Studies of Contemporary and Novel Membrane Raft Fluorescent Probes" Molecules 19, no. 7: 10230-10241. https://doi.org/10.3390/molecules190710230
APA StyleBlake, H. L., & Robinson, D. (2014). QM/MM Studies of Contemporary and Novel Membrane Raft Fluorescent Probes. Molecules, 19(7), 10230-10241. https://doi.org/10.3390/molecules190710230