Relative Quantitation of Glycopeptides Based on Stable Isotope Labeling Using MALDI-TOF MS

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
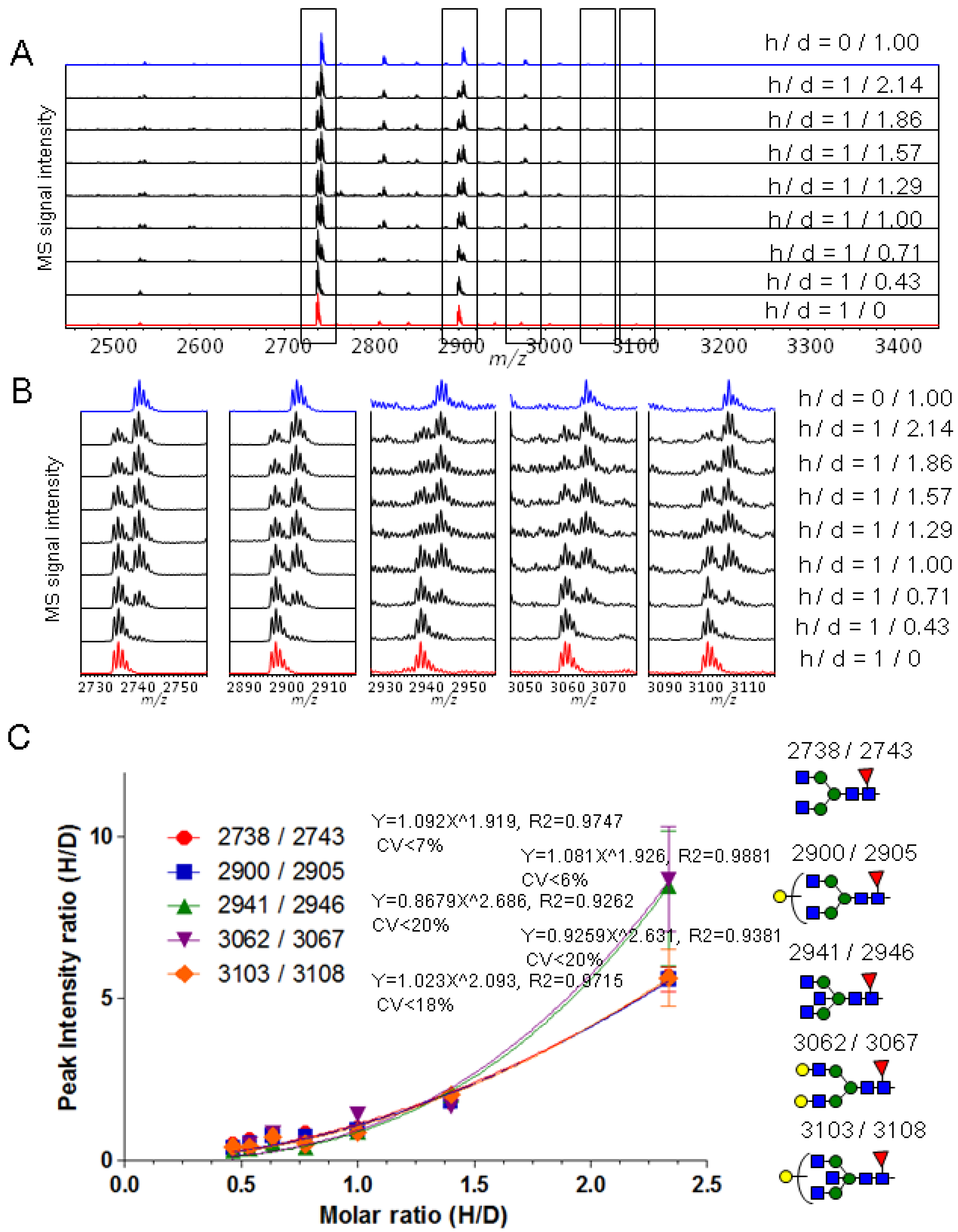
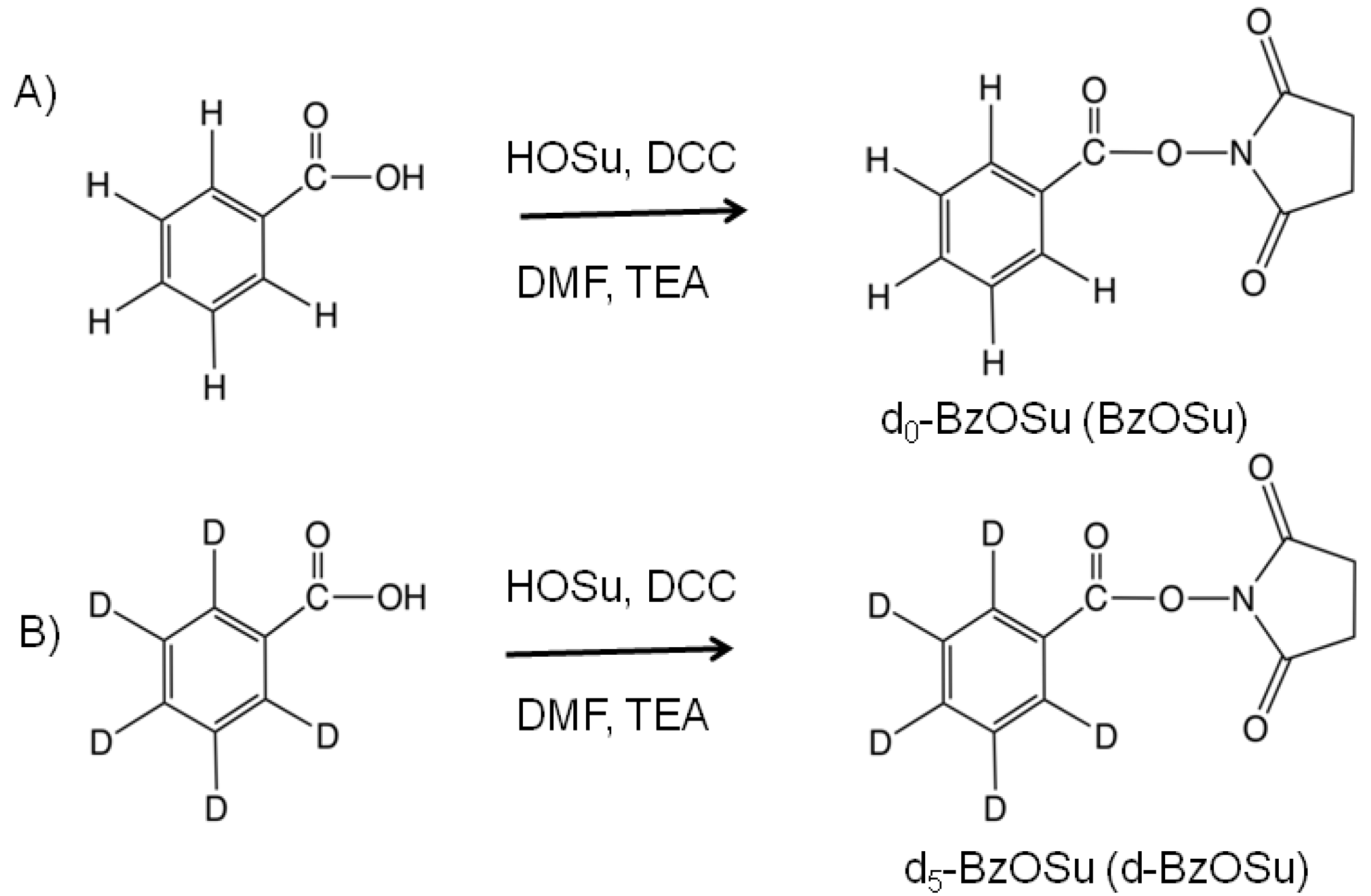
2.1. Synthesis and Evaluation of Stable Isotope-Labeled Amine-Reactive Mass Tag Reagent for Glycopeptides



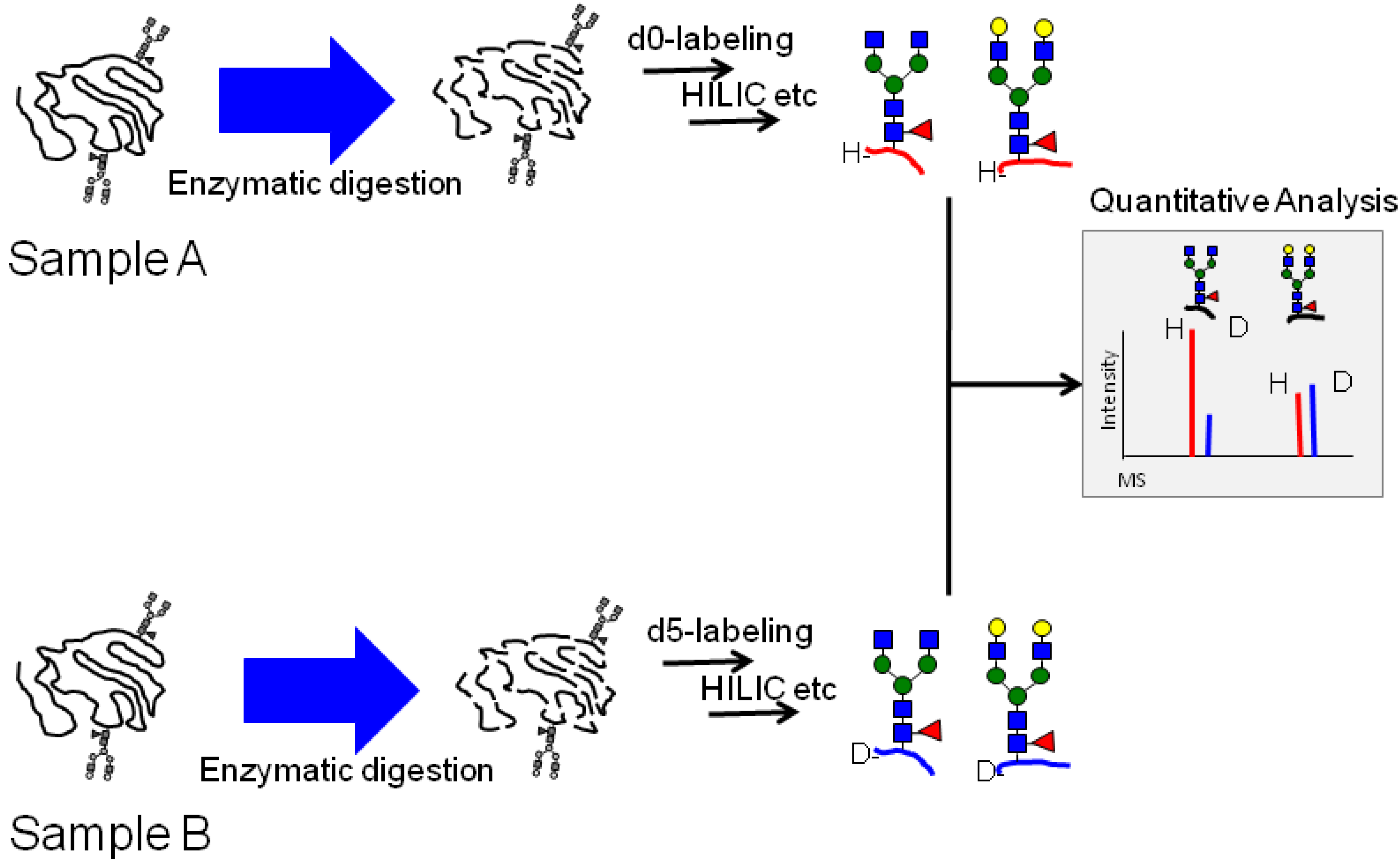
2.2. Calibration of IgG1 Glycopeptides Using Stable Isotope Labeling

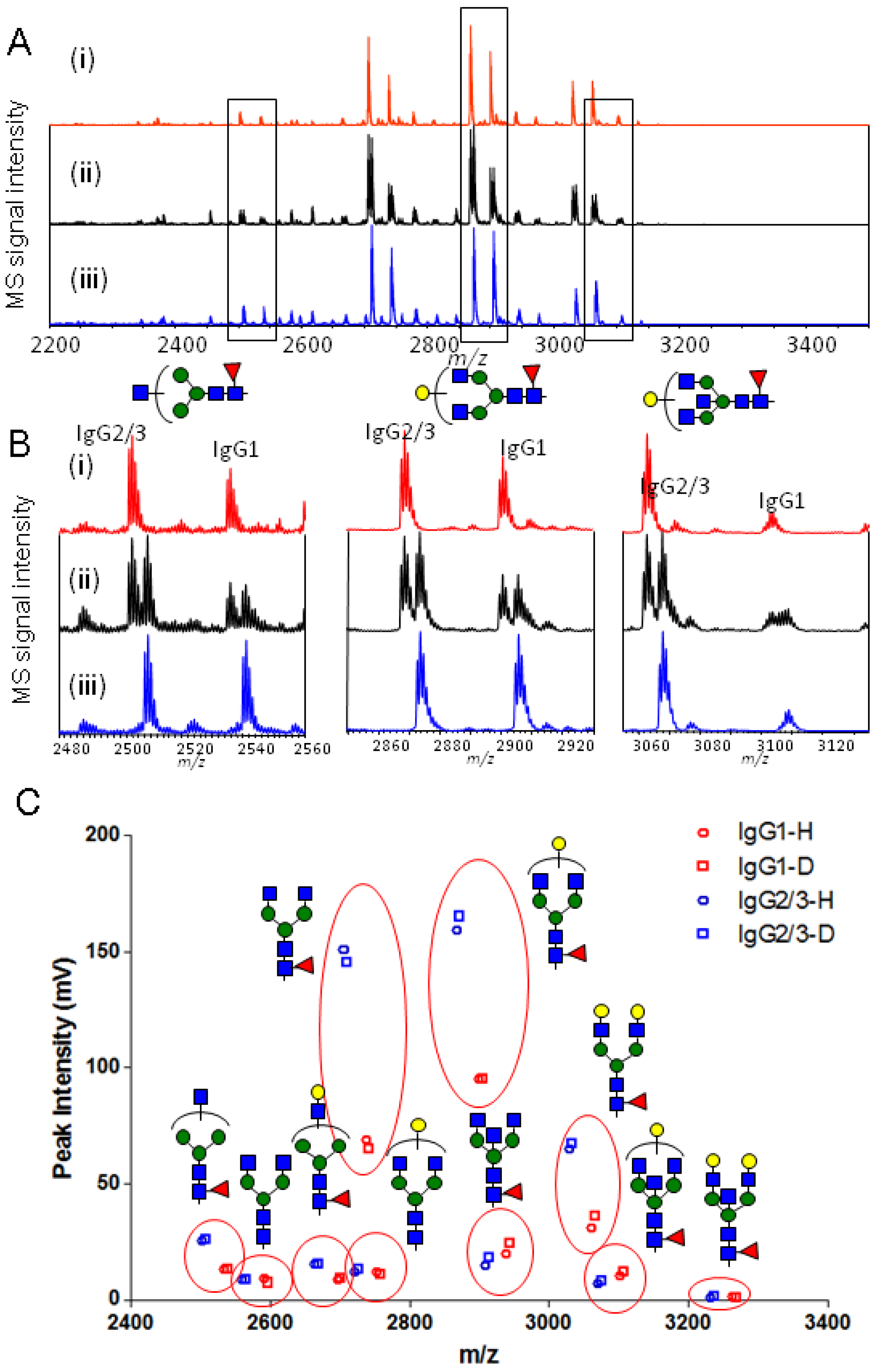
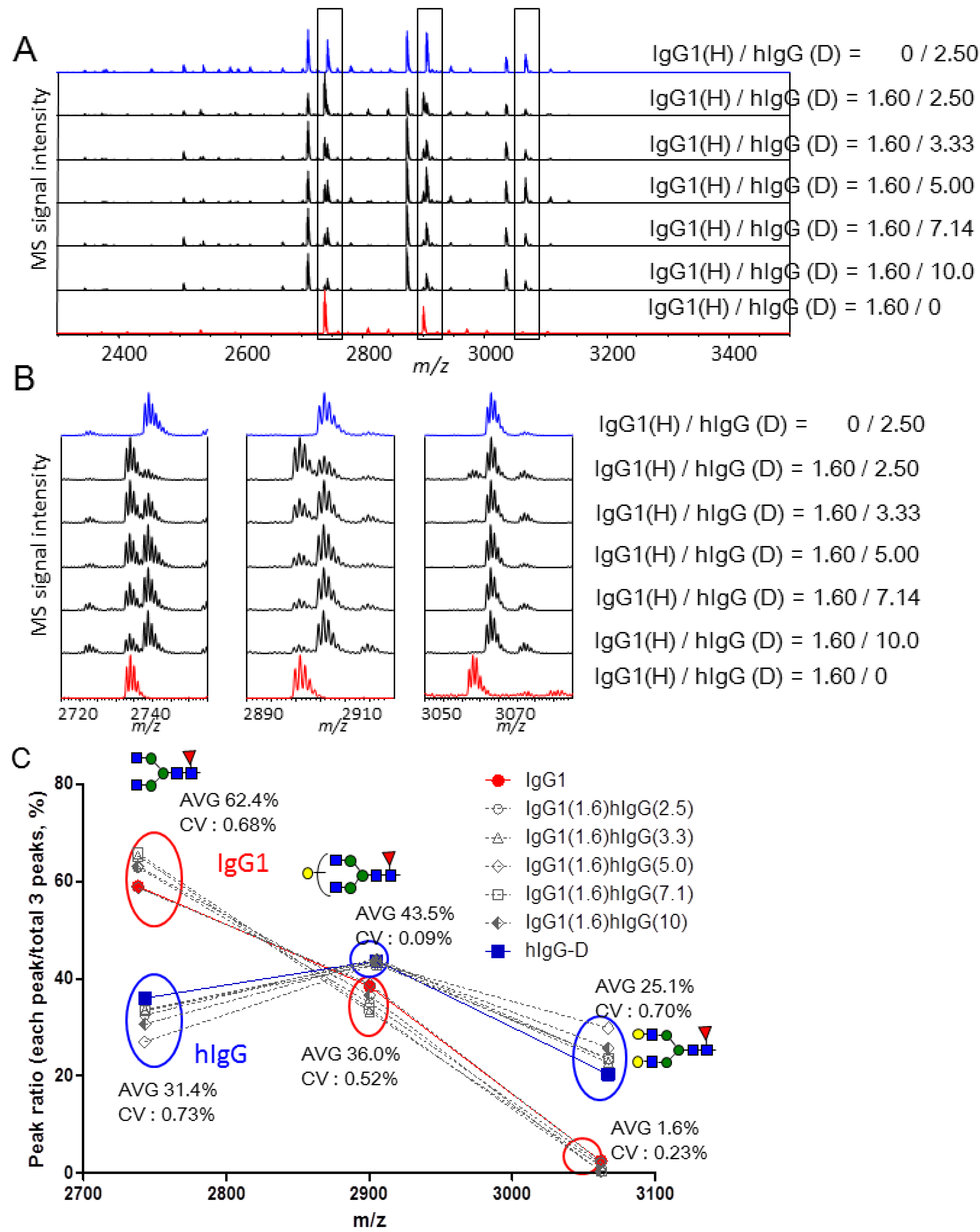
2.3. Quantitative Comparison of Glycopeptides from Human Serum IgG and IgG1 κ from Myeloma Plasma

2.4. Quantitative Monitoring of Glycopeptides for Enzymatic Reaction using Stable Isotope Labeling
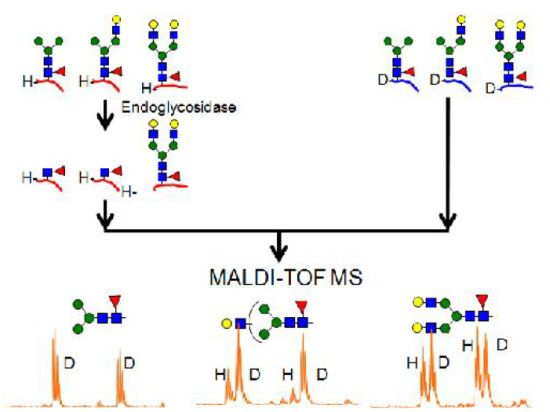
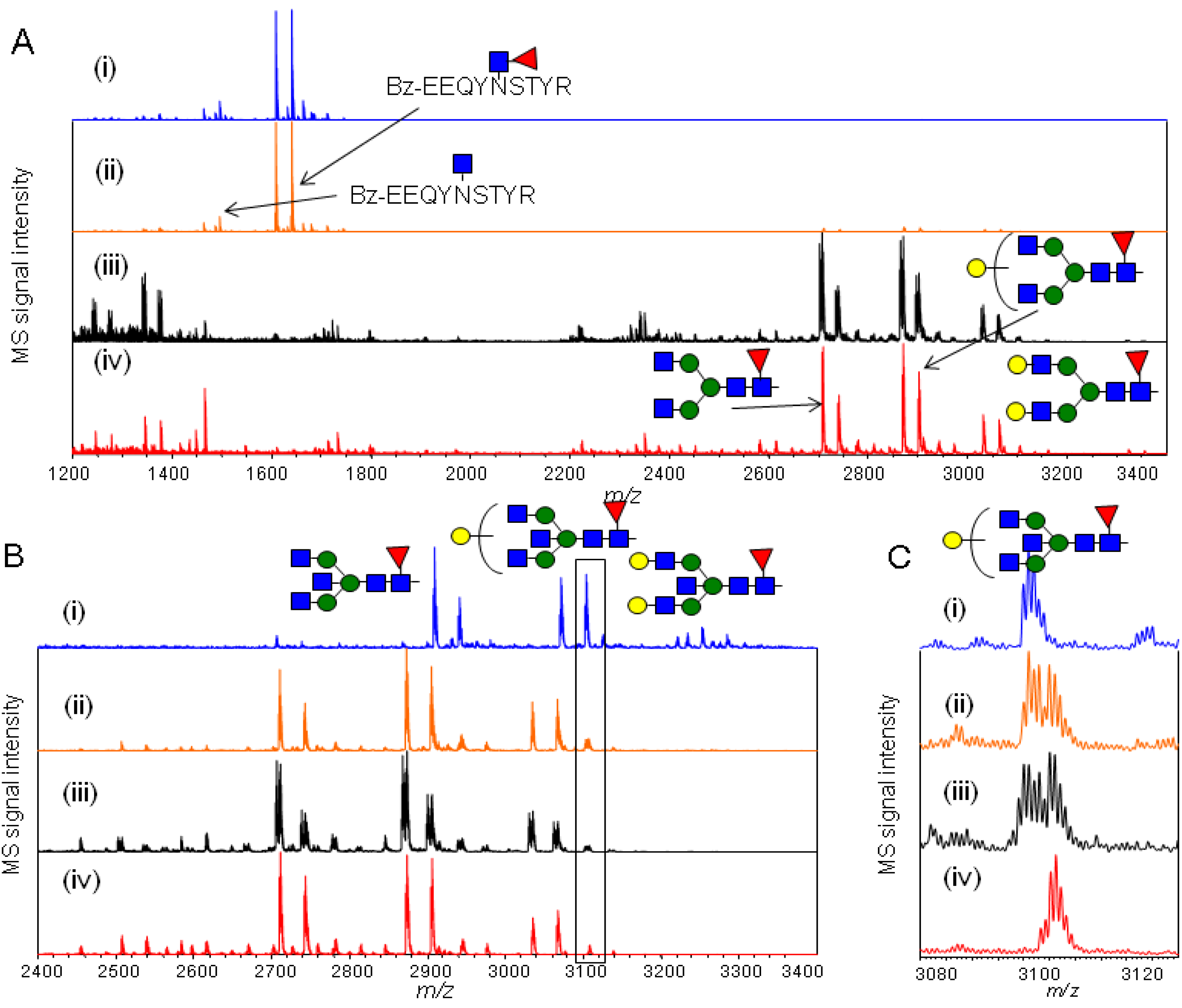
2.4.1. Substrate Specificity of Endo-β-N-acetylglucosaminidase from Streptococcus Pyogenes (endoS) for hIgG Glycopeptides
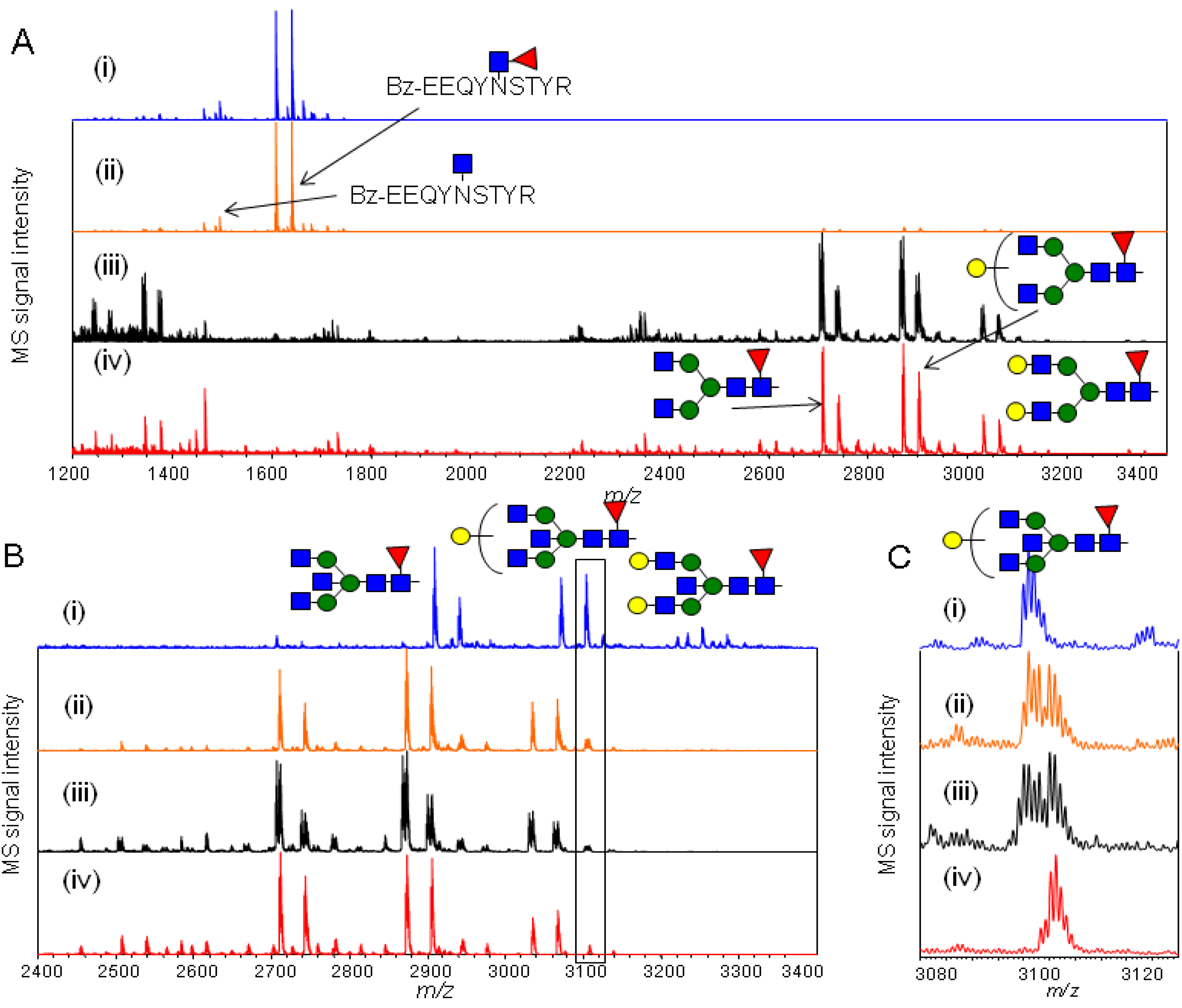
2.4.2. Characterization of the hIgG Glycopeptide Isoforms by endo-β-N-Acetylglucosaminidase from Streptococcus pneumoniae (endo-D) Combined with exo-β-N-Acetylglucosaminidase (β-GlcNAc’ase)

3. Experimental Section
3.1. General Information
3.2. Synthesis of Benzoic Acid N-Succinimidyl Ester (Bz and d-Bz Labeling Reagents)
3.3. Preparation of hIgG and IgG1 Glycopeptides
3.4. Stable Isotope Labeling of Glycopeptides
3.5. Enzymatic Reaction of Bz-Labeled Glycopeptides with EndoS
3.6. Enzymatic Reaction of Labeled Glycopeptides by β-N-Acetylglucosaminidase (β-GlcNAc’ase)
