Combined Kinetic Studies and Computational Analysis on Kojic Acid Analogs as Tyrosinase Inhibitors

 , , ,
, , ,
Abstract
:1. Introduction


| Inhibitor | 2D Structure | Ki Value |
|---|---|---|
| INH1 |  | 1 mM |
| INH2 |  | 145 µM |
| INH3 |  | ND * |
| INH4 |  | 64 µM |
| KA |  | 5 µM |
| Tropolone |  | 0.8 µM ** |
2. Results and Discussion
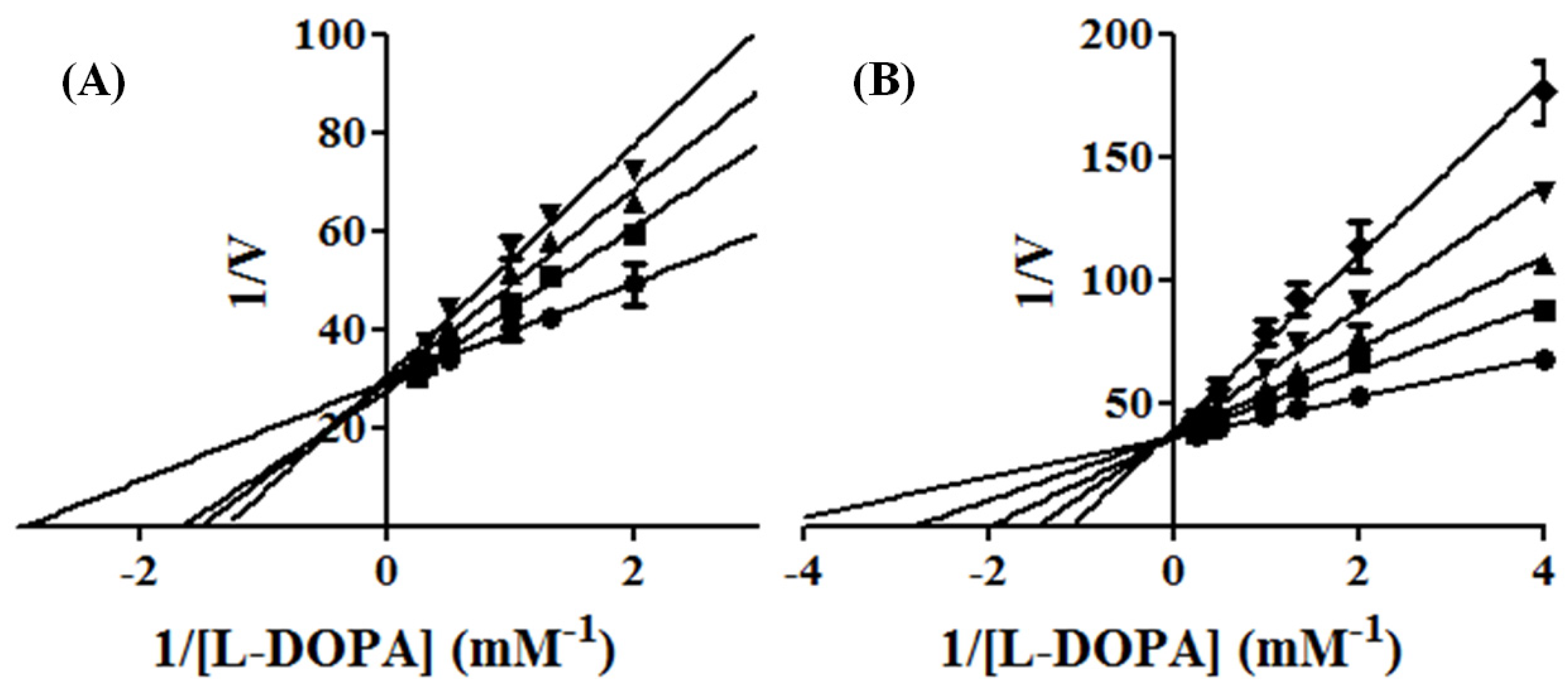
2.1. Experimental Kinetic Assays of Tyrosinase and Inhibition by KA Analogs


2.2. Molecular Docking
| KA Analogue | Atom | AbTYR Atom | Distance |
|---|---|---|---|
| INH1 | O1 H2 | Cu2+ A OD1 (Asn260) | 3.69 2.91 |
| INH2 | 3.50 1.82 | ||
| INH3 | 3.84 2.04 | ||
| INH4 | 3.57 1.74 |
| Inhibitor | MolDock Score (kcal·mol−1) | Type of Inhibition |
|---|---|---|
| KA | −11.81 | Competitive |
| INH1 | −12.05 | Mix |
| INH2 | −14.10 | Competitive |
| INH3 | −11.33 | ND * |
| INH4 | −13.08 | Competitive |
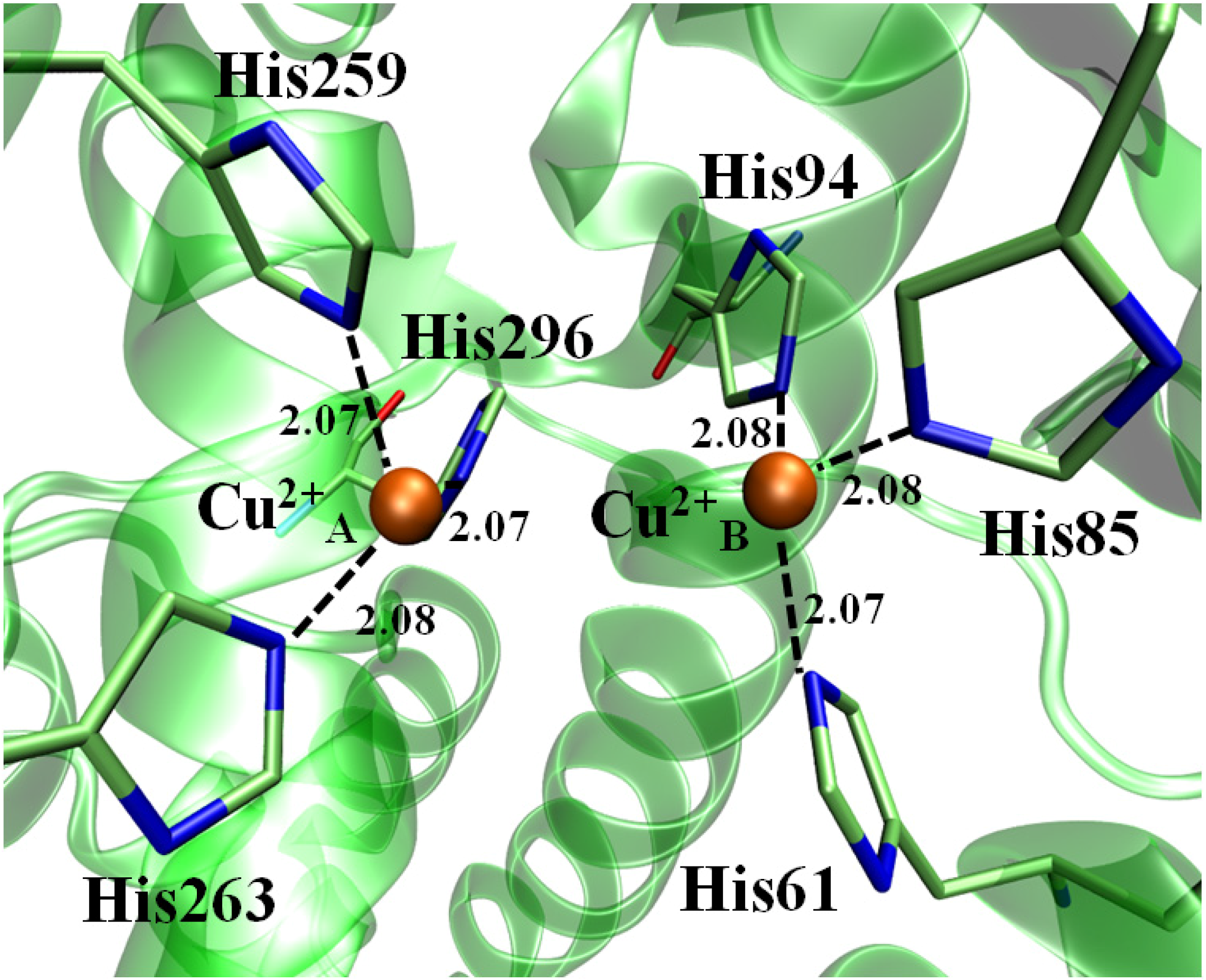
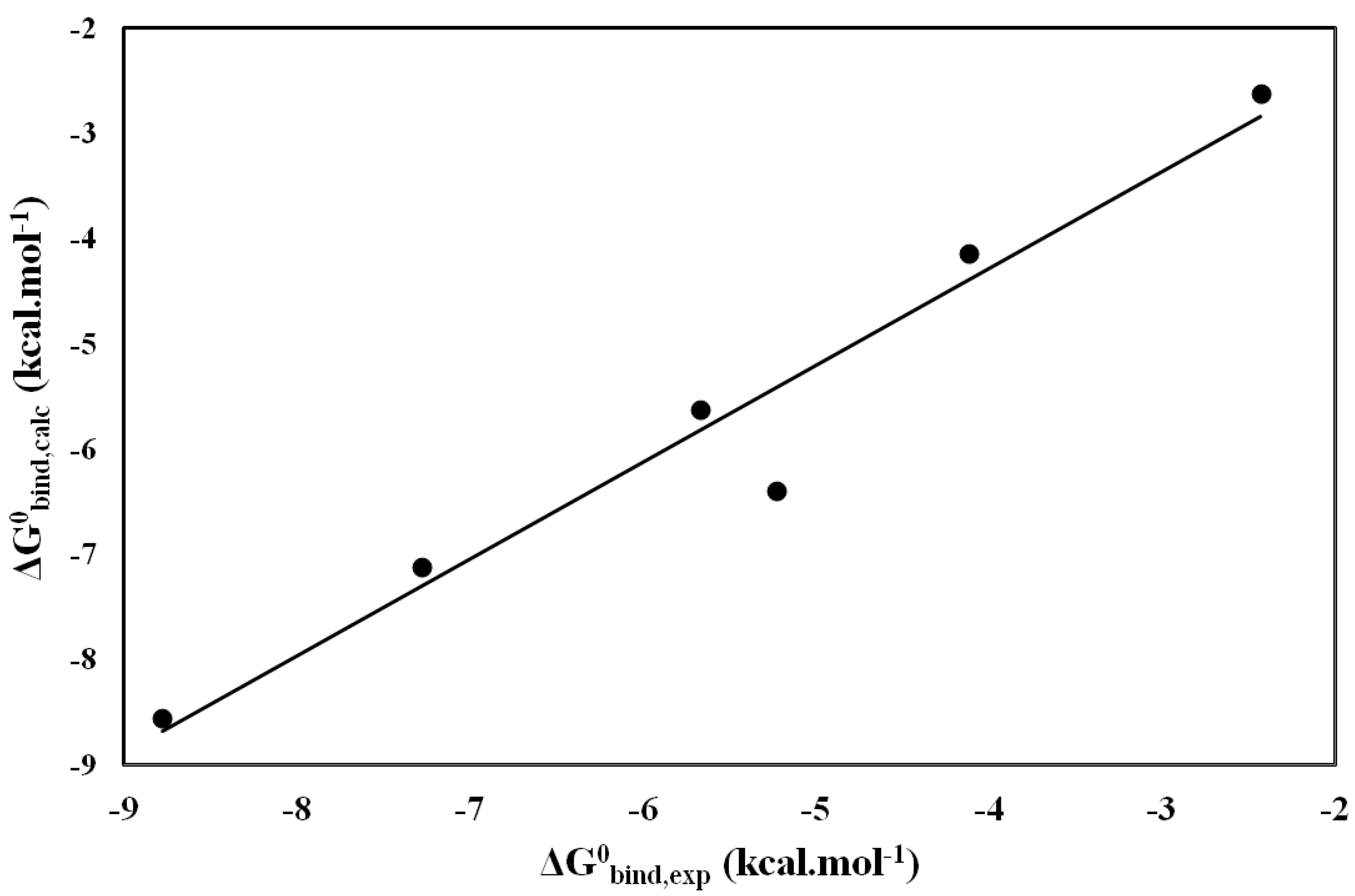
2.3. Molecular Dynamics (MD) and LIE
| His Residue | Cu2+ ion | Atomic Distance |
|---|---|---|
| His60(NE2) | Cu2+ A | 2.07 |
| His84(NE2) | 2.08 | |
| His93(NE2) | 2.08 | |
| His258(NE2) | Cu2+ B | 2.07 |
| His262(NE2) | 2.07 | |
| His295(NE2) | 2.08 |
| Inhibitors |  bind bind | free | bind | free |  |  |
|---|---|---|---|---|---|---|
| Tropolone | −17.89 | −10.39 | −55.12 | −35.68 | −8.78 | −8.55 |
| KA | −22.9 | −9.57 | −47.13 | −32.86 | −7.28 | −7.12 |
| INH1 | −18.85 | −12.77 | −26.54 | −18.32 | −4.12 | −4.14 |
| INH2 | −21.79 | −9.71 | −52.83 | −40.08 | −5.23 | −6.40 |
| INH3 | −18.14 | −11.08 | −23.21 | −19.59 | −2.43 | −2.62 |
| INH4 | −19.53 | −11.84 | −42.23 | −30.79 | −5.76 | −5.62 |
 and
and  (energy values in kcal·mol−1).
(energy values in kcal·mol−1).
3. Experimental
3.1. Experimental Section
3.1.1. Tyrosinase Activity
