1. Introduction
Gastrin-releasing peptide receptor (GRPR) is a cell membrane receptor expressed in almost all the prostate cancer (PC) primary tumors and, frequently, in metastases [
1,
2]. The receptor expression was observed in 85.7% of lymph node metastases and 52.9% of bone metastases of PC patients [
1]. Since normal prostate tissue as well as benign prostate tumors are mostly GRPR-negative, this receptor emerged as an attractive target for new diagnostic approaches and radionuclide therapeutic applications for PC patients [
2].
The presence of GRPRs has also been documented in breast cancer (BC) [
3].
In vitro receptor autoradiography on tissue sections has shown that 62% of the primary breast tumors are expressing GRPRs and when the receptor-positive breast cancers spread to local lymph nodes, the expression of GRPRs has been kept in the metastatic deposit [
4]. It has been shown that all the metastases of the receptor-positive primary tumors are GRPR-positive and the expression density of primary tumors and metastases are generally high [
4]. It has been also shown that these receptors contribute to the metastatic process by increasing cellular migration in BC [
5]. The prevalence of these receptors in BC has led to develop GRPR-targeted diagnostic and therapeutic peptide-based pharmaceuticals [
6,
7].
Bombesin (BN), a linear amphibian tetradecapeptide, is an analog of the mammalian gastrin-releasing peptide (GRP) and binds to GRPRs with high affinity and selectivity. Because of the poor
in vivo stability of GRP, appreciable efforts have been made in the development of BN analogs for targeting of GRPR [
8]. Different BN analogues were labeled with cytotoxic beta- and alpha-particle emitting nuclides
90Y [
9],
177Lu [
9,
10] and
212/213Bi [
11] for radionuclide therapy, and with gamma- and positron-emitting radionuclides
111In [
9],
99mTc[
12,
13,
14],
68Ga [
13] and
64Cu [
13,
15] for visualization of GRPR-expressing tumors. BN shows high structural and functional homology with GRP. They share seven amino acids amidated C-terminus sequence homology, Trp
-Ala-Val-Gly-His-Leu-Met-NH
2. It has been shown that these C-terminal seven amino acids are responsible for binding to the GRPRs and any modification near C-terminus of BN analogs decreases the binding affinity [
16]. Therefore, the N-terminus of these analogs was coupled to the chelators for loading with radiometals or modified for labeling with other radionuclides [
13,
15].
It has been shown that direct coupling of the radiometal-chelator complex to the N-terminal of the BN analogs decreases the receptor-binding affinity [
17], while the application of a spacer prevents the interference of the radiometal-chelator complex with the binding site of the targeting vector to the GRPR [
18]. The analog containing no spacer (X = 0) for
111In-DOTA-X-BN[
7,
8,
9,
10,
11,
12,
13,
14]NH
2 has exhibited 100-fold lower binding affinity to GRPR in comparison with a 8-carbon aliphatic spacer containing analog [
18]. Lys(sha)-βAla-βAla spacer for coupling of retro[N
α-carboxymethyl-histidine] to BN analogs for
99mTc(CO)
3 labeling improved the affinity of conjugates by factor 23 [
12]. It has also been demonstrated that excessive increase of the lipophilicity of the linker reduces the affinity [
18]. Significantly higher liver uptake for hydrophobic spacer-containing analogs adds to the problem of the suboptimal pharmacokinetics of these analogs [
18]. To improve the pharmacokinetic profiles for BN analogs, more hydrophilic spacer moieties were introduced. Schweinsberg
et al. have shown that hydrophilic carbohydrate groups introduced into the linker sequence of
99mTc-labeled BN analogs decreased liver uptake significantly [
19]. Increasing the charge of the spacer by insertion of negatively charged β
3hGlu, demonstrated favorable biodistribution for a BN analog labeled using
99mTc-tricarbonyl core [
20].
The polyethylene glycol (PEG) has been widely used for modification of therapeutic peptides and proteins [
21]. PEGylation, the covalent attachment of the PEG to the biologically active molecule, is a modification methodology with the main purpose of reducing immunogenicity and rapid enzymatic degradation of the proteins [
22]. The PEG
4 spacer has been introduced in BN agonist (DOTA-PESIN) with the aim to increase its metabolic stability and to improve its tumour accumulation [
23]. This has been found to be successful, moreover, an appreciable switch of elimination pathway from hepatic to renal was observed. PEGylation can also increase the overall hydrophilicity of the modified peptides and proteins [
24,
25]. However, introduction of PEG
3 spacer into fluorinated RGD-based integrin-targeted peptide did not affected its hepatic uptake [
26].