3.7. Enzymatic Reaction of Bz-Labeled Glycopeptides by Endo-D
3.8. MALDI Sample Preparation and Measurement
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar]
- Roth, J. Protein N-Glycosylation along the secretory pathway: Relationship to organelle topography and function, protein quality control, and cell interactions. Chem. Rev. 2002, 102, 285–303. [Google Scholar]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochimi. Biophys. Acta-Gen. Subj. 1999, 1473, 4–8. [Google Scholar]
- Budnik, B.A.; Lee, R.S.; Steen, J.A. Global methods for protein glycosylation analysis by mass spectrometry. Biochim. Biophys. Acta 2006, 1764, 1870–1880. [Google Scholar]
- Geng, M.; Zhang, X.; Bina, M.; Regnier, F. Proteomics of glycoprotein’s based on affinity selection of glycopeptides from tryptic digests. J. Chromatogr. B Biomed. Sci. Appl. 2001, 752, 293–306. [Google Scholar]
- Kaji, H.; Saito, H.; Yamauchi, Y.; Shinkawa, T.; Taoka, M.; Hirabayashi, J.; Kasai, K.; Takahashi, N.; Isobe, T. Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoprotein’s. Nat. Biotechnol. 2003, 21, 667–672. [Google Scholar]
- Zhang, H.; Li, X.J.; Martin, D.B.; Aebersold, R. Identification and quantification of N-linked glycoprotein’s using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003, 21, 660–666. [Google Scholar]
- Kurogochi, M.; Amano, M.; Fumoto, M.; Takimoto, A.; Kondo, H.; Nishimura, S.-I. Reverse glycoblotting allows rapid enrichment glycoproteomics of biopharmaceuticals and disease-related biomarkers. Angew. Chem. Int. Ed. Engl. 2007, 46, 8808–8813. [Google Scholar]