3.1.2. Determination of Inhibition Constant (Ki)
- Competitive Inhibition:
![Molecules 19 09591 i007]()
- Mixed Inhibition:
![Molecules 19 09591 i008]()


{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.2. Computational Section
3.2.1. Molecular Docking


3.2.2. Molecular Dynamics Simulations and Free Energy Binding


4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gupta, S.; Mukhtar, H. Chemoprevention of skin cancer through natural agents. Skin Pharmacol. Appl. Skin Physiol. 2001, 14, 373–385. [Google Scholar] [CrossRef]
- Rao, A.R.; Sindhuja, H.N.; Dharmesh, S.M.; Sankar, K.U.; Sarada, R.; Ravishankar, G.A. Effective Inhibition of skin cancer, tyrosinase, and antioxidative properties by astaxanthin and astaxanthin esters from the green alga Haematococcus pluvialis. J. Agric. Food Chem. 2013, 61, 3842–3851. [Google Scholar] [CrossRef]
- Yin, S.-J.; Si, Y.-X.; Qian, G.-Y. Inhibitory Effect of phthalic Acid on tyrosinase: The mixed-type inhibition and docking simulations. Enzym. Res. 2011, 2011, 294724. [Google Scholar]
- Jimbow, K.; Park, J.S.; Kato, F.; Hirosaki, K.; Toyofuku, K.; Hua, C.; Yamashita, T. Assembly, target-signaling and intracellular transport of tyrosinase gene family proteins in the initial stage of melanosome biogenesis. Pigment. Cell Res. 2000, 13, 222–229. [Google Scholar]
- Olivares, C.; Solano, F. New insights into the active site structure and catalytic mechanism of tyrosinase and its related proteins. Pigment Cell Melanoma Res. 2009, 22, 750–760. [Google Scholar] [CrossRef]
- Kanost, M.R.; Jiang, H.; Yu, X.-Q. Innate immune responses of a lepidopteran insect, Manduca sexta. Immunol. Rev. 2004, 198, 97–105. [Google Scholar] [CrossRef]