PEGylation might improve the pharmacokinetic properties of targeting molecules by appreciable reduction of the hepatic uptake and/or hepatobiliary excretion of the radiotracers. For example, a replacing the lipophilic 6-aminocaporoic acid (CA) linker with more hydrophilic PEG
4 linker could enhance the clearance kinetics of
99mTc-labeled cyclic RGD peptide with minor impact on binding affinity to GPIIb/IIIa receptors [
27]. The PEGylated BN(7–14) analog, where PEG with the length of 5–20 kDa was conjugated to the BN radiolabeled using
99mTc(CO)
3, exhibited higher
in vitro and
in vivo stability [
14]. It has also been shown that PEGylation did not affect the binding affinity although slower
in vitro kinetics was found for PEG-conjugated BN analogs. Fast blood clearance via renal elimination and decreased hepatobiliary excretion led to higher tumor-to-non-tumor ratios for PEG-modified (5 kDa) BN in comparison with non-PEGylated analogs [
14].
It should be taken into consideration that small modifications in the bioactive molecular targeting vectors can influence their biodistribution pattern and tumor targeting properties. The number of monomer units of polymer plays significant role in the biological behavior of PEG-modified conjugates. Because of the flexibility of the PEG chain, there is a risk of masking the biologically active receptor recognition site of the peptide and subsequently impairment of binding affinity by using long PEG spacers or chains [
28]. There are some examples in the literature where PEGylated proteins exhibited unexpected behavior of increased activity or PEGylation changed the biological effects [
29,
30]. Asparaginase, an enzyme of therapeutic interest, exhibited increased activity after modification with mPEG
2-COOSu [
29]. It has further been shown that PEGvisomant, the PEGylated form of growth hormone receptor antagonist, showed lower antagonistic function compared with its non-PEGylated core molecule B2036 [
30]. Thus, PEG modification can affect the pharmacological activity of drugs and therefore the balance between the degree of PEG modification and the molecular size of the drug should be considered [
31].This influence could be even more pronounced for short peptides. Hence, modification in the length of the spacer moiety might influence the BN binding affinity to GRPRs and its
in vivo kinetics.
We have previously investigated an antagonist analog of BN (D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH
2, RM26) conjugated to 1,4,7-triazacyclononane-
N,
N',
N''-triacetic acid (NOTA) via a diethylene glycol (PEG
2) spacer (NOTA-PEG
2-RM26) labeled with
68Ga,
111In and Al
18F [
32,
33]. This conjugate showed favorable properties for
in vivo imaging of GRPR-expression in PC.
In this study we investigated the possibility to modulate the lipophilicity and thus liver uptake of the conjugate by introducing extended mini-PEG linkers. Since the liver is one of the major metastatic sites for BC, high radioactivity accumulation in the liver is an undesirable property for the imaging agents.
For this purpose, a series of NOTA-PEGn-RM26 (n = 2, 3, 4 and 6) conjugates were synthesized. The peptides were labeled with a short-lived generator-produced radionuclide 68Ga (T½ = 67.8 min) for imaging using positron emission tomography (PET). In vitro and in vivo studies were performed using PC-3 (PC cells) and BT-474 (BC cells). More long-lived 111In (T½ = 2.8 days) was used as a label in some in vitro experiments requiring longer time than it is permissible by the short half-life of 68Ga.
3. Experimental Section
3.1. Peptide Synthesis
NOTA-PEG
n-[D-Phe
6, Sta
13, Leu
14]Bombesin[
6,
7,
8,
9,
10,
11,
12,
13,
14] (
n = 2, 3, 4 and 6), NOTA-PEG
n-RM26 (
Figure 4) were synthesized by manual solid-phase peptide synthesis (SPPS) using standard Fmoc/
t-Bu conditions as described earlier [
32].
Figure 4.
Structure of NOTA-PEG
n-[D-Phe
6, Sta
13, Leu
14]-Bombesin[
6,
7,
8,
9,
10,
11,
12,
13,
14], NOTA-PEG
n-RM26, (
n = 2, 3, 4 and 6).
Figure 4.
Structure of NOTA-PEG
n-[D-Phe
6, Sta
13, Leu
14]-Bombesin[
6,
7,
8,
9,
10,
11,
12,
13,
14], NOTA-PEG
n-RM26, (
n = 2, 3, 4 and 6).