- Nilsson, J.; Ruetshi, U.; Halim, A.; Hesse, C.; Carlsohn, E.; Brinkmalm, G.; Larson, G. Enrichment of glycopeptides for glycan structure and attachment site identification. Nat. Methods 2009, 6, 809–811. [Google Scholar]
- Kurogochi, M.; Matsushita, T.; Amano, M.; Furukawa, J.-I.; Shinohara, Y.; Aoshima, M.; Nishimura, S.-I. Sialic acid-focused quantitative mouse serum glycoproteomics by Multiple Reaction Monitoring Assay. Mol. Cell. Proteomics 2010, 9, 2354–2368. [Google Scholar]
- Sparbier, K.; Koch, S.; Kessler, I.; Wenzel, T.; Kostrzewa, M. Selective isolation of glycoproteins and glycopeptides for MALDI-TOF MS detection supported by magnetic particles. J. Biomol. Tech. 2005, 16, 407–413. [Google Scholar]
- Sparbier, K.; Wenzel, T.; Kostrzewa, M. Exploring the binding profiles of ConA, boronic acid and WGA by MALDI-TOF/TOF MS and magnetic particles. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2006, 840, 29–36. [Google Scholar]
- Wada, Y.; Tajiri, M.; Yoshida, S. Hydrophilic affinity isolation and MALDI multiple-stage tandem mass spectrometry of glycopeptides for glycoproteomics. Anal. Chem. 2004, 76, 6560–6565. [Google Scholar]
- Hägglund, P.; Bunkenborg, J.; Elortza, F.; Jensen, O.N.; Roepstorff, P. A new strategy for identification of N-glycosylated proteins and unambiguous assignment of their glycosylation sites using HILIC enrichment and partial deglycosylation. J. Proteome Res. 2004, 3, 556–566. [Google Scholar]
- Segu, Z.M.; Mechref, Y. Characterizing protein glycosylation sites through higher-energy C-trap dissociation. Rapid Commun. Mass Spectrom. 2010, 24, 1217–1225. [Google Scholar]