- Lai, S.-C.; Chen, C.-C.; Hou, R.F. Immunolocalization of prophenoloxidase in the process of wound healing in the mosquito Armigeres subalbatus (Diptera: Culicidae). J. Med. Entomol. 2002, 39, 266–274. [Google Scholar] [CrossRef]
- Yoon, J.; Fujii, S.; Solomon, E.I. Geometric and electronic structure differences between the type 3 copper sites of the multicopper oxidases and hemocyanin/tyrosinase. Proc. Natl. Acad. Sci. USA 2009, 106, 6585–6590. [Google Scholar] [CrossRef]
- Si, Y.-X.; Wang, Z.-J.; Park, D.; Chung, H.Y.; Wang, S.-F.; Yan, L.; Yang, J.-M.; Qian, G.-Y.; Yin, S.-J.; Park, Y.-D. Effect of hesperetin on tyrosinase: Inhibition kinetics integrated computational simulation study. Int. J. Biol. Macromol. 2012, 50, 257–262. [Google Scholar] [CrossRef]
- Chen, Q.-X.; Kubo, I. Kinetics of mushroom tyrosinase inhibition by quercetin. J. Agric. Food Chem. 2002, 50, 4108–4112. [Google Scholar] [CrossRef]
- Prezioso, J.A.; Epperly, M.W.; Wang, N.; Bloomer, W.D. Effects of tyrosinase activity on the cytotoxicity of 4-S-cysteaminylphenol and N-acetyl-4-S-cysteaminylphenol in melanoma cells. Cancer Lett. 1992, 63, 73–79. [Google Scholar] [CrossRef]
- Greggio, E.; Bergantino, E.; Carter, D.; Ahmad, R.; Costin, G.-E.; Hearing, V.J.; Clarimon, J.; Singleton, A.; Eerola, J.; Hellström, O. Tyrosinase exacerbates dopamine toxicity but is not genetically associated with Parkinson’s disease. J. Neurochem. 2005, 93, 246–256. [Google Scholar] [CrossRef]
- Kim, D.; Park, J.; Kim, J.; Han, C.; Yoon, J.; Kim, N.; Seo, J.; Lee, C. Flavonoids as mushroom tyrosinase Inhibitors: A fluorescence quenching study. J. Agric. Food Chem. 2006, 54, 935–941. [Google Scholar] [CrossRef]
- Kanade, S.R.; Suhas, V.L.; Chandra, N.; Gowda, L.R. Functional interaction of diphenols with polyphenol oxidase: Molecular determinants of substrate/inhibitor specificity. FEBS J. 2007, 274, 4177–4187. [Google Scholar] [CrossRef]