3.2. Radiolabeling and Stability Test
The
111In-,
68Ga- and
natGa-labeling and quality controls were performed based on protocols presented earlier [
32]. Buffers for labeling were purified from metal contamination by Chelex 100 resin (Bio-Rad Laboratories, Hercules, CA). For
68Ga labeling, 10 µL (aqueous solution of 10 nmol) of NOTA-PEG
n-RM26 buffered with 250 µL of sodium acetate (Merck, Darmstadt, Germany) to obtain pH 3.9 was incubated with 1 mL
68Ga-containing eluate (600 MBq) for 10 min at 75 °C. For cold isotope loading, 14 nmol of
natGaCl
3, aq. solution (50 µL), (Sigma-Aldrich, St. Louis, Missouri, MO, USA) were added to 10 nmol of NOTA-PEG
n-RM26 using the same protocols as for radioactive isotopes.
The reaction mixtures of 68Ga-NOTA-PEGn-RM26 were purified using solid phase extraction. Briefly, the reaction mixtures were diluted with 3 mL of deionized water and passed through a 1 mL Oasis HLB cartridge (Waters, Milford, MA, USA). The cartridge was then washed with 5 mL of deionized water to remove any unreacted 68Ga. The radiolabeled product was eluted with 1 mL of 1:1 EtOH/water. The chemical and radiochemical purity of 68Ga-NOTA-PEGn-RM26 after purification was checked by UV-radio-HPLC. The conditions were as follows: A = 10 mM TFA; B = 70% acetonitrile (MeCN), 30% H2O, and 10 mM TFA with UV-detection at 220 nm; gradient elution: 0–2 min at 35% B, 2–9 min at 35 to 100% B, 9–12 min at 100% B; and flow rate was 2.0 mL/min. The analytes were separated using an analytical column with a stationary phase consisting of covalently bonded pentylsilane (Discovery BIO Wide Pore C5; 5 cm × 4.6 mm).
For routine control, radiochemical purity of 68Ga- and 111In-NOTA-PEGn-RM26 was analyzed by ITLC (Biodex Medical Systems, Shirley, NY, USA) using 0.2 M citric acid (pH 2.0) as the mobile phase. The method was cross-calibrated with HPLC (see above). In this system free indium, gallium and their complexes migrate with the solvent front (Rf = 1.0), while peptides remain at the origin (Rf = 0.0).
For 111In labeling, the pH of 10 µL (aqueous solution of 10 nmol) of NOTA-PEGn-RM26 was adjusted to 5.5 using 80 µL of 0.2 M ammonium acetate (Merck) and mixed with 80–150 µL (~60 MBq) of 111In (Covidien, Dublin, Ireland). The mixture was incubated at 80 °C for 30 min.
To evaluate labeling stability, 68Ga-NOTA-PEGn-RM26 or 111In -NOTA-PEGn-RM26 were challenged with 1000-fold molar excess of disodium salt of EDTA (Sigma) and incubated for 1 h at room temperature (RT).
For determination of logD, n-octanol (750 µL) was added to a tube with 750 μL of PBS (pH 7.4) containing 0.1 nmol of a 68Ga-labeled conjugate, the tube was vigorously vortexed and then centrifuged (14,000 rpm) for 4 min. Aliquots of 50 μL were taken from each phase, and their radioactivity was measured in an automated gamma-counter with 3-inch NaI(Tl) detector (1480 WIZARD, Wallac OyTurku, Finland). Each measurement was repeated nine times. The distribution coefficient was calculated as the average log of a ratio of the radioactivity in the octanol and the PBS fractions.
3.3. In Vitro Studies
GRPR expressing human PC cell line PC-3 and human BC cell line BT-474 (ATCC) were cultured in RPMI media complemented with 10% FBS, 2 mM l-glutamine and PEST (penicillin 100 IU/mL) (all from Biochrom AG, Berlin, Germany). This media is referred as complete media in the text. In all in vitro experiments, cells were incubated in complete medium and detached using trypsin-EDTA solution (0.25% trypsin, 0.02% EDTA in buffer; Biochrom AG). All experiments were performed in triplicate and 1 × 106 cells/dish were seeded one day before the experiment.