- Håkansson, K.; Chalmers, M.J.; Quinn, J.P.; McFarland, M.A.; Hendrickson, C.L.; Marshall, A.G. Combined electron capture and infrared multiphoton dissociation for multistage MS/MS in a Fourier transform ion cyclotron resonance mass spectrometer. Anal. Chem. 2003, 75, 3256–3262. [Google Scholar]
- Håkansson, K.; Cooper, H.J.; Emmett, M.R.; Costello, C.E.; Marshall, A.G.; Nilsson, C.L. Electron capture dissociation and infrared multiphoton dissociation MS/MS of an N-glycosylated tryptic peptic to yield complementary sequence information. Anal. Chem. 2001, 73, 4530–4536. [Google Scholar]
- Haselmann, K.F.; Budnik, B.A.; Olsen, J.V.; Nielsen, M.L.; Reis, C.A.; Clausen, H.; Johnsen, A.H.; Zubarev, R.A. Advantages of external accumulation for electron capture dissociation in Fourier transform mass spectrometry. Anal. Chem. 2001, 3, 2998–3005. [Google Scholar]
- Kjeldsen, F.; Haselmann, K.F.; Sørensen, E.S.; Zubarev, R.A. Distinguishing of Ile/Leu amino acid residues in the PP3 protein by (hot) electron capture dissociation in Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2003, 75, 1267–1274. [Google Scholar]
- Mirgorodskaya, E.; Roepstorff, P.; Zubarev, R.A. Localization of O-glycosylation sites in peptides by electron capture dissociation in a Fourier transform mass spectrometer. Anal. Chem. 1999, 71, 4431–4436. [Google Scholar]
- Alley, W.R., Jr.; Mechref, Y.; Novotny, M.V. Characterization of glycopeptides by combining collision-induced dissociation and electron-transfer dissociation mass spectrometry data. Rapid Commun. Mass Spectrom. 2009, 23, 161–170. [Google Scholar]