- Yokota, T.; Nishio, H.; Kubota, Y.; Mizoguchi, M. The inhibitory effect of glabridin from licorice extracts on melanogenesis and inflammation. Pigment Cell Res. 1998, 11, 355–361. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kawano, Y.; Yamanaka, A.; Maruyama, S. N-[(Dihydroxyphenyl)acyl]serotonins as potent inhibitors of tyrosinase from mouse and human melanoma cells. Bioorganic Med. Chem. Lett. 2009, 19, 4178–4182. [Google Scholar]
- Yan, Q.; Cao, R.; Yi, W.; Yu, L.; Chen, Z.; Ma, L.; Song, H. Synthesis and evaluation of 5-benzylidene(thio)barbiturate-β-d-glycosides as mushroom tyrosinase inhibitors. Bioorg. AMP Med. Chem. Lett. 2009, 19, 4055–4058. [Google Scholar] [CrossRef]
- Shiino, M.; Watanabe, Y.; Umezawa, K. Synthesis of N-substituted N-nitrosohydroxylamines as inhibitors of Mushroom Tyrosinase. Bioorg. AMP Med. Chem. 2001, 9, 1233–1240. [Google Scholar] [CrossRef]
- Ismaya, W.T.; Rozeboom, H.T.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef]
- Muñoz-Muñoz, J.L.; Garcia-Molina, F.; Varon, R.; Garcia-Ruíz, P.A.; Tudela, J.; Garcia-Cánovas, F.; Rodríguez-López, J.N. Suicide inactivation of the diphenolase and monophenolase activities of tyrosinase. IUBMB Life 2010, 62, 539–547. [Google Scholar] [CrossRef]
- Si, Y.-X.; Yin, S.-J.; Park, D.; Chung, H.Y.; Yan, L.; Lü, Z.-R.; Zhou, H.-M.; Yang, J.-M.; Qian, G.-Y.; Park, Y.-D. Tyrosinase inhibition by isophthalic acid: Kinetics and computational simulation. Int. J. Biol. Macromol. 2011, 48, 700–704. [Google Scholar] [CrossRef]
- Espin, J.C.; Wichers, H.J. Slow-binding inhibition of mushroom (Agaricus bisporus) tyrosinase isoforms by tropolone. J. Agric. Food Chem. 1999, 47, 2638–2644. [Google Scholar]
- Chang, T.-S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef]