3.3.1. In Vitro Binding Specificity Assay
The binding specificity of 68Ga- and 111In-labeled NOTA-PEGn-RM26 was tested on PC-3 and BT-474 cells. The cells were incubated with 1 nM concentration of 68Ga- and 111In-NOTA-PEGn-RM26 solution for 1 h at 37 °C. One set of dishes in each experiment was pre-saturated with 100-fold excess of unlabeled peptide, added 5 min before the addition of the radiolabeled compound. After being washed twice with serum free media, cells were detached by treatment with 0.5 mL trypsin-EDTA solution. Cell associated radioactivity was measured in the gamma-counter and presented as percentage from added radioactivity.
3.3.2. Real-Time Ligand Binding Kinetics: KD and Bmax Determination
The kinetics of binding of
111In-NOTA-PEG
n-RM26 to the PC-3 cells was measured in real-time at RT using LigandTracer instruments (Ridgeview Instruments AB, Uppsala, Sweden), as described previously [
42]. In addition, binding of the most promising candidate,
111In-NOTA-PEG
3-RM26, was measured using BT-474 cells. Two radioligand concentrations were used: 0.3 and 10 nM. These concentrations were chosen to receive a clear increase in signal by addition of the second concentration. Uptake was monitored for 200 min and retention for 1200 min. Interaction analysis and calculation of equilibrium dissociation constant (K
D) was performed with TracerDrawer software (Ridgeview Instruments AB). To determine the number of binding sites per cell, B
max, cells were incubated with 20 nM of
111In-NOTA-PEG
3-RM26 at RT. When radioactivity uptake reached saturation, cells were washed twice with serum-free media and after being trypsinized, detached cells were counted and collected for radioactivity measurement. Data were used for calculation of the B
max, on PC-3 and BT-474 cells.
3.3.3. In Vitro Competitive Binding Assay: IC50 Determination
An in vitro competition experiment was performed on PC-3 cells using 125I-Tyr4-BBN (Perkin Elmer). The half maximal inhibitory concentration (IC50) was determined for non-radioactive natGa-loaded conjugates. Cell monolayers were incubated with natGa-NOTA-PEGn-RM26 (0–395 nM) in the presence of 0.1 pmol (~100,000 cpm) 125I-Tyr4-BBN for 3 h at 4 °C. After the incubation, the cells were collected, and cell-associated radioactivity was determined as described above. The IC50 values were determined using GraphPad software.
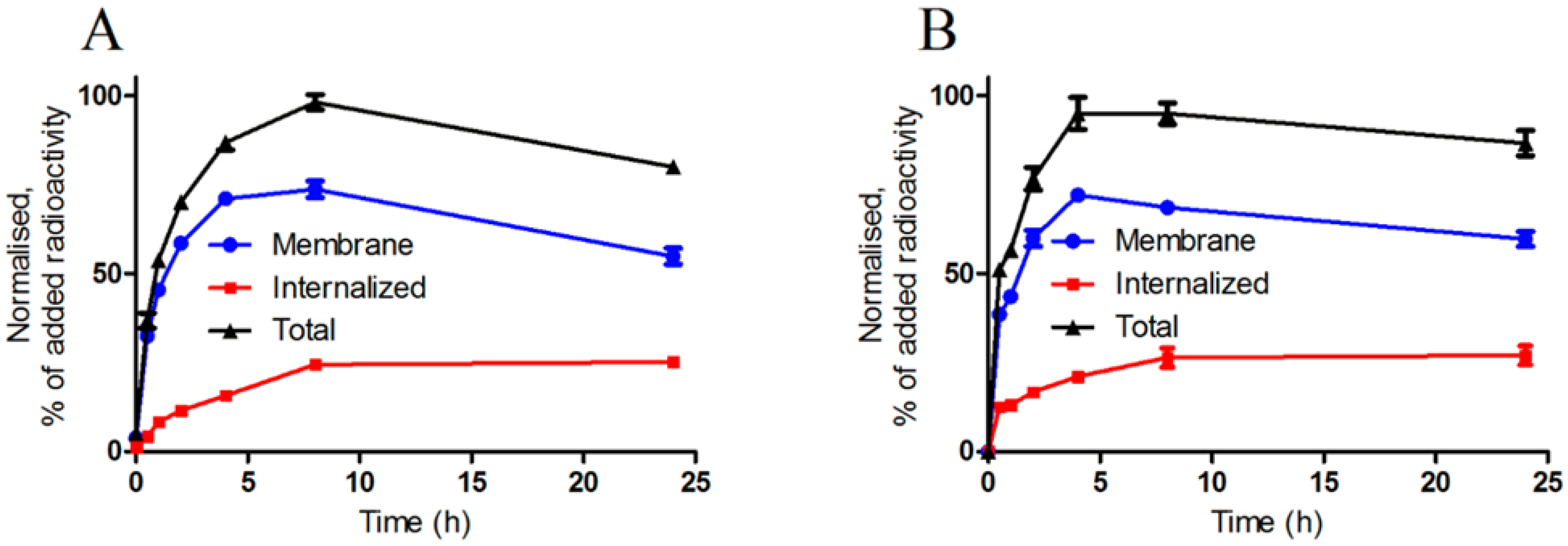
3.3.4. Cellular Uptake and Internalization Assay
PC-3 cells were incubated with 2 nM of
111In-NOTA-PEG
n-RM26 at 37 °C for 24 h. At predetermined time points, the incubation media was discarded, the cells were washed and the membrane-bound and internalized radioactivity were collected using the method described earlier [
43]. In addition, the uptake and internalization of
111In-NOTA-PEG
3-RM26 was studied on BT-474 cells.