- Snovida, S.I.; Bodnar, E.D.; Viner, R.; Saba, J.; Perreault, H. A simple cellulose column procedure for selective enrichment of glycopeptides and characterization by nano LC coupled with electron-transfer and high-energy collisional-dissociation tandem mass spectrometry. Carbohydr. Res. 2010, 345, 792–801. [Google Scholar]
- Eng, J.K.; McCormack, A.L.; Yates, J.R., III. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar]
- Craig, R.; Beavis, R.C. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar]
- Deutsch, E.W.; Lam, H.; Aebersold, R. PeptideAtlas: A resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008, 9, 429–434. [Google Scholar]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. [Google Scholar]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [Google Scholar]
- Mirgorodskaya, O.A.; Kozmin, Y.P.; Titov, M.I.; Körner, R.; Sönksen, C.P.; Roepstorff, P. Quantitation of peptides and proteins by matrix-assisted laser desorption/ionization mass spectrometry using (18)O-labeled internal standards. Rapid Commun. Mass Spectrom. 2000, 14, 1226–1232. [Google Scholar]
- Anderson, L.; Hunter, C.L. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 2006, 5, 573–588. [Google Scholar]
- Uematsu, R.; Furukawa, J.; Nakagawa, H.; Shinohara, Y.; Deguchi, K.; Monde, K.; Nishimura, S. High throughput quantitative glycomics and glycoform-focused proteomics of murine dermis and epidermis. Mol. Cell. Proteomics 2005, 4, 1977–1989. [Google Scholar]