- Ha, Y.M.; Chung, S.W.; Song, S.; Lee, H.; Suh, H.; Chung, H.Y. 4-(6-Hydroxy-2-naphthyl)-1,3-bezendiol: A potent, new tyrosinase inhibitor. Biol. Pharm. Bull. 2007, 30, 1711–1715. [Google Scholar] [CrossRef]
- Moto, M.; Mori, T.; Okamura, M.; Kashida, Y.; Mitsumori, K. Absence of liver tumor-initiating activity of kojic acid in mice. Arch. Toxicol. 2006, 80, 299–304. [Google Scholar] [CrossRef]
- Rodrigues, A.P.D.; Carvalho, A.S.C.; Santos, A.S.; Alves, C.N.; do Nascimento, J.L.M.; Silva, E.O. Kojic acid, a secondary metabolite from Aspergillus sp., acts as an inducer of macrophage activation. Cell Biol. Int. 2011, 35, 335–343. [Google Scholar] [CrossRef]
- Liu, X.-X.; Sun, S.-Q.; Wang, Y.-J.; Xu, W.; Wang, Y.-F.; Park, D.; Zhou, H.-M.; Han, H.-Y. Kinetics and computational docking studies on the inhibition of tyrosinase induced by oxymatrine. Appl. Biochem. Biotechnol. 2013, 169, 145–158. [Google Scholar]
- Hu, W.-J.; Yan, L.; Park, D.; Jeong, H.O.; Chung, H.Y.; Yang, J.-M.; Ye, Z.M.; Qian, G.-Y. Kinetic, structural and molecular docking studies on the inhibition of tyrosinase induced by arabinose. Int. J. Biol. Macromol. 2012, 50, 694–700. [Google Scholar] [CrossRef]
- Gąsowska, B.; Kafarski, P.; Wojtasek, H. Interaction of mushroom tyrosinase with aromatic amines, o-diamines and o-aminophenols. Biochim. Biophys. Acta Gen. Subj. 2004, 1673, 170–177. [Google Scholar] [CrossRef]
- Khatib, S.; Nerya, O.; Musa, R.; Shmuel, M.; Tamir, S.; Vaya, J. Chalcones as potent tyrosinase inhibitors: The importance of a 2,4-substituted resorcinol moiety. Bioorg. AMP Med. Chem. 2005, 13, 433–441. [Google Scholar] [CrossRef]
- Munoz-Munoz, J.L.; Garcia-Molina, F.; Garcia-Ruiz, P.A.; Molina-Alarcon, M.; Tudela, J.; Garcia-Canovas, F.; Rodriguez-Lopez, J.N. Phenolic substrates and suicide inactivation of tyrosinase: Kinetics and mechanism. Biochem. J. 2008, 416, 431–440. [Google Scholar] [CrossRef]