3.4. In Vivo Studies
All animal experiments were planned and performed according to the Swedish national legislation on the protection of laboratory animals, and the study plans were approved by the local committee for animal research ethics. The biodistribution of 68Ga-labeled NOTA-PEGn-RM26 was evaluated in female NMRI mice (weight: 30 ± 3 g). BALB/c nu/nu female mice (weight: 19.8 ± 0.7 g) bearing PC-3 or BT-474 xenografts (10 × 106 cells/mouse, implanted 3 weeks before the experiment) were used for in vivo binding specificity and tumor targeting study using 68Ga-labeled NOTA-PEG3-RM26. The mice for BT-474 xenografts were implanted subcutaneously with 17β-estradiol pellets (0.05 mg/21-day release, Innovative Research of America, Sarasota, FL, USA) before tumor cell inoculation and cells were inoculated in Matrigel/PBS (1:1). The average tumor size for both cell lines was 0.4 ± 0.1 g at the time of the experiment.
3.4.1. Biodistribution in NMRI Mice
Female NMRI mice (3 mice per data point) were injected with 45 pmol of 68Ga-NOTA-PEGn-RM26 (350 or 700 kBq, 100 µL in PBS) into the tail vein. The administrated peptide dose was adjusted to 45 pmol by dilution with non-labeled NOTA-PEGn-RM26. The mice were euthanized at 1 and 2 h p.i. by intraperitoneal injection of a Ketalar-Rompun solution (10 mg/mL Ketalar and 1 mg/mL Rompun; 20 µL of solution per gram of body weight). Blood samples were collected by heart puncture. The organs of interest were collected, weighed, and their radioactivity content was measured in the gamma-counter. The organ uptake values are expressed as %ID/g except for gastrointestinal (GI) tract (with content) and carcass in which data are presented as %ID per whole sample.
3.4.2. Biodistribution and in vivo Binding Specificity Test in BALB/c nu/nu Mice Bearing PC-3 Prostate Cancer and BT-474 Breast Cancer Xenografts
To study tumor targeting, female BALB/c nu/nu mice bearing PC-3 and BT-474 xenografts (4 mice per data point) were intravenously injected with 45 pmol of 68Ga-NOTA-PEG3-RM26 (700 kBq, 100 µL in PBS). The mice were euthanized at 2 h p.i. and the organ radioactivity content was measured and evaluated as described above. To test the in vivo binding specificity, a group of animals was co-injected with 20 nmol of unlabeled peptide, and biodistribution was measured at 2 h p.i.