- Xia, B.; Feasley, C.L.; Sachdev, G.P.; Smith, D.F.; Cummings, R.D. Glycan reductive isotope labeling for quantitative glycomics. Anal. Biochem. 2009, 387, 162–170. [Google Scholar]
- Amano, J.; Nishikaze, T.; Tougasaki, F.; Jinmei, H.; Sugimoto, I.; Sugawara, S.; Fujita, M.; Osumi, K.; Mizuno, M. Derivatization with 1-pyrenyldiazomethane enhances ionization of glycopeptides but not peptides in matrix-assisted laser desorption ionization mass spectrometry. Anal. Chem. 2010, 82, 8738–8743. [Google Scholar]
- Taniguchi, K.; Kuyama, H.; Kajihara, S.; Tanaka, K. MALDI mass spectrometry-based sequence analysis of arginine-containing glycopeptides: Improved fragmentation of glycan and peptide chains by modifying arginine residue. J. Mass Spectrom. 2013, 48, 951–960. [Google Scholar]
- Huang, Z.H.; Wu, J.; Roth, K.D.; Yang, Y.; Gage, D.A.; Watson, J.T. A picomole-scale method for charge derivatization of peptides for sequence analysis by mass spectrometry. Anal. Chem. 1997, 69, 137–144. [Google Scholar]
- Dayon, L.; Hainard, A.; Licker, V.; Turck, N.; Kuhn, K.; Hochstrasser, D.F.; Burkhard, P.R.; Sanchez, J.C. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal. Chem. 2008, 80, 2921–2931. [Google Scholar]
- Hahne, H.; Neubert, P.; Kuhn, K.; Etienne, C.; Bomgarden, R.; Rogers, J.C.; Kuster, B. Carbonyl-reactive tandem mass tags for the proteome-wide quantification of N-linked glycans. Anal. Chem. 2012, 84, 3716–3724. [Google Scholar]
- Hsu, J.L.; Huang, S.Y.; Chow, N.H.; Chen, S.H. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 2003, 75, 6843–6852. [Google Scholar]