- Alencar, N.A.N.D.; Sousa, P.R.M.; Silva, J.R.A.; Lameira, J.; Alves, C.N.; Martí, S.; Moliner, V. Computational analysis of human OGA Structure in complex with PUGNAc and NAG-thiazoline derivatives. J. Chem. Inf. Model. 2012, 52, 2775–2783. [Google Scholar] [CrossRef]
- Gutiérrez-de-Terán, H.; Nervall, M.; Dunn, B.M.; Clemente, J.C.; Åqvist, J. Computational analysis of plasmepsin IV bound to an allophenylnorstatine inhibitor. FEBS Lett. 2006, 580, 5910–5916. [Google Scholar] [CrossRef]
- Kubo, I.; Kinst-Hori, I.; Chaudhuri, S.K.; Kubo, Y.; Sanchez, Y.; Ogura, T. Flavonols from Heterotheca inuloides: Tyrosinase inhibitory activity and structural criteria. Bioorg. Med. Chem. 2000, 8, 1749–1755. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Gehlhaar, D.; Bouzida, D.; Rejto, P. Fully automated and rapid flexible docking of inhibitors covalently bound to serine proteases. In Evolutionary Programming VII; Porto, V.W., Saravanan, N., Waagen, D., Eiben, A.E., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; Volume 1447, pp. 449–461. [Google Scholar]
- Gehlhaar, D.K.; Verkhivker, G.; Rejto, P.A.; Fogel, D.B.; Fogel, L.J.; Freer, S.T. Docking conformationally Flexible small molecules into a protein binding site through evolutionary programming. In Evolutionary Programming; Morgan Kaufmann: San Mateo, CA, USA, 1995; pp. 615–627. [Google Scholar]
- Yang, J.-M.; Chen, C.-C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins Struct. Funct. Bioinform. 2004, 55, 288–304. [Google Scholar] [CrossRef]
- Hansson, T.; Marelius, J.; Åqvist, J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Aided Mol. Des. 1998, 12, 27–35. [Google Scholar] [CrossRef]