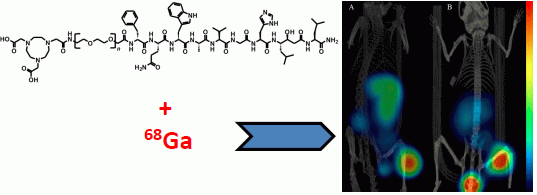
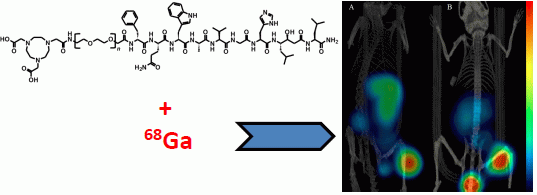
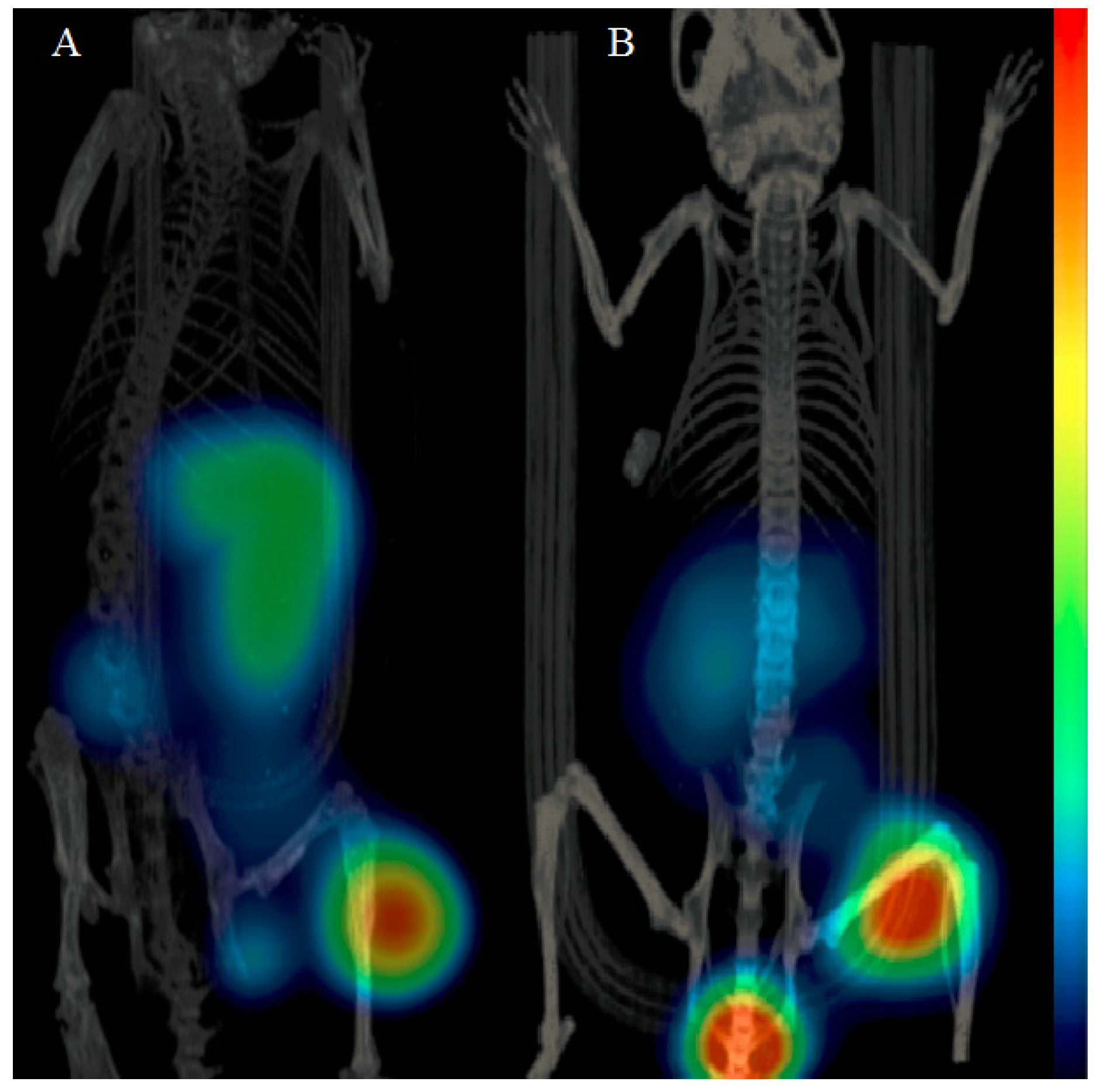
3.5. Imaging Studies
The mice bearing PC-3 or BT-474 tumors were injected with 45 pmol of 68Ga-NOTA-PEG3-RM26. The animals were sacrificed 2 h p.i. and scanned with a micro-PET/CT (Gamma Medica Inc., Salem, NH, USA). The PET and CT data were fused and analyzed using PMOD v3.13 (PMOD Technologies Ltd., Zurich, Switzerland).
3.6. Statistics
Statistical analyses were performed by un-paired, two-tailed t-test using Prism (version 4.00; GraphPad Software, La Jolla, CAP value below 0.05 was considered significant.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}