- Ji, C.; Guo, N.; Li, L. Differential dimethyl labeling of N-termini of peptides after guanidination for proteome analysis. J. Proteome Res. 2005, 4, 2099–2108. [Google Scholar]
- Lin, C.Y.; Ma, Y.C.; Pai, P.J.; Her, G.R. A comparative study of glycoprotein concentration, glycoform profile and glycosylation site occupancy using isotope labeling and electrospray linear ion trap mass spectrometry. Anal. Chim. Acta 2012, 728, 49–56. [Google Scholar]
- Morell, A.; Skvaril, F.; Hitzig, W.H.; Barandun, S. IgG subclasses: Development of the serum concentrations in "normal" infants and children. J. Pediatr. 1972, 80, 960–964. [Google Scholar]
- Wuhrer, M.; Stam, J.C.; van de Geijn, F.E.; Koeleman, C.A.; Verrips, C.T.; Dolhain, R.J.; Hokke, C.H.; Deelder, A.M. Glycosylation profiling of immunoglobulin G (IgG) subclasses from human serum. Proteomics 2007, 7, 4070–4081. [Google Scholar]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar]
- Anderson, N.L.; Razavi, M.; Pearson, T.W.; Kruppa, G.; Paape, R.; Suckau, D. Precision of heavy-light peptide ratios measured by maldi-tof mass spectrometry. J. Proteome Res. 2012, 11, 1868–1878. [Google Scholar]
- Collin, M.; Olsén, A. EndoS, a novel secreted protein from Streptococcus pyogenes with endoglycosidase activity on human IgG. EMBO J. 2001, 20, 3046–3055. [Google Scholar]
- Dixon, E.V.; Claridge, J.K.; Harvey, D.J.; Baruah, K.; Yu, X.; Vasiljevic, S.; Mattick, S.; Pritchard, L.K.; Krishna, B.; Scanlan, C.N.; et al. Fragments of bacterial endoglycosidase S and immunoglobulin G reveal subdomains of each that contribute to deglycosylation. J. Biol. Chem. 2014, 289, 13876–13889. [Google Scholar]