- Antosiewicz, J.; McCammon, J.A.; Gilson, M.K. Prediction of Ph-dependent properties of proteins. J. Mol. Biol. 1994, 238, 415–436. [Google Scholar] [CrossRef]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins Struct. Funct. Bioinform. 2005, 61, 704–721. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- King, G.; Warshel, A. A surface constrained all-atom solvent model for effective simulations of polar solutions. J. Chem. Phys. 1989, 91, 3647–3661. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Lee, F.S.; Warshel, A. A local reaction field method for fast evaluation of long-range electrostatic interactions in molecular simulations. J. Chem. Phys. 1992, 97, 3100–3107. [Google Scholar] [CrossRef]
- Jorgensen, W.; Maxwell, D.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- qvist, J.; Medina, C.; Samuelsson, J.-E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, C.R.; Silva, J.R.A.; De Tássia Carvalho Cardoso, É.; Silva, E.O.; Lameira, J.; Do Nascimento, J.L.M.; Do Socorro Barros Brasil, D.; Alves, C.N. Combined Kinetic Studies and Computational Analysis on Kojic Acid Analogs as Tyrosinase Inhibitors. Molecules 2014, 19, 9591-9605. https://doi.org/10.3390/molecules19079591
Lima CR, Silva JRA, De Tássia Carvalho Cardoso É, Silva EO, Lameira J, Do Nascimento JLM, Do Socorro Barros Brasil D, Alves CN. Combined Kinetic Studies and Computational Analysis on Kojic Acid Analogs as Tyrosinase Inhibitors. Molecules. 2014; 19(7):9591-9605. https://doi.org/10.3390/molecules19079591
Chicago/Turabian StyleLima, Carlyle Ribeiro, José Rogério A. Silva, Érica De Tássia Carvalho Cardoso, Edilene O. Silva, Jerônimo Lameira, José Luiz Martins Do Nascimento, Davi Do Socorro Barros Brasil, and Cláudio N. Alves. 2014. "Combined Kinetic Studies and Computational Analysis on Kojic Acid Analogs as Tyrosinase Inhibitors" Molecules 19, no. 7: 9591-9605. https://doi.org/10.3390/molecules19079591