- Tomiya, N.; Kurono, M.; Ishihara, H.; Tejima, S.; Endo, S.; Arata, Y.; Takahashi, N. Structural analysis of N-linked oligosaccharides by a combination of glycopeptidase, exoglycosidases, and high-performance liquid chromatography. Anal. Biochem. 1987, 163, 489–499. [Google Scholar]
- Takahashi, N.; Ishii, I.; Ishihara, H.; Mori, M.; Tejima, S.; Jefferis, R.; Endo, S.; Arata, Y. Comparative structural study of the N-linked oligosaccharides of human normal and pathological immunoglobulin G. Biochemistry 1987, 26, 1137–1144. [Google Scholar]
- Tai, T.; Yamashita, K.; Ogata-Arakawa, M.; Koide, N.; Muramatsu, T.; Iwashita, S.; Inoue, Y.; Kobata, A. Structural studies of two ovalbumin glycopeptides in relation to the endo-beta-N-acetylglucosaminidase specificity. J. Biol. Chem. 1975, 250, 8569–8575. [Google Scholar]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurogochi, M.; Amano, J. Relative Quantitation of Glycopeptides Based on Stable Isotope Labeling Using MALDI-TOF MS. Molecules 2014, 19, 9944-9961. https://doi.org/10.3390/molecules19079944
Kurogochi M, Amano J. Relative Quantitation of Glycopeptides Based on Stable Isotope Labeling Using MALDI-TOF MS. Molecules. 2014; 19(7):9944-9961. https://doi.org/10.3390/molecules19079944
Chicago/Turabian StyleKurogochi, Masaki, and Junko Amano. 2014. "Relative Quantitation of Glycopeptides Based on Stable Isotope Labeling Using MALDI-TOF MS" Molecules 19, no. 7: 9944-9961. https://doi.org/10.3390/molecules19